
Microbial Community Structure and Metabolic Potential Shape Soil-Mediated Resistance Against Fruit Flesh Spongy Tissue Disorder of Peach


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site and Sample Collection
2.2. Soil Physicochemical Analysis
2.3. Separation of Root-Associated Microbes
2.4. 16S rRNA Gene Amplicon Sequencing and Bioinformatic Analysis
2.5. Shotgun Metagenomic Sequencing and Metagenomic Binning
2.6. Data Visualization
3. Results
3.1. Physicochemical Differences in Soils with and Without Fruit Flesh Spongy Tissue Disorder of Peach
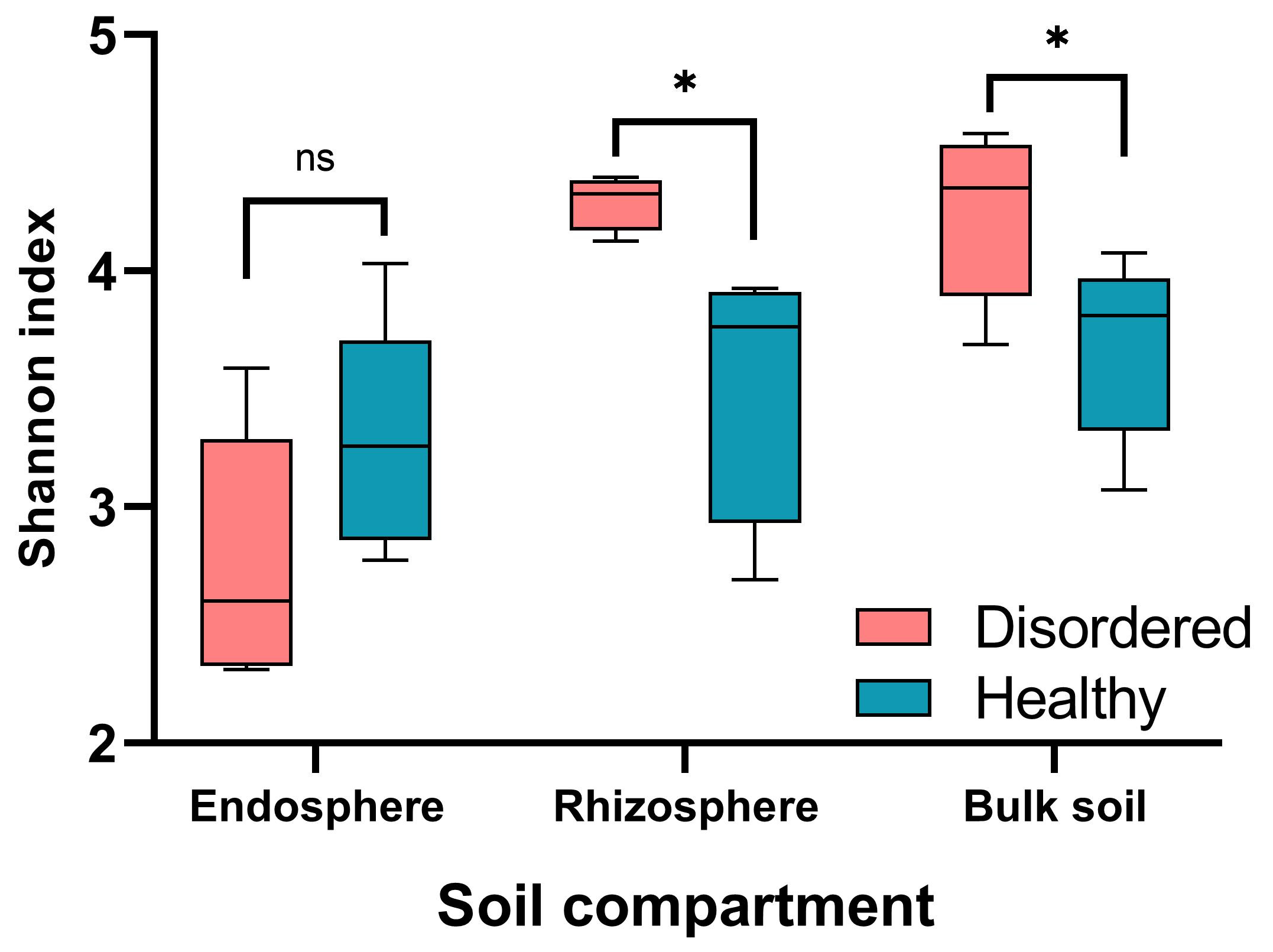
3.2. Diversity and Composition of Root-Associated Microbial Communities
3.3. Identification of Core Differential Bacteria in Rhizosphere
3.4. Co-Occurrence Networks of Rhizosphere Microbes
3.5. Metabolic Potential of Rhizosphere Microbes
3.6. Metabolic Mechanisms of Key Taxa Revealed by Metagenome Binning
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liao, Y.; Wang, L.; Zhang, Y.; Huang, S.; Li, C.; Liu, W. Investigation on Disease Condition of Spongy Tissue in Yingzui Peach. J. Anhui Agri. Sci. 2016, 44, 123–124. (In Chinese) [Google Scholar] [CrossRef]
- Lu, J.; Lin, X.; Liao, Y. Transcriptome sequencing analysis of differentially-expressed genes involved in the spongy tissue of Olecranon peach (Prunus persica L.). J. Fruit Sci. 2023, 40, 2524–2535. (In Chinese) [Google Scholar] [CrossRef]
- Ma, X.W.; Liu, B.; Zhang, Y.H.; Su, M.Q.; Zheng, B.; Wang, S.B.; Wu, H.X. Unraveling correlations between calcium deficiency and spongy tissue in mango fruit flesh. Sci. Hortic. 2023, 309, 111694. [Google Scholar] [CrossRef]
- Mirás-Avalos, J.M.; Alcobendas, R.; Alarcón, J.J.; Valsesia, P.; Génard, M.; Nicolás, E. Assessment of the water stress effects on peach fruit quality and size using a fruit tree model, QualiTree (vol 128C, pg 1, 2013). Agric. Water Manag. 2013, 130, 178. [Google Scholar] [CrossRef]
- Rahimi-Moghaddam, S.; Eyni-Nargeseh, H.; Ahmadi, S.A.K.; Azizi, K. Towards withholding irrigation regimes and drought-resistant genotypes as strategies to increase canola production in drought-prone environments: A modeling approach. Agric. Water Manag. 2021, 243, 106487. [Google Scholar] [CrossRef]
- Hartmann, M.; Six, J. Soil structure and microbiome functions in agroecosystems. Nat. Rev. Earth Environ. 2023, 4, 4–18. [Google Scholar] [CrossRef]
- Oak, P.; Jha, V.; Deshpande, A.; Tanpure, R.; Dawkar, V.; Mundhe, S.; Ghuge, S.; Prabhudesai, S.; Krishanpal, A.; Jere, A.; et al. Transcriptional and translational perturbation in abiotic stress induced physiological activities and metabolic pathway networks in spongy tissue disorder of mango fruit. Postharvest Biol. Technol. 2022, 188, 111880. [Google Scholar] [CrossRef]
- Manganaris, G.A.; Vicente, A.R.; Crisosto, C.H.; Labavitch, J.M. Cell wall modifications in chilling-injured plum fruit (Prunus salicina). Postharvest Biol. Technol. 2008, 48, 77–83. [Google Scholar] [CrossRef]
- Campi, P.; Gaeta, L.; Mastrorilli, M.; Losciale, P. Innovative Soil Management and Micro-Climate Modulation for Saving Water in Peach Orchards. Front. Plant Sci. 2020, 11, 1052. [Google Scholar] [CrossRef]
- Shi, Y.N.; Li, B.J.; Grierson, D.; Chen, K.S. Insights into cell wall changes during fruit softening from transgenic and naturally occurring mutants. Plant Physiol. 2023, 192, 1671–1683. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, Z.; Wang, Y.; Wang, J.; Zhai, R.; Lin-Wang, K.; Espley, R.; Ma, F.; Li, P. Competition between anthocyanin and kaempferol glycosides biosynthesis affects pollen tube growth and seed set of Malus. Hortic. Res. 2021, 8, 173. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Albizua, A.; Williams, A.; Hedlund, K.; Pascual, U. Crop rotations including ley and manure can promote ecosystem services in conventional farming systems. Appl. Soil Ecol. 2015, 95, 54–61. [Google Scholar] [CrossRef]
- Kong, Q.S.; Gao, L.Y.; Cao, L.; Liu, Y.; Saba, H.; Huang, Y.; Bie, Z.L. Assessment of Suitable Reference Genes for Quantitative Gene Expression Studies in Melon Fruits. Front. Plant Sci. 2016, 7, 1178. [Google Scholar] [CrossRef]
- Long, Q.X.; Yan, R.; Hu, J.L.; Cai, D.W.; Mitra, B.; Kim, E.S.; Marchetti, A.; Zhang, H.; Wang, S.J.; Liu, Y.J.; et al. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog. 2017, 13, e1006784. [Google Scholar] [CrossRef]
- Raval, S.S.; Mahatma, M.K.; Chakraborty, K.; Bishi, S.K.; Singh, A.L.; Rathod, K.J.; Jadav, J.K.; Sanghani, J.M.; Mandavia, M.K.; Gajera, H.P.; et al. Metabolomics of groundnut (Arachis hypogaea L.) genotypes under varying temperature regimes. Plant Growth Regul. 2018, 84, 493–505. [Google Scholar] [CrossRef]
- Wu, W.K.; Panyod, S.; Liu, P.Y.; Chen, C.C.; Kao, H.L.; Chuang, H.L.; Chen, Y.H.; Zou, H.B.; Kuo, H.C.; Kuo, C.H.; et al. Characterization of TMAO productivity from carnitine challenge facilitates personalized nutrition and microbiome signatures discovery. Microbiome 2020, 8, 162. [Google Scholar] [CrossRef]
- Shamrikova, E.V.; Kondratenok, B.M.; Tumanova, E.A.; Vanchikova, E.V.; Lapteva, E.M.; Zonova, T.V.; Lu-Lyan-Min, E.I.; Davydova, A.P.; Libohova, Z.; Suvannang, N. Transferability between soil organic matter measurement methods for database harmonization. Geoderma 2022, 412, 115547. [Google Scholar] [CrossRef]
- Elsgaard, L. Dynamics of mineral nitrogen, water-soluble carbon and potential nitrification in band-steamed arable soil. Biol. Fertil. Soils 2010, 46, 883–889. [Google Scholar] [CrossRef]
- Milham, P.J.; Jill, K.C.; and Holford, P. Selective Measurement of Phosphate in 0.5 M Sodium Bicarbonate Soil Extracts. Commun. Soil Sci. Plan 2024, 55, 529–535. [Google Scholar] [CrossRef]
- Ullah, R.; Abbas, Z.; Bilal, M.; Habib, F.; Iqbal, J.; Bashir, F.; Noor, S.; Qazi, M.A.; Niaz, A.; Baig, K.S.; et al. Method development and validation for the determination of potassium (K2O) in fertilizer samples by flame photometry technique. J. King Saud Univ. Sci. 2022, 34, 102070. [Google Scholar] [CrossRef]
- Caravajal, G.S.; Mahan, K.I.; Goforth, D.; Leyden, D.E. Evaluation of methods based on acid extraction and atomic absorption spectrometry for multi-element determinations in rivers sidements. Anal. Chim. Acta 1983, 147, 133–150. [Google Scholar] [CrossRef]
- Gao, H.B.; Guo, Z.H.; Xu, R.; He, X.; Fernio, J.U.; Li, S.K.; Liu, X.C.; Liu, H.X.; Xue, W.J. Chemolithoautotrophic Antimonite Oxidation Coupled Nitrogen Fixation in the Rhizosphere of Local Plant in Antimony Tailing Area. Environ. Sci. Technol. 2025, 59, 12703–12716. [Google Scholar] [CrossRef]
- Xu, Y.M.; Zhang, X.Y.; Yang, H.; Lu, D.L. Effects of Exogenous Brassinolide Application at the Silking Stage on Nutrient Accumulation, Translocation and Remobilization of Waxy Maize Under Post-Silking Heat Stress. Agriculture 2022, 12, 572. [Google Scholar] [CrossRef]
- Guo, Z.H.; Cao, J.; Xu, R.; Zhang, H.L.; He, L.L.; Gao, H.B.; Zhu, L.N.; Jia, M.Y.; Yang, Z.H.; Xiong, W.P. Novel Photoelectron-Assisted Microbial Reduction of Arsenate Driven by Photosensitive Dissolved Organic Matter in Mine Stream Sediments. Environ. Sci. Technol. 2024, 58, 22170–22182. [Google Scholar] [CrossRef]
- Hall, M.; Beiko, R.G. 16S rRNA Gene Analysis with QIIME2. In Microbiome Analysis: Methods and Protocols; Beiko, R.G., Hsiao, W., Parkinson, J., Eds.; Springer: New York, NY, USA, 2018; pp. 113–129. [Google Scholar]
- Chong, J.; Liu, P.; Zhou, G.Y.; Xia, J.G. Using Microbiome Analyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Serino, L.; Reimmann, C.; Visca, P.; Beyeler, M.; Chiesa, V.D.; Haas, D. Biosynthesis of pyochelin and dihydroaeruginoic acid requires the iron-regulated pchDCBA operon in Pseudomonas aeruginosa. J. Bacteriol. 1997, 179, 248–257. [Google Scholar] [CrossRef]
- Davidson, A.L.; Chen, J. ATP-binding cassette transporters in bacteria. Annu. Rev. Biochem. 2004, 73, 241–268. [Google Scholar] [CrossRef]
- Laville, J.; Blumer, C.; Von Schroetter, C.; Gaia, V.; Défago, G.; Keel, C.; Haas, D. Characterization of the hcnABC Gene Cluster Encoding Hydrogen Cyanide Synthase and Anaerobic Regulation by ANR in the Strictly Aerobic Biocontrol Agent Pseudomonas fluorescens CHA0. J. Bacteriol. 1998, 180, 3187–3196. [Google Scholar] [CrossRef]
- Peypoux, F.; Bonmatin, J.M.; Wallach, J. Recent trends in the biochemistry of surfactin. Appl. Microbiol. Biot. 1999, 51, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yao, S.; Shimizu, K. Effect of poxB gene knockout on metabolism in Escherichia coli based on growth characteristics and enzyme activities. World J. Microbiol. Biotechnol. 2007, 23, 573–580. [Google Scholar] [CrossRef]
- Wang, M.; Fu, J.; Zhang, X.; Chen, T. Metabolic engineering of Bacillus subtilis for enhanced production of acetoin. Biotechnol. Lett. 2012, 34, 1877–1885. [Google Scholar] [CrossRef]
- Kim, B.; Lee, S.; Yang, J.; Jeong, D.; Shin, S.H.; Kook, J.H.; Yang, K.S.; Lee, J. The influence of budA deletion on glucose metabolism related in 2,3-butanediol production by Klebsiella pneumoniae. Enzym. Microb. Technol. 2015, 73–74, 1–8. [Google Scholar] [CrossRef]
- Kanekura, K.; Hashimoto, Y.; Niikura, T.; Aiso, S.; Matsuoka, M.; Nishimoto, I. Alsin, the Product of ALS2 Gene, Suppresses SOD1 Mutant Neurotoxicity Through RhoGEF Domain by Interacting with SOD1 Mutants. J. Biol. Chem. 2004, 279, 19247–19256. [Google Scholar] [CrossRef]
- Ji, X.J.; Huang, H.; Ouyang, P.K. Microbial 2,3-butanediol production: A state-of-the-art review. Biotechnol. Adv. 2011, 29, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Kempf, B.; Bremer, E. Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Arch. Microbiol. 1998, 170, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Rath, H.; Reder, A.; Hoffmann, T.; Hammer, E.; Seubert, A.; Bremer, E.; Völker, U.; Mäder, U. Management of Osmoprotectant Uptake Hierarchy in Bacillus subtilis via a SigB-Dependent Antisense RNA. Front. Microbiol. 2020, 11, 622. [Google Scholar] [CrossRef]
- Hsieh, Y.J.; Wanner, B.L. Global regulation by the seven-component Pi signaling system. Curr. Opin. Microbiol. 2010, 13, 198–203. [Google Scholar] [CrossRef]
- Cox, G.B.; Webb, D.; Rosenberg, H. Specific amino acid residues in both the PstB and PstC proteins are required for phosphate transport by the Escherichia coli Pst system. J. Bacteriol. 1989, 171, 1531–1534. [Google Scholar] [CrossRef]
- Braibant, M.; De Wit, L.; Peirs, P.; Kalai, M.; Ooms, J.; Drowart, A.; Huygen, K.; Content, J. Structure of the Mycobacterium tuberculosis antigen 88, a protein related to the Escherichia coli PstA periplasmic phosphate permease subunit. Infect. Immun. 1994, 62, 849–854. [Google Scholar] [CrossRef]
- Malhotra, M.; Srivastava, S. An ipdC gene knock-out of Azospirillum brasilense strain SM and its implications on indole-3-acetic acid biosynthesis and plant growth promotion. Antonie Van Leeuwenhoek 2008, 93, 425–433. [Google Scholar] [CrossRef]
- Tikhonova, E.B.; Zgurskaya, H.I. AcrA, AcrB, and TolC of Escherichia coli Form a Stable Intermembrane Multidrug Efflux Complex. J. Biol. Chem. 2004, 279, 32116–32124. [Google Scholar] [CrossRef]
- Krithika, R.; Marathe, U.; Saxena, P.; Ansari, M.Z.; Mohanty, D.; Gokhale, R.S. A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 2069–2074. [Google Scholar] [CrossRef]
- Lamont, I.L.; Beare, P.A.; Ochsner, U.; Vasil, A.I.; Vasil, M.L. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2002, 99, 7072–7077. [Google Scholar] [CrossRef]
- Müller, K.; Matzanke, B.F.; Schünemann, V.; Trautwein, A.X.; Hantke, K. FhuF, an iron-regulated protein of Escherichia coli with a new type of [2Fe-2S] center. Eur. J. Biochem. 1998, 258, 1001–1008. [Google Scholar] [CrossRef]
- Shen, J.; Meldrum, A.; Poole, K. FpvA Receptor Involvement in Pyoverdine Biosynthesis in Pseudomonas aeruginosa. J. Bacteriol. 2002, 184, 3268–3275. [Google Scholar] [CrossRef]
- Bitter, W.; van Leeuwen, I.S.; de Boer, J.; Zomer, H.W.M.; Koster, M.C.; Weisbeek, P.J.; Tommassen, J. Localization of functional domains in the Escherichia coli coprogen receptor FhuE and the Pseudomonas putida ferric-pseudobactin 358 receptor PupA. Mol. Gen. Genet. MGG 1994, 245, 694–703. [Google Scholar] [CrossRef]
- Ma, Y.; Oliveira, R.S.; Nai, F.; Rajkumar, M.; Luo, Y.; Rocha, I.; Freitas, H. The hyperaccumulator Sedum plumbizincicola harbors metal-resistant endophytic bacteria that improve its phytoextraction capacity in multi-metal contaminated soil. J. Environ. Manag. 2015, 156, 62–69. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Braig, K.; Otwinowski, Z.; Hegde, R.; Boisvert, D.C.; Joachimiak, A.; Horwich, A.L.; Sigler, P.B. The crystal structure of the bacterial chaperonln GroEL at 2.8 Å. Nature 1994, 371, 578–586. [Google Scholar] [CrossRef]
- Michaux, C.; Holmqvist, E.; Vasicek, E.; Sharan, M.; Barquist, L.; Westermann, A.J.; Gunn, J.S.; Vogel, J. RNA target profiles direct the discovery of virulence functions for the cold-shock proteins CspC and CspE. Proc. Natl. Acad. Sci. USA 2017, 114, 6824–6829. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Zheng, W.; Crooke, E.; Wang, Y.-H.; Inouye, M. CspD, a novel DNA replication inhibitor induced during the stationary phase in Escherichia coli. Mol. Microbiol. 2001, 39, 1572–1584. [Google Scholar] [CrossRef]
- Jiang, W.; Hou, Y.; Inouye, M. CspA, the Major Cold-shock Protein of Escherichia coli, Is an RNA Chaperone. J. Biol. Chem. 1997, 272, 196–202. [Google Scholar] [CrossRef]
- Keyer, K.; Imlay, J.A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 1996, 93, 13635–13640. [Google Scholar] [CrossRef]
- Imlay, K.R.; Imlay, J.A. Cloning and analysis of sodC, encoding the copper-zinc superoxide dismutase of Escherichia coli. J. Bacteriol. 1996, 178, 2564–2571. [Google Scholar] [CrossRef]
- Newton, S.M.C.; Igo, J.D.; Scott, D.C.; Klebba, P.E. Effect of loop deletions on the binding and transport of ferric enterobactin by FepA. Mol. Microbiol. 1999, 32, 1153–1165. [Google Scholar] [CrossRef]
- Crosa Jorge, H.; Walsh Christopher, T. Genetics and Assembly Line Enzymology of Siderophore Biosynthesis in Bacteria. Microbiol. Mol. Biol. Rev. 2002, 66, 223–249. [Google Scholar] [CrossRef]
- Yap, W.H.; Li, X.; Soong, T.W.; Davies, J.E. Genetic diversity of soil microorganisms assessed by analysis of hsp70 (dnaK) sequences. J. Ind. Microbiol. 1996, 17, 179–184. [Google Scholar] [CrossRef]
- Beatrice, C.; Linthorst, J.M.H.; Cinzia, F.; Luca, R. Enhancement of PR1 and PR5 gene expressions by chitosan treatment in kiwifruit plants inoculated with Pseudomonas syringae pv. actinidiae. Eur. J. Plant Pathol. 2017, 148, 163–179. [Google Scholar] [CrossRef]
- Sikorski, M.M.; Biesiadka, J.; Kasperska, A.E.; Kopcińska, J.; Łotocka, B.; Golinowski, W.; Legocki, A.B. Expression of genes encoding PR10 class pathogenesis-related proteins is inhibited in yellow lupine root nodules. Plant Sci. 1999, 149, 125–137. [Google Scholar] [CrossRef]
- He, X.N.; Su, F.; Lou, Z.Z.; Jia, W.Z.; Song, Y.L.; Chang, H.Y.; Wu, Y.H.; Lan, J.; He, X.Y.; Zhang, Y. Ipr1 Gene Mediates RAW 264.7 Macrophage Cell Line Resistance to Mycobacterium bovis. Scand. J. Immunol. 2011, 74, 438–444. [Google Scholar] [CrossRef]
- Kizilova, A.K.; Titova, L.V.; Kravchenko, I.K.; Iutinskaya, G.A. Evaluation of the diversity of nitrogen-fixing bacteria in soybean rhizosphere by nifH gene analysis. Microbiology 2012, 81, 621–629. [Google Scholar] [CrossRef]
- Wärngård, L.; Fransson, R.; Drakenberg, T.B.; Flodström, S.; Ahlborg, U.G. Calmodulin involvement in TPA and DDT induced inhibition of intercellular communication. Chem. Biol. Interact. 1988, 65, 41–49. [Google Scholar] [CrossRef]
- Dong, J.; Signo, K.S.L.; Vanderlinde, E.M.; Yost, C.K.; Dahms, T.E.S. Atomic force microscopy of a ctpA mutant in Rhizobium leguminosarum reveals surface defects linking CtpA function to biofilm formation. Microbiology 2011, 157, 3049–3058. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; Paulitz, T.C.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- White, P.J.; Broadley, M.R. Calcium in Plants. Ann. Bot. 2003, 92, 487–511. [Google Scholar] [CrossRef]
- Liang, C.; Schimel, J.P.; Jastrow, J.D. The importance of anabolism in microbial control over soil carbon storage. Nat. Microbiol. 2017, 2, 17105. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Vismans, G.; Yu, K.; Song, Y.; de Jonge, R.; Burgman, W.P.; Burmolle, M.; Herschend, J.; Bakker, P.A.H.M.; Pieterse, C.M.J. Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 2018, 12, 1496–1507. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.M.; Singh, B.K. Plant-microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, H.F.; Chen, Y.; Wang, R.; Guo, S.L. Thirty-year dryland crop rotation improves soil multifunctionality and shifts soil fungal community. Plant Soil 2025, 507, 11–24. [Google Scholar] [CrossRef]
- Levy, A.; Gonzalez, I.S.; Mittelviefhaus, M.; Clingenpeel, S.; Paredes, S.H.; Miao, J.M.; Wang, K.R.; Devescovi, G.; Stillman, K.; Monteiro, F.; et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 2018, 50, 138. [Google Scholar] [CrossRef]
- Fu, X.P.; Liu, S.; Ru, J.R.; Tang, B.Y.; Zhai, Y.J.; Wang, Z.G.; Wang, L.C. Biological control of potato late blight by sp. FXP04 and potential role of secondary metabolites. Biol. Control 2022, 169, 104891. [Google Scholar] [CrossRef]
- Paredes-Páliz, K.; Rodríguez-Vázquez, R.; Duarte, B.; Caviedes, M.A.; Mateos-Naranjo, E.; Redondo-Gómez, S.; Caçador, M.I.; Rodríguez-Llorente, I.D.; Pajuelo, E. Investigating the mechanisms underlying phytoprotection by plant growth-promoting rhizobacteria in under metal stress. Plant Biol. 2018, 20, 497–506. [Google Scholar] [CrossRef]
- Cao, Y.F.; Shen, Z.Z.; Zhang, N.; Deng, X.H.; Thomashow, L.S.; Lidbury, I.; Liu, H.J.; Li, R.; Shen, Q.R.; Kowalchuk, G.A. Phosphorus availability influences disease-suppressive soil microbiome through plant-microbe interactions. Microbiome 2024, 12, 185. [Google Scholar] [CrossRef]
- Mazzola, M.; Manici, L.M. Apple Replant Disease: Role of Microbial Ecology in Cause and Control. Annu. Rev. Phytopathol. 2012, 50, 45–65. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Tang, D.; Huang, J.; Yang, Y.; Zhang, L. Microbial Community Structure and Metabolic Potential Shape Soil-Mediated Resistance Against Fruit Flesh Spongy Tissue Disorder of Peach. Agronomy 2025, 15, 1697. https://doi.org/10.3390/agronomy15071697
Chen W, Tang D, Huang J, Yang Y, Zhang L. Microbial Community Structure and Metabolic Potential Shape Soil-Mediated Resistance Against Fruit Flesh Spongy Tissue Disorder of Peach. Agronomy. 2025; 15(7):1697. https://doi.org/10.3390/agronomy15071697
Chicago/Turabian StyleChen, Weifeng, Dan Tang, Jia Huang, Yu Yang, and Liangbo Zhang. 2025. "Microbial Community Structure and Metabolic Potential Shape Soil-Mediated Resistance Against Fruit Flesh Spongy Tissue Disorder of Peach" Agronomy 15, no. 7: 1697. https://doi.org/10.3390/agronomy15071697
APA StyleChen, W., Tang, D., Huang, J., Yang, Y., & Zhang, L. (2025). Microbial Community Structure and Metabolic Potential Shape Soil-Mediated Resistance Against Fruit Flesh Spongy Tissue Disorder of Peach. Agronomy, 15(7), 1697. https://doi.org/10.3390/agronomy15071697