
Full-Length Transcriptome Analysis of Sesbania cannabina Stem Response to Waterlogging Stress


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Design
2.2. RNA Extraction and SMRT Sequencing
2.3. Data Processing and Basic Annotation
2.4. Prediction and Analysis of CDS, SSR, TF, LncRNA, AS Event, Functional Unit
2.5. Data Statistical Analysis
3. Results
3.1. General Properties of Sesbania Transcriptome
3.2. Analysis of CDS, TF, SSR, LncRNA, AS Event, Protein Domain Architecture, and Functional Motif
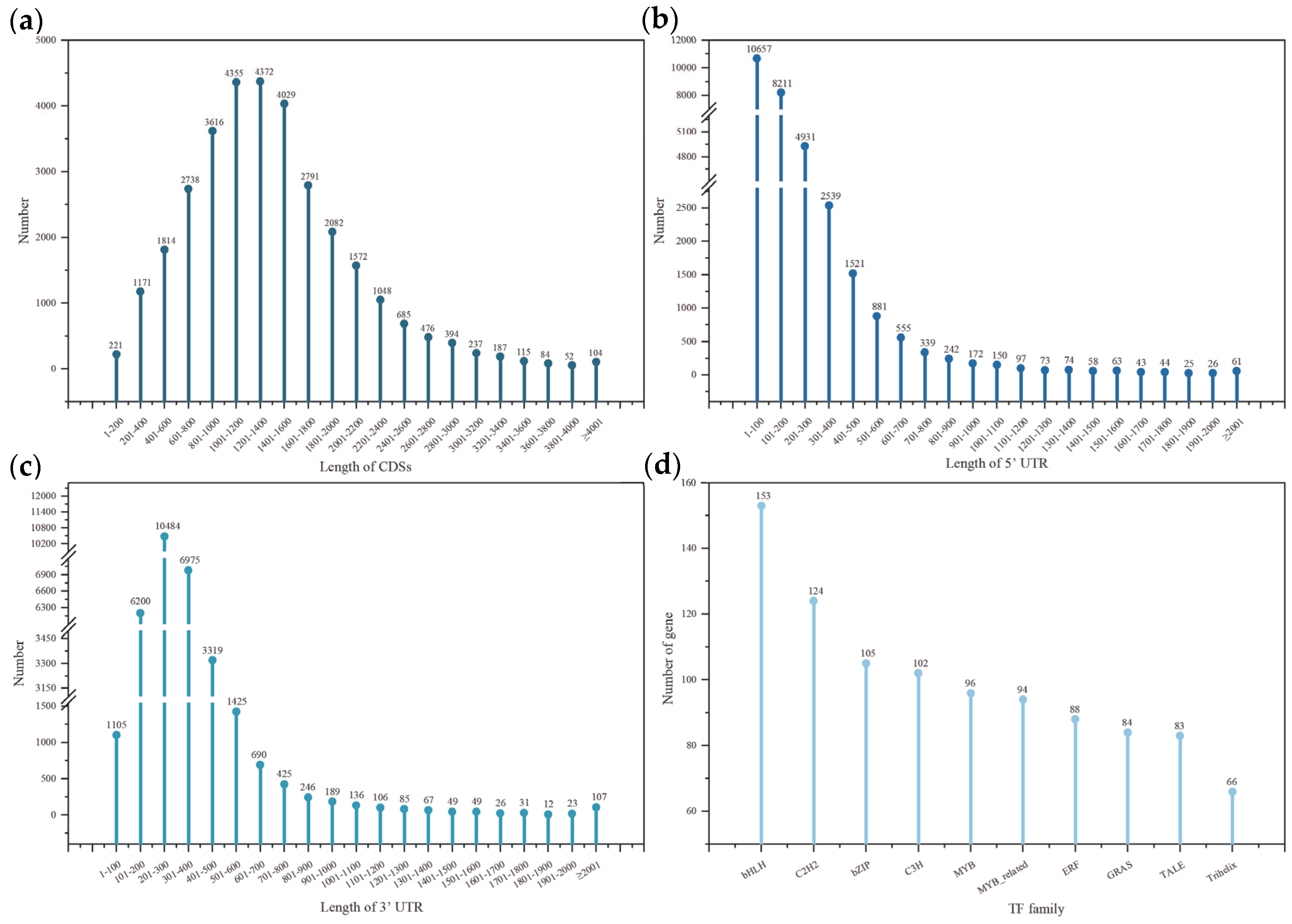
3.2.1. CDS
3.2.2. TF
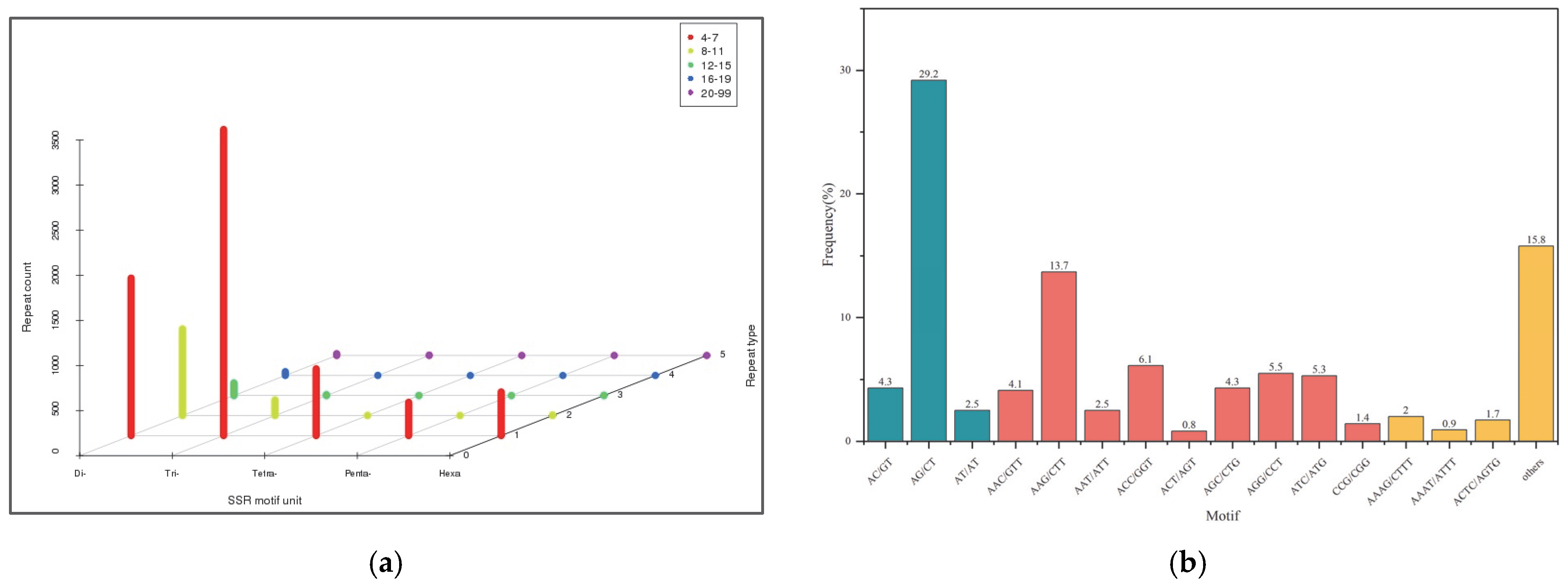
3.2.3. SSR
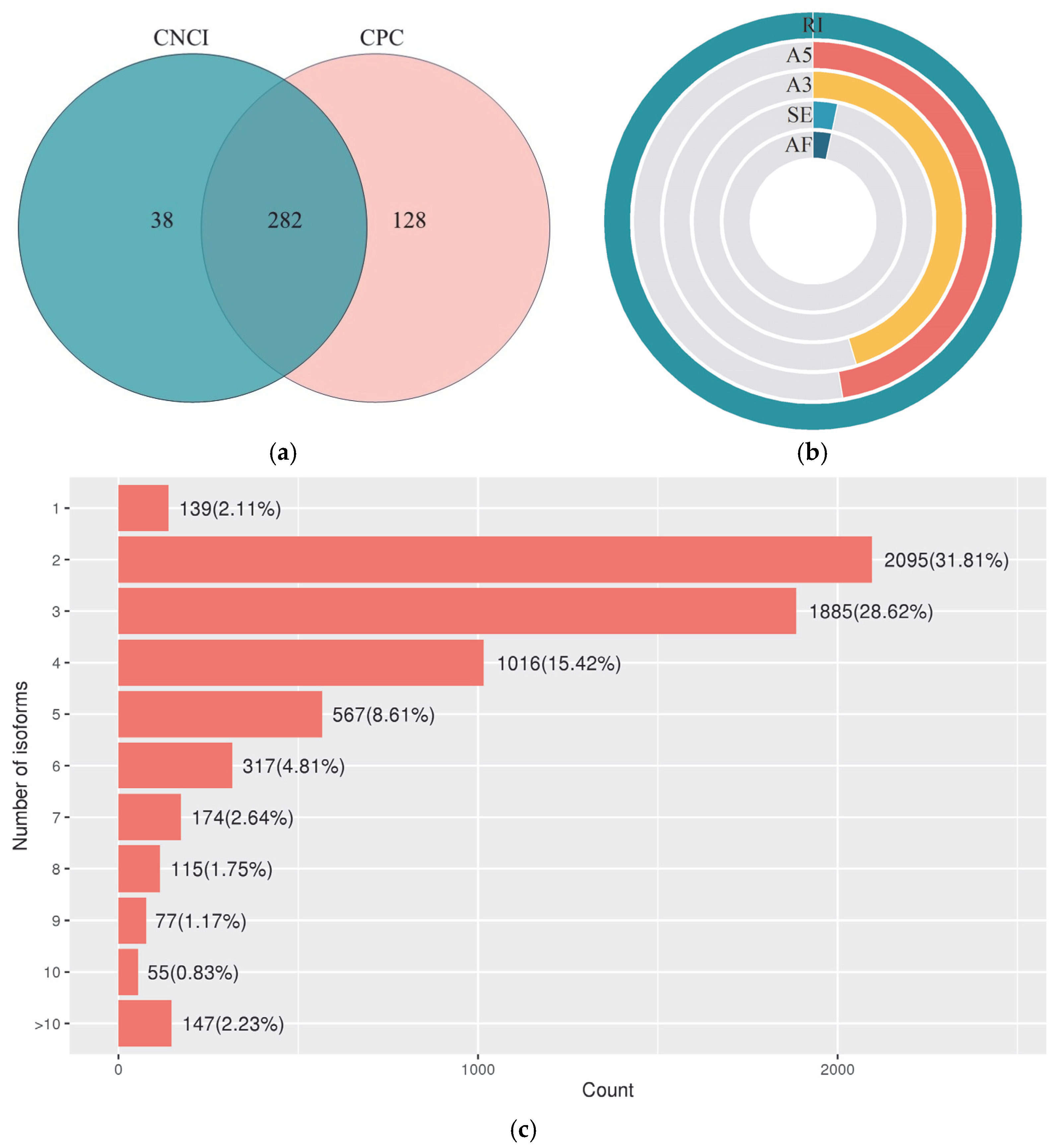
3.2.4. LncRNA and AS Event
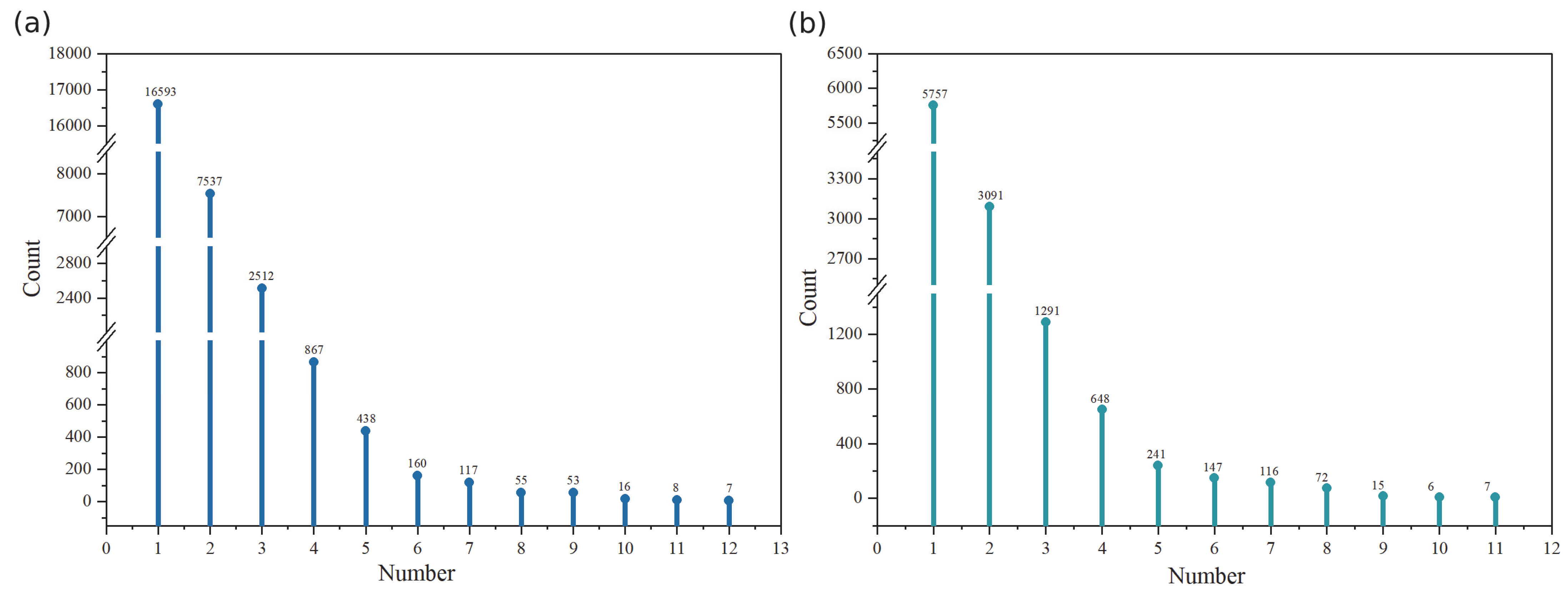
3.2.5. Functional Unit Prediction and Analysis
3.3. Functional Annotation
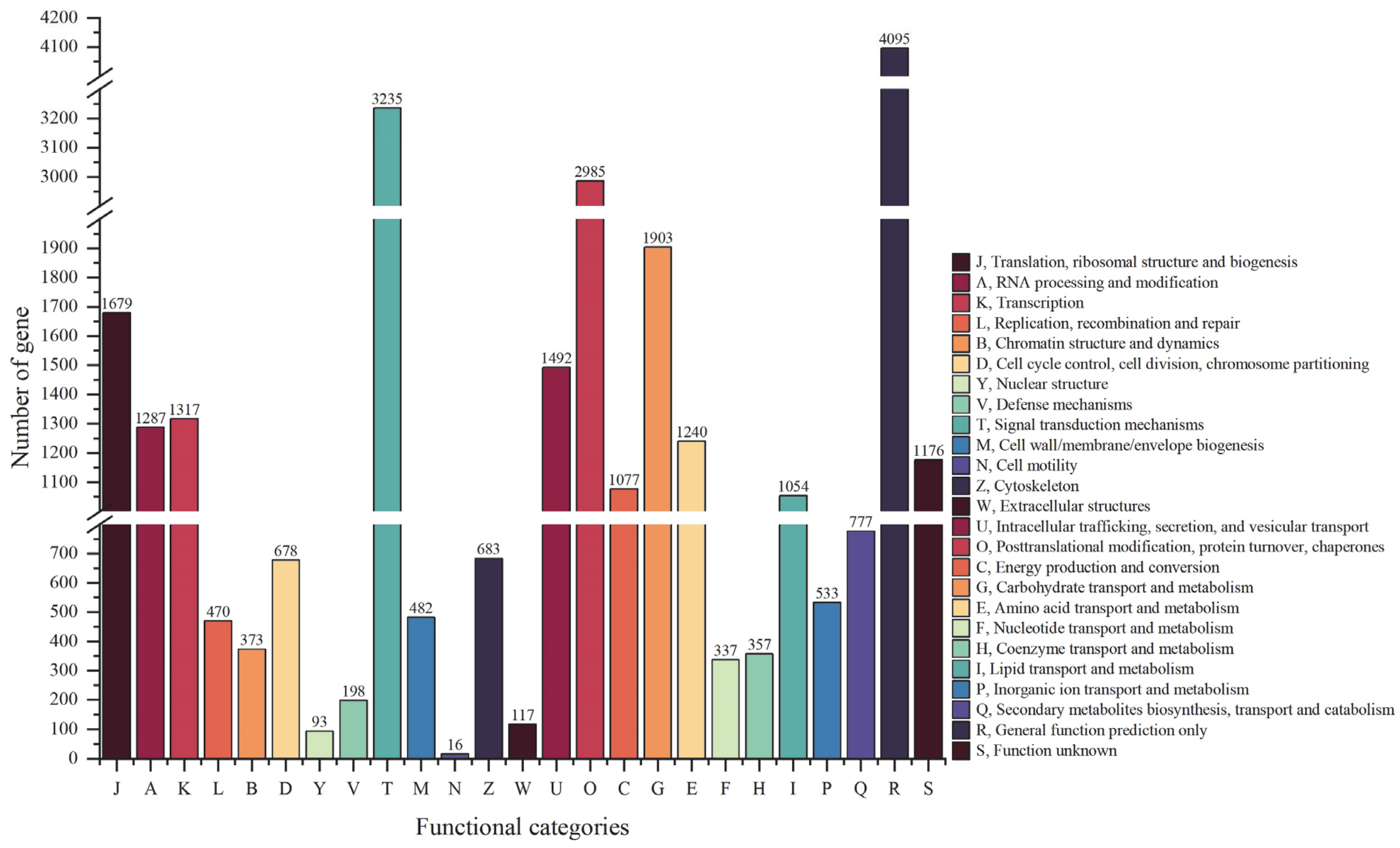
3.4. KOG Annotation
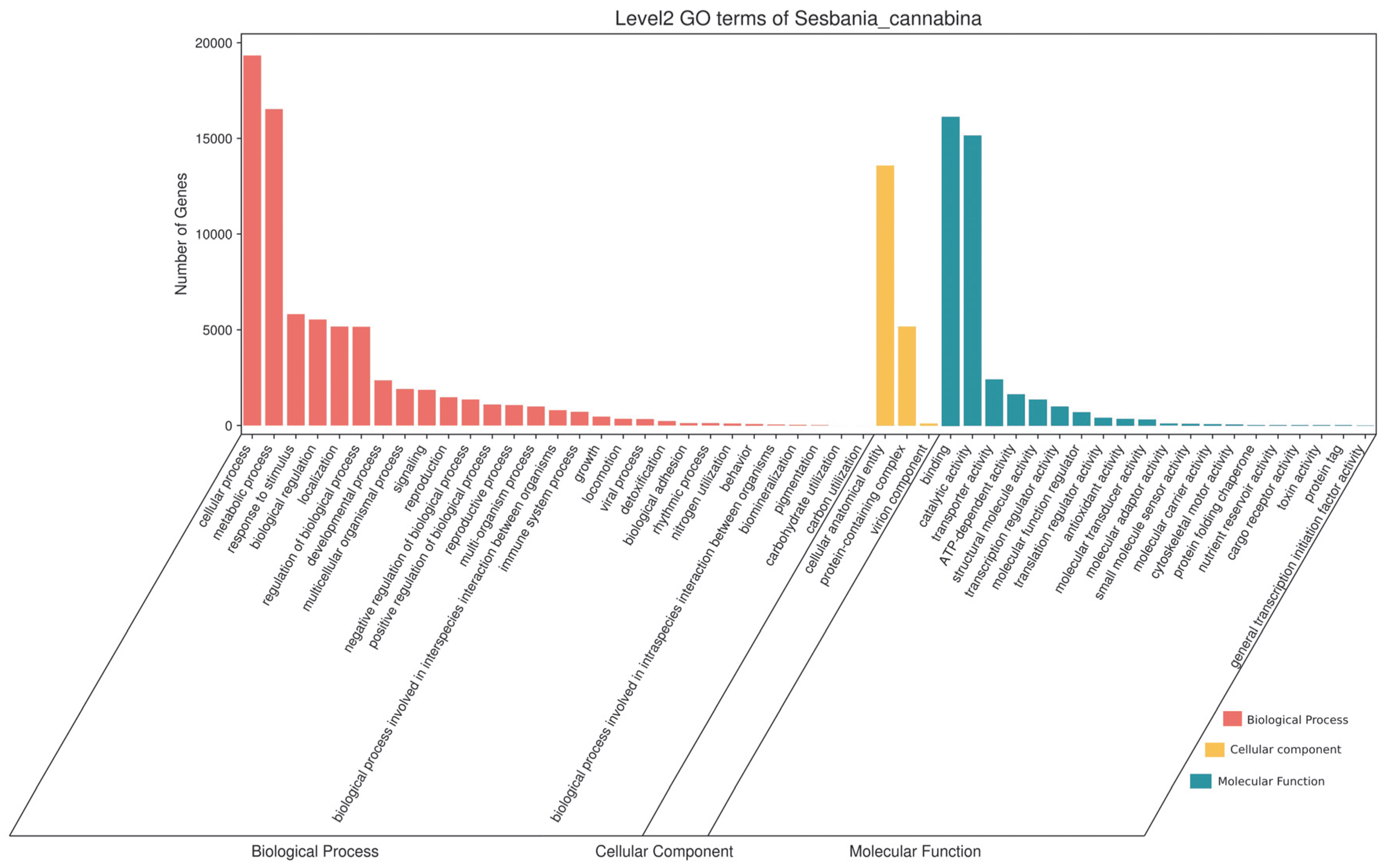
3.5. GO Annotation
3.6. KEGG Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, K.; Gera, R.; Sharma, R.; Maithani, D.; Chandra, D.; Bhat, M.A.; Kumar, R.; Bhatt, P. Mechanism and application of Sesbania root-nodulating bacteria: An alternative for chemical fertilizers and sustainable development. Arch. Microbiol. 2021, 203, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Bunma, S.; Balslev, H. A review of the economic botany of Sesbania (Leguminosae). Bot. Rev. 2019, 85, 185–251. [Google Scholar] [CrossRef]
- Jiang, P.; Tang, H.; Li, Y.; Liu, X. Effect of particle size of Sesbania gum on its modification, structure and performances. Int. J. Biol. Macromol. 2024, 262 Pt 2, 129719. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.; Hartman, S. How plant roots respond to waterlogging. J. Exp. Bot. 2024, 75, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Olorunwa, O.J.; Adhikari, B.; Shi, A.; Barickman, T.C. Screening of cowpea (Vigna unguiculata (L.) Walp.) genotypes for waterlogging tolerance using morpho-physiological traits at early growth stage. Plant Sci. 2022, 315, 111136. [Google Scholar] [CrossRef]
- Molla, M.; Rohman, M.; Islam, M.; Hasanuzzaman, M.; Hassan, L. Screening and evaluation of chilli (Capsicum annuum L.) genotypes for waterlogging tolerance at seedling stage. Biocell 2022, 46, 1613–1627. [Google Scholar] [CrossRef]
- Rajendran, A.; Ramlal, A.; Harika, A.; Subramaniam, S.; Raju, D.; Lal, S.K. Waterlogging stress mechanism and membrane transporters in soybean (Glycine max (L.) Merr.). Plant Physiol. Biochem. 2025, 220, 109579. [Google Scholar] [CrossRef]
- Krishnan, H.B.; Oehrle, N.W.; Alaswad, A.A.; Stevens, W.G.; Mari John, K.M.; Luthria, D.L.; Natarajan, S.S. Biochemical and anatomical investigation of Sesbania herbacea (Mill.) McVaugh nodules grown under flooded and non–flooded conditions. Int. J. Mol. Sci. 2019, 20, 1824. [Google Scholar] [CrossRef]
- Ren, C.G.; Kong, C.C.; Yan, K.; Zhang, H.; Luo, Y.M.; Xie, Z.H. Elucidation of the molecular responses to waterlogging in Sesbania cannabina roots by transcriptome profiling. Sci. Rep. 2017, 7, 9256. [Google Scholar] [CrossRef]
- Geng, S.; Lin, Z.; Xie, S.; Xiao, J.; Wang, H.; Zhao, X.; Zhou, Y.; Duan, L. Ethylene enhanced waterlogging tolerance by changing root architecture and inducing aerenchyma formation in maize seedlings. J. Plant Physiol. 2023, 287, 154042. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, C.; Pang, J.; Niu, Y.; Liu, H.; Zhang, W.; Zhou, M. Genome-wide association study reveals quantitative trait loci for waterlogging-triggered adventitious roots and aerenchyma formation in common wheat. Front. Plant Sci. 2022, 13, 1066752. [Google Scholar] [CrossRef] [PubMed]
- Elena, L.; Hans, V.; Pierdomenico, P. Plant responses to fooding stress. Curr. Opin. Plant Biol. 2016, 33, 64–71. [Google Scholar] [CrossRef]
- Ren, C.G.; Kong, C.C.; Xie, Z.H. Role of abscisic acid in strigolactone-induced salt stress tolerance in arbuscular mycorrhizal Sesbania cannabina seedlings. BMC Plant Biol. 2018, 18, 74. [Google Scholar] [CrossRef]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Hu, X.F.; Jin, M.J.; Gong, Z.X.; Lin, Z.L.; Zhang, L.Z.; Zeng, Z.J.; Wang, Z.L. Full-length transcriptome profile of Apis cerana revealed by nanopore sequencing. Int. J. Mol. Sci. 2024, 25, 10833. [Google Scholar] [CrossRef]
- Xiong, Y.; Yang, J.; Xiong, Y.; Zhao, J.; Liu, L.; Liu, W.; Sha, L.; Zhou, J.; You, M.; Li, D.; et al. Full-length transcriptome sequencing analysis and characterization, development and validation of microsatellite markers in Kengyilia melanthera. Front. Plant Sci. 2022, 13, 959042. [Google Scholar] [CrossRef]
- Liao, T.; Zhang, L.; Wang, Y. Full-length transcriptome characterization of Platycladus orientalis based on the PacBio platform. Front. Genet. 2024, 18, 1345039. [Google Scholar] [CrossRef]
- Wang, L.; Sa, W.; Yin, W.; Wei, W.; Wang, Y.; Sa, W.; Liang, J. Single-molecule real-time sequencing of the full-length transcriptome of purple garlic (Allium sativum L. cv. Leduzipi) and identification of serine O-acetyltransferase family proteins involved in cysteine biosynthesis. J. Sci. Food Agric. 2022, 102, 2864–2873. [Google Scholar] [CrossRef]
- Zhang, X.; Li, G.; Zhou, J.; Lv, M.; Li, L.; Chen, J. Full-length gonad transcriptome analysis of Amur sturgeon Dmrt family genes: Identification, characterization, and expression patterns during gonadal differentiation. Fish Physiol. Biochem. 2022, 48, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Xu, Q.T.; Yang, L.; Zhou, Z.Q.; Mei, F.Z.; Qu, L.H.; Zhou, G.S. Process of aerenchyma formation and reactive oxygen species induced by waterlogging in wheat seminal roots. Planta 2013, 238, 969–982. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Tuan, P.A.; Mukherjee, S.; Son, S.; Ayele, B.T. Hormonal regulation in adventitious roots and during their emergence under waterlogged conditions in wheat. J. Exp. Bot. 2018, 69, 4065–4082. [Google Scholar] [CrossRef]
- Nagai, K.; Ashikari, M. Molecular mechanism of internode elongation in rice. Breed Sci. 2023, 73, 108–116. [Google Scholar] [CrossRef]
- Xu, X.; Liu, M.; Hu, Q.; Yan, W.; Pan, J.; Yan, Y.; Chen, X. A CsEIL3-CsARN6.1 module promotes waterlogging-triggered adventitious root formation in cucumber by activating the expression of CsPrx5. Plant J. 2023, 114, 824–835. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, J.; Wang, Z.; Li, X.; Kan, J.; Li, J. SMRT sequencing of full-length transcriptome of birch-leaf pear (Pyrus betulifolia Bunge) under drought stress. J. Genet. 2021, 100, 29. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, L.; Yang, L.; Hao, Z.; Chen, J.; Cheng, T. The full-length transcriptome sequencing and identification of Na+/H+ antiporter genes in halophyte Nitraria tangutorum Bobrov. Genes 2021, 12, 836. [Google Scholar] [CrossRef]
- He, W.; Luo, L.; Xie, R.; Chai, J.; Wang, H.; Wang, Y.; Chen, Q.; Wu, Z.; Yang, S.; Li, M.; et al. Genome-Wide identification and functional analysis of the AP2/ERF transcription factor family in citrus rootstock under waterlogging stress. Int. J. Mol. Sci. 2023, 24, 8989. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, P.; Zhou, T.; Wu, Y.; Yuan, M.; Zhang, X.; Liu, Y. Genome-wide characterization and expression analysis of the bHLH gene family in response to abiotic stresses in Zingiber officinale Roscoe. BMC Genom. 2025, 26, 143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Yang, L.; Niu, J.; Huang, R.; Yuan, F.; Liang, Q. Development of SSR and SNP markers for identifying opium poppy. Int. J. Legal Med. 2022, 136, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Z.; Hu, A.; Wu, H.; Zhu, J.; Wang, F.; Cao, P.; Yang, X.; Zhang, H. Full-length transcriptome analysis of the halophyte Nitraria sibirica Pall. Genes 2022, 13, 661. [Google Scholar] [CrossRef]
- Tian, R.; Sun, X.; Liu, C.; Chu, J.; Zhao, M.; Zhang, W.H. A Medicago truncatula lncRNA MtCIR1 negatively regulates response to salt stress. Planta 2023, 257, 32. [Google Scholar] [CrossRef]
- Zhu, Z.; Dong, Q.; Bing, J.; Songbuerbatu; Zheng, L.; Dorjee, T.; Liu, Q.; Zhou, Y.; Gao, F. Combined lncRNA and mRNA expression profiles identified the lncRNA-miRNA-mRNA modules regulating the cold stress response in Ammopiptanthus nanus. Int. J. Mol. Sci. 2023, 24, 6502. [Google Scholar] [CrossRef]
- Tong, C.; Hill, C.B.; Zhou, G.; Zhang, X.Q.; Jia, Y.; Li, C. Opportunities for improving waterlogging tolerance in cereal crops-physiological traits and genetic mechanisms. Plants 2021, 10, 1560. [Google Scholar] [CrossRef]
- Lin, C.; Sauter, M. Polar auxin transport determines adventitious root emergence and growth in rice. Front. Plant Sci. 2019, 10, 444. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Annotated Transcripts |
|---|---|
| Nr | 32,002 |
| KEGG | 31,831 |
| KOG | 21,555 |
| SwissProt | 27,811 |
| annotation isoforms | 32,022 |
| without annotation isoform | 481 |
| Total transcripts | 32,503 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, T.; Sun, G.; He, S.; Zhang, Z.; Dong, J.; Zhu, X.; Dai, J.; Wang, K.; Xing, J. Full-Length Transcriptome Analysis of Sesbania cannabina Stem Response to Waterlogging Stress. Agronomy 2025, 15, 1197. https://doi.org/10.3390/agronomy15051197
He T, Sun G, He S, Zhang Z, Dong J, Zhu X, Dai J, Wang K, Xing J. Full-Length Transcriptome Analysis of Sesbania cannabina Stem Response to Waterlogging Stress. Agronomy. 2025; 15(5):1197. https://doi.org/10.3390/agronomy15051197
Chicago/Turabian StyleHe, Tingting, Guoli Sun, Sunan He, Zhenhua Zhang, Jing Dong, Xiaomei Zhu, Jinying Dai, Kai Wang, and Jincheng Xing. 2025. "Full-Length Transcriptome Analysis of Sesbania cannabina Stem Response to Waterlogging Stress" Agronomy 15, no. 5: 1197. https://doi.org/10.3390/agronomy15051197
APA StyleHe, T., Sun, G., He, S., Zhang, Z., Dong, J., Zhu, X., Dai, J., Wang, K., & Xing, J. (2025). Full-Length Transcriptome Analysis of Sesbania cannabina Stem Response to Waterlogging Stress. Agronomy, 15(5), 1197. https://doi.org/10.3390/agronomy15051197