Genome-Wide Identification of Insertion and Deletion Markers in Chinese Commercial Rice Cultivars, Based on Next-Generation Sequencing Data

Abstract
:1. Introduction
2. Results
2.1. Genome Re-Sequencing and Analysis
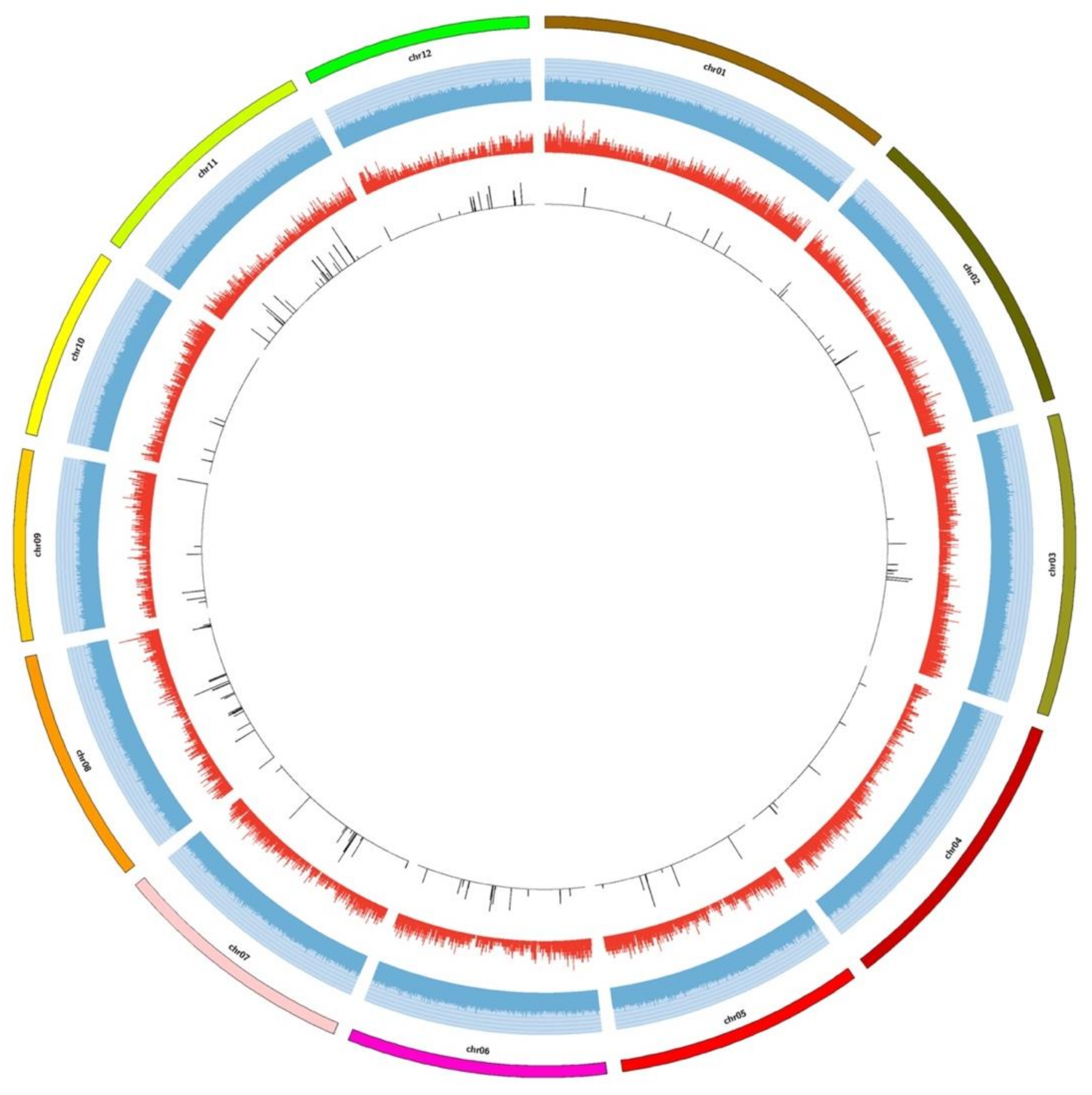
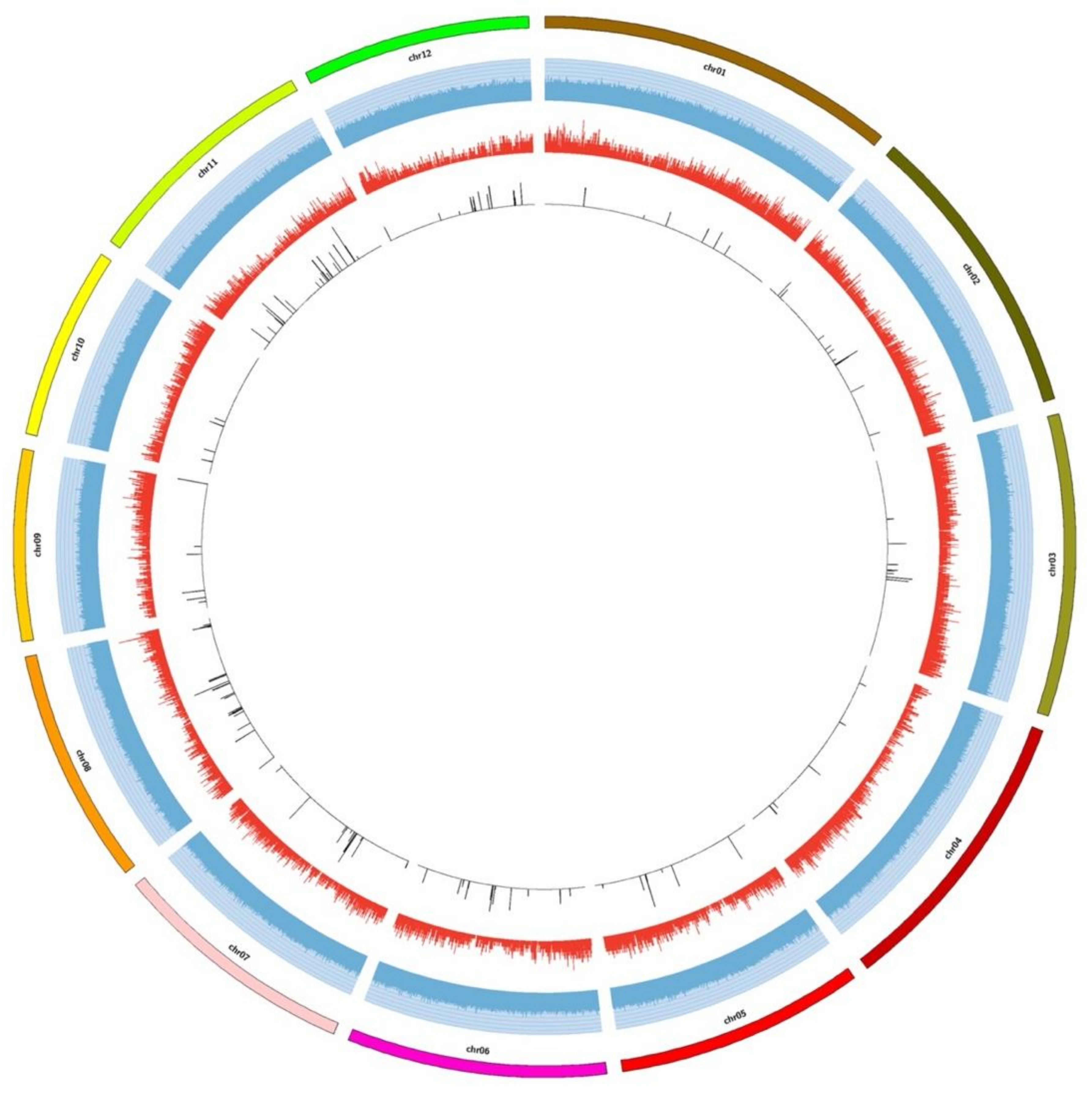
2.2. Distribution of SNP and InDel Markers
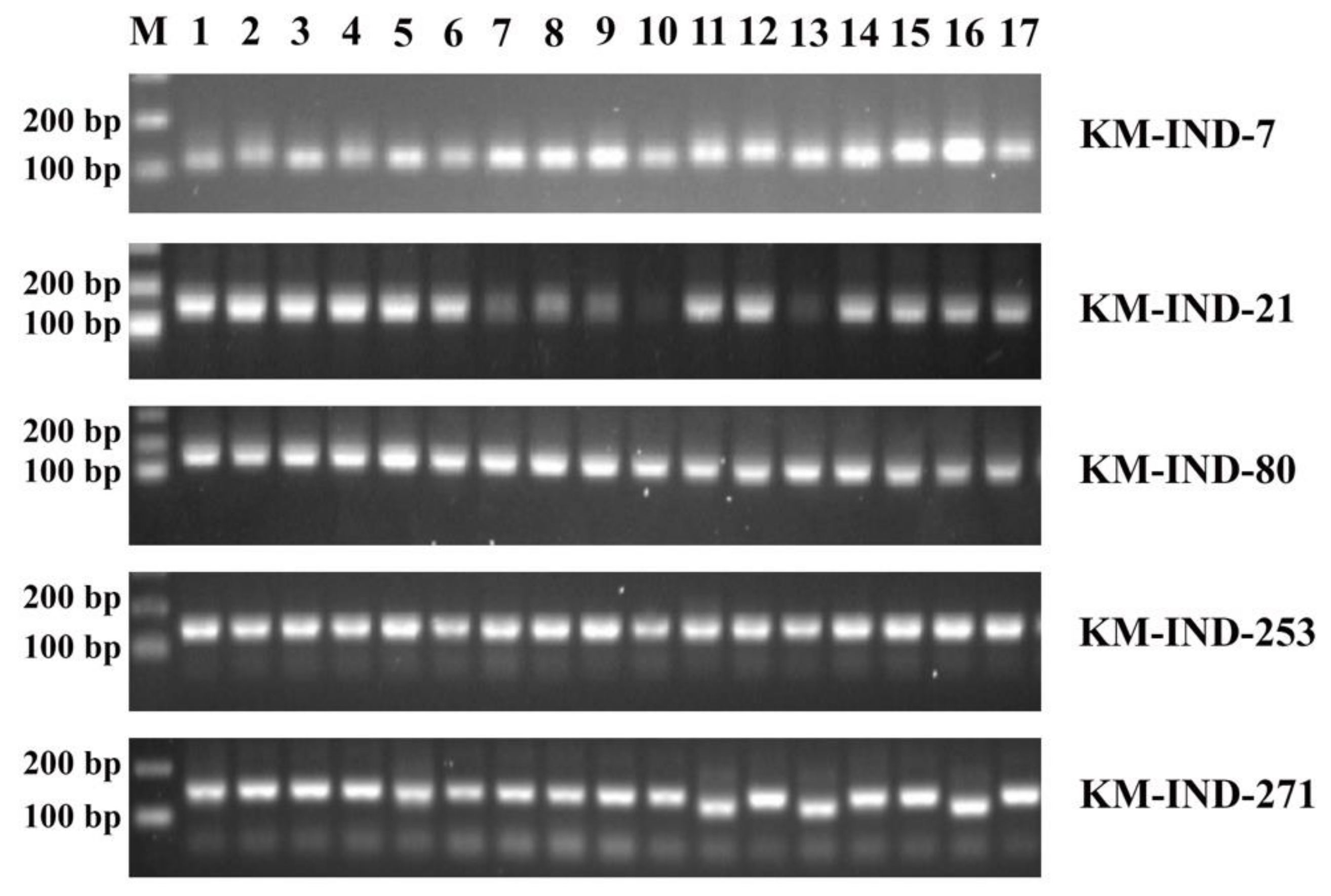
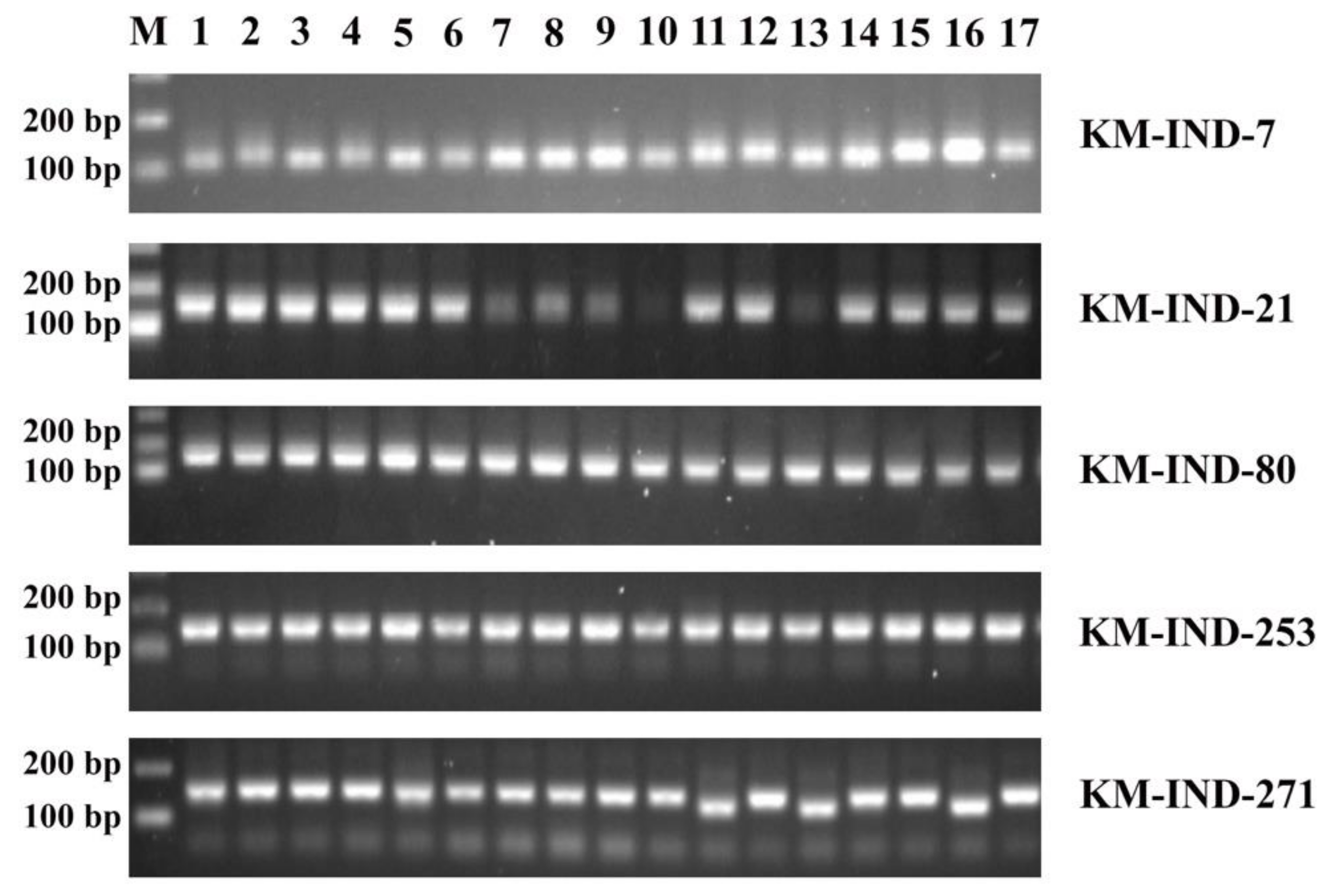
2.3. Experimental Validation of Five InDel Markers
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Isolation and Genome Sequencing
4.3. Mapping of Reads to the Reference
4.4. Detection of SNPs and InDels
4.5. Primer Design for Common InDel Markers
4.6. PCR Validation
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vemireddy, L.R.; Satyavathi, V.; Siddiq, E.; Nagaraju, J. Review of methods for the detection and quantification of adulteration of rice: Basmati as a case study. J. Food Sci. Technol. 2015, 52, 3187–3202. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.M.; Shabir, G.; Aslam, K.; Sabar, M.; Arif, M. Identification of SSR markers to find adulteration in elite basmati rice varieties. Environ. Plant Syst. 2015, 1, 4–15. [Google Scholar]
- Ghoshray, A. Asymmetric adjustment of rice export prices: The case of Thailand and Vietnam. Int. J. Appl. Econ. 2008, 5, 80–91. [Google Scholar]
- Vemireddy, L.R.; Noor, S.; Satyavathi, V.; Srividhya, A.; Kaliappan, A.; Parimala, S.; Bharathi, P.M.; Deborah, D.A.; Rao, K.S.; Shobharani, N. Discovery and mapping of genomic regions governing economically important traits of Basmati rice. BMC Plant Biol. 2015, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Colyer, A.; Macarthur, R.; Lloyd, J.; Hird, H. Comparison of calibration methods for the quantification of Basmati and non-Basmati rice using microsatellite analysis. Food Addit. Contam. 2008, 25, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S. Food adulteration affecting the nutrition and health of human beings. J. Biol. Sci. Med. 2016, 1, 65–70. [Google Scholar]
- Vlachos, A.; Arvanitoyannis, I.S. A review of rice authenticity/adulteration methods and results. Crit. Rev. Food Sci. Nutr. 2008, 48, 553–598. [Google Scholar] [CrossRef] [PubMed]
- Lees, M. Food Authenticity and Traceability; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Fontanesi, L. Genetic authentication and traceability of food products of animal origin: New developments and perspectives. Ital. J. Anim. Sci. 2009, 8 (Suppl. 2), 9–18. [Google Scholar] [CrossRef]
- Cho, K.-S.; Hong, S.-Y.; Yun, B.-K.; Won, H.-S.; Yoon, Y.-H.; Kwon, K.-B.; Mekapogu, M. Application of InDel Markers Based on the Chloroplast Genome Sequences for Authentication and Traceability of Tartary and Common Buckwheat. Czech J. Food Sci. 2017, 35, 122–130. [Google Scholar]
- Wen, J.; Zeng, L.; Sun, Y.; Chen, D.; Xu, Y.; Luo, P.; Zhao, Z.; Yu, Z.; Fan, S. Authentication and traceability of fish maw products from the market using DNA sequencing. Food Control 2015, 55, 185–189. [Google Scholar] [CrossRef]
- Chuang, H.-Y.; Lur, H.-S.; Hwu, K.-K.; Chang, M.-C. Authentication of domestic Taiwan rice varieties based on fingerprinting analysis of microsatellite DNA markers. Bot. Stud. 2011, 52, 393–405. [Google Scholar]
- Choi, S.; Dyck, J.; Childs, N. The Rice Market in South Korea; US Department of Agriculture: Washington, DC, USA, 2016. [Google Scholar]
- Chen, H.; Xie, W.; He, H.; Yu, H.; Chen, W.; Li, J.; Yu, R.; Yao, Y.; Zhang, W.; He, Y. A high-density SNP genotyping array for rice biology and molecular breeding. Mol. Plant 2014, 7, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Wang, J.; Zeigler, R.S. The 3000 rice genomes project: new opportunities and challenges for future rice research. GigaScience 2014, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-H.; Wu, H.-P.; Wang, C.-S.; Tseng, H.-Y.; Hwu, K.-K. Genome-wide InDel marker system for application in rice breeding and mapping studies. Euphytica 2013, 192, 131–143. [Google Scholar] [CrossRef]
- Yamaki, S.; Ohyanagi, H.; Yamasaki, M.; Eiguchi, M.; Miyabayashi, T.; Kubo, T.; Kurata, N.; Nonomura, K.-I. Development of INDEL markers to discriminate all genome types rapidly in the genus Oryza. Breed. Sci. 2013, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Wen, Z.; Ma, L.; Ji, Z.; Li, X.; Yang, C. Development of 1047 insertion-deletion markers for rice genetic studies and breeding. Genet. Mol. Res. 2013, 12, 5226–5235. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; Krueger, F.; Seconds-Pichon, A.; Biggins, F.; Wingett, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2012. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Strömberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucl. Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| No. | Chinese Rice Brand | Sequencing Data from All Reads | Mapped Sequence Data within the Genome | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sequence Reads | Clean Reads | Mapped Reads | Coverage (%) | Unique Reads | Unique Rate (%) | Average Depth | Coverage 30× (%) | ||
| 1 | Su Jung Mi | 105,072,772 | 97,876,196 | 93,872,807 | 97.19 | 79,787,991 | 82.6 | 25.09 | 19.28 |
| 2 | Do Hwa Hyang | 119,758,074 | 114,440,050 | 107,607,456 | 96.65 | 90,245,722 | 81.06 | 28.81 | 34.46 |
| 3 | Kum Gaeng Do | 107,139,768 | 102,746,548 | 96,570,831 | 96.61 | 81,492,231 | 81.53 | 25.86 | 22.14 |
| 4 | Saeng Tae Mi | 112,162,556 | 107,100,578 | 101,215,436 | 97.03 | 86,086,576 | 82.52 | 27.1 | 29.43 |
| 5 | Won Rip Gaeng Mi | 116,136,382 | 111,158,652 | 104,855,221 | 96.89 | 88,455,622 | 81.74 | 28.09 | 30.66 |
| 6 | Jang Rip Gaeng Mi | 131,387,698 | 125,670,080 | 118,530,326 | 97.02 | 100,048,014 | 81.89 | 31.74 | 49.55 |
| 7 | Cheang Hang Do | 126,197,574 | 121,035,598 | 113,822,239 | 96.72 | 96,046,869 | 81.62 | 30.48 | 42.86 |
| 8 | Geum Yong Eo-Kum Dea Mi | 120,031,414 | 114,699,600 | 108,137,537 | 96.75 | 91,889,823 | 82.22 | 28.94 | 38.27 |
| 9 | Hae Jeon-Kum Dea Mi | 122,675,242 | 117,511,332 | 110,851,176 | 96.9 | 94,164,685 | 82.31 | 29.68 | 41.92 |
| 10 | Mi-Kum Ju-Kum Dea Mi | 114,614,342 | 109,943,262 | 103,868,290 | 96.97 | 88,517,369 | 82.64 | 27.81 | 33.88 |
| 11 | Hang Dea Mi | 111,689,172 | 107,197,300 | 101,449,431 | 97.16 | 86,253,129 | 82.61 | 27.17 | 30.82 |
| 12 | TeaJin Ju Mi | 107,910,318 | 102,761,240 | 96,597,250 | 96.69 | 81,906,982 | 81.99 | 25.85 | 23.68 |
| 13 | Kum Do Jean DongBuk Dea Mi | 118,302,364 | 112,489,548 | 105,661,252 | 96.18 | 90,882,775 | 82.73 | 28.29 | 36.03 |
| 14 | Wu Ju-DongBuk De Mi | 128,450,312 | 122,723,200 | 115,388,796 | 96.89 | 97,534,245 | 81.89 | 30.89 | 45.24 |
| 15 | Jung Rang | 122,307,360 | 117,089,646 | 111,105,640 | 97.43 | 95,651,329 | 83.87 | 29.78 | 42.25 |
| 16 | MMA | 108,964,330 | 104,319,578 | 97,686,547 | 96.49 | 82,451,900 | 81.44 | 26.16 | 22.46 |
| 17 | Sunrice-Sushi-rice | 120,588,040 | 115,051,754 | 108,964,490 | 97.33 | 92,837,652 | 82.92 | 29.2 | 37.83 |
| No. | Chinese Rice Brand | SNPs | InDels |
|---|---|---|---|
| 1 | Su Jung Mi | 785,394 | 106,624 |
| 2 | Do Hwa Hyang | 990,095 | 118,942 |
| 3 | Kum Gaeng Do | 902,411 | 106,352 |
| 4 | Saeng Tae Mi | 932,382 | 107,665 |
| 5 | Won Rip Gaeng Mi | 998,599 | 132,142 |
| 6 | Jang Rip Gaeng Mi | 878,930 | 105,025 |
| 7 | Cheang Hang Do | 1,039,436 | 132,780 |
| 8 | Geum Yong Eo-Kum Dea Mi | 1,006,468 | 120,292 |
| 9 | Hae Jeon-Kum Dea Mi | 999,562 | 112,545 |
| 10 | Mi-Kum Ju-Kum Dea Mi | 1,019,042 | 112,787 |
| 11 | Hang Dea Mi | 862,603 | 101,055 |
| 12 | TeaJin Ju Mi | 1,019,123 | 117,282 |
| 13 | Kum Do Jean DongBuk Dea Mi | 1,184,780 | 156,768 |
| 14 | Wu Ju-DongBuk De Mi | 991,872 | 118,880 |
| 15 | Jung Rang | 440,306 | 63,885 |
| 16 | MMA | 990,558 | 117,703 |
| 17 | Sunrice-Sushi-rice | 567,065 | 64,613 |
| No. | Primer ID | Forward Primer | Reverse Primer |
|---|---|---|---|
| 1 | KM-IND-7 | TCCCTTGTAGGCTCCTATCT | TCTCTCACGAGTGGAAAAAGCA |
| 2 | KM-IND-21 | CCCTTCTTCCTCTTCTTTCTTCCTA | ATTAAGGACGGAAATGTGGCAG |
| 3 | KM-IND-80 | CAGATGTGATGCGCAAGGC | TCATGGATTCCTGGTGCAAGTT |
| 4 | KM-IND-253 | GCTAATCTGCAACGGGTACATG | TGGAGCCCGAAAAGTGTTCATA |
| 5 | KM-IND-271 | CTCTGCTGCTGCTGCTGGAA | CGTCAAATCTCGACGAGCTCTT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markkandan, K.; Yoo, S.-i.; Cho, Y.-C.; Lee, D.W. Genome-Wide Identification of Insertion and Deletion Markers in Chinese Commercial Rice Cultivars, Based on Next-Generation Sequencing Data. Agronomy 2018, 8, 36. https://doi.org/10.3390/agronomy8040036
Markkandan K, Yoo S-i, Cho Y-C, Lee DW. Genome-Wide Identification of Insertion and Deletion Markers in Chinese Commercial Rice Cultivars, Based on Next-Generation Sequencing Data. Agronomy. 2018; 8(4):36. https://doi.org/10.3390/agronomy8040036
Chicago/Turabian StyleMarkkandan, Kesavan, Seung-il Yoo, Young-Chan Cho, and Dong Woo Lee. 2018. "Genome-Wide Identification of Insertion and Deletion Markers in Chinese Commercial Rice Cultivars, Based on Next-Generation Sequencing Data" Agronomy 8, no. 4: 36. https://doi.org/10.3390/agronomy8040036
APA StyleMarkkandan, K., Yoo, S.-i., Cho, Y.-C., & Lee, D. W. (2018). Genome-Wide Identification of Insertion and Deletion Markers in Chinese Commercial Rice Cultivars, Based on Next-Generation Sequencing Data. Agronomy, 8(4), 36. https://doi.org/10.3390/agronomy8040036