
A Comprehensive Analysis of the Multiple AP2/ERF Regulatory Network Unveils Putative Components of the Fatty Acid Pathway for Environmental Adaptation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Genomes and Transcriptomes Mining
2.2. Systematic Identification of the AP2/ERF Gene Family
2.3. Orthogroups Identification of Multispecies AP2/ERFs
2.4. Sequence Alignment, TimeTree, and Phylogenetic Tree Construction
2.5. Systematic Analysis of AP2/ERF Gene Structure
2.6. Chromosome Distribution, Syntenic Analysis, and Comparative Genomics
2.7. Identification of AP2/ERFs Gene Types and Development-Related Phenotypic Analysis
2.8. Gene Ontology (GO) Analysis and Correlation Network Construction
2.9. Gene-Module Association Determination (G-MAD)
2.10. Construction of CqAP2/ERFs Expression Profiles
3. Results
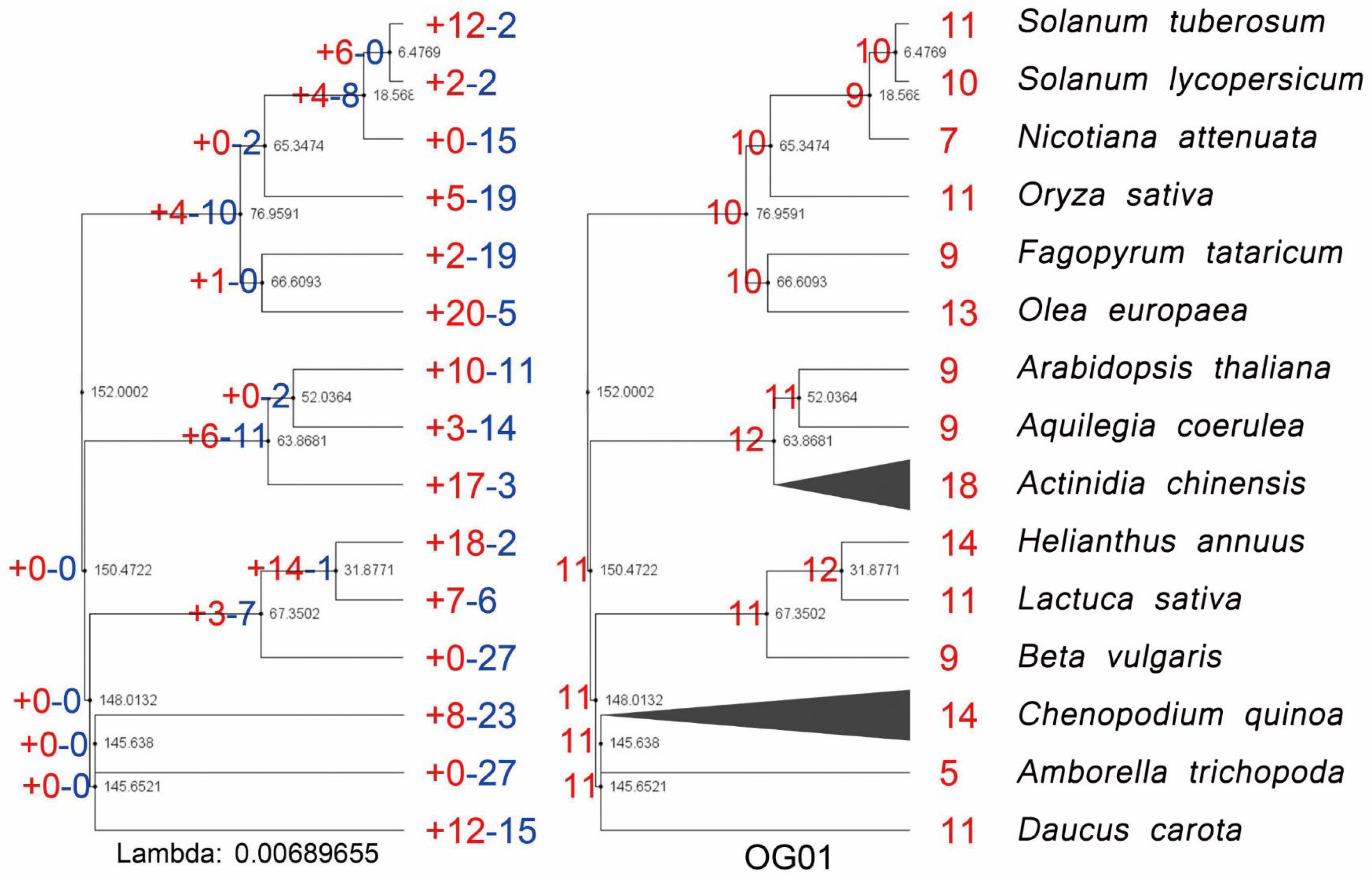
3.1. Phylogenetic Identification, Evolutionary Expansion, and Contraction of AP2/ERFs in Important Plants
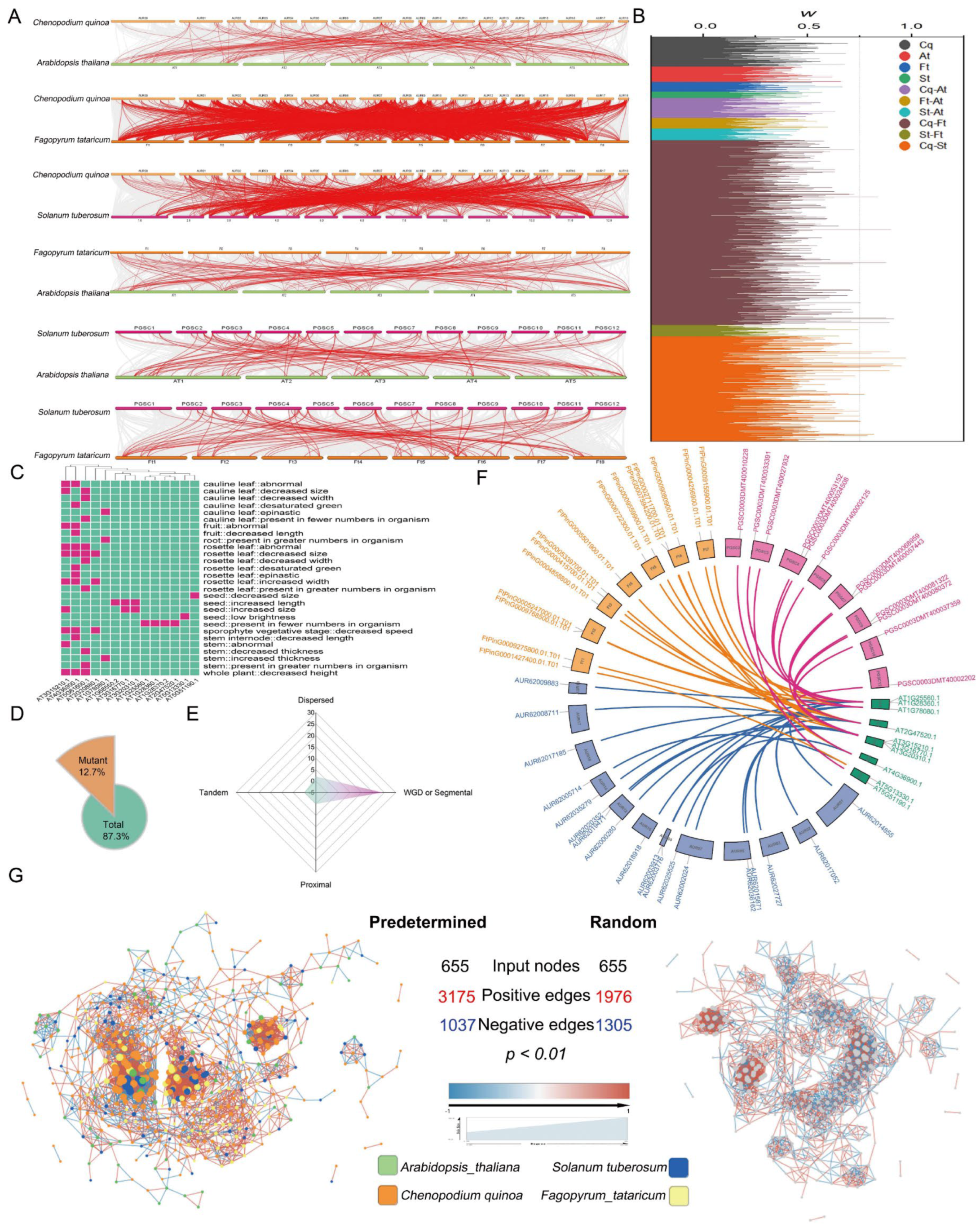
3.2. The Expansion of AP2/ERFs in Important Plants Caused by Recursive Amplification
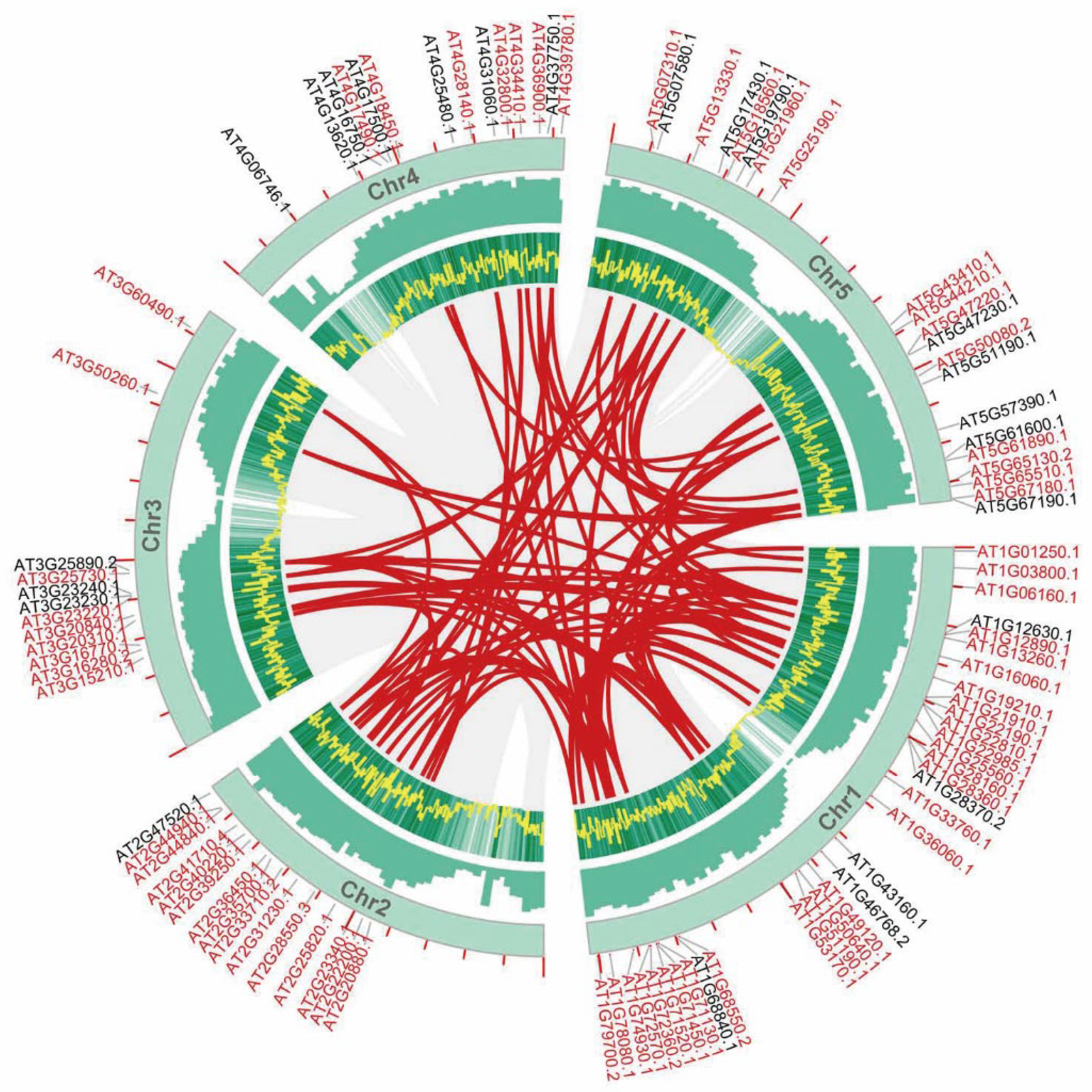
3.3. Genomic Synteny Conservation of AP2/ERFs in Important Plants
3.4. Phenotypic Analysis of AtAP2/ERFs and Mapping to Important AP2/ERFs in Other Species
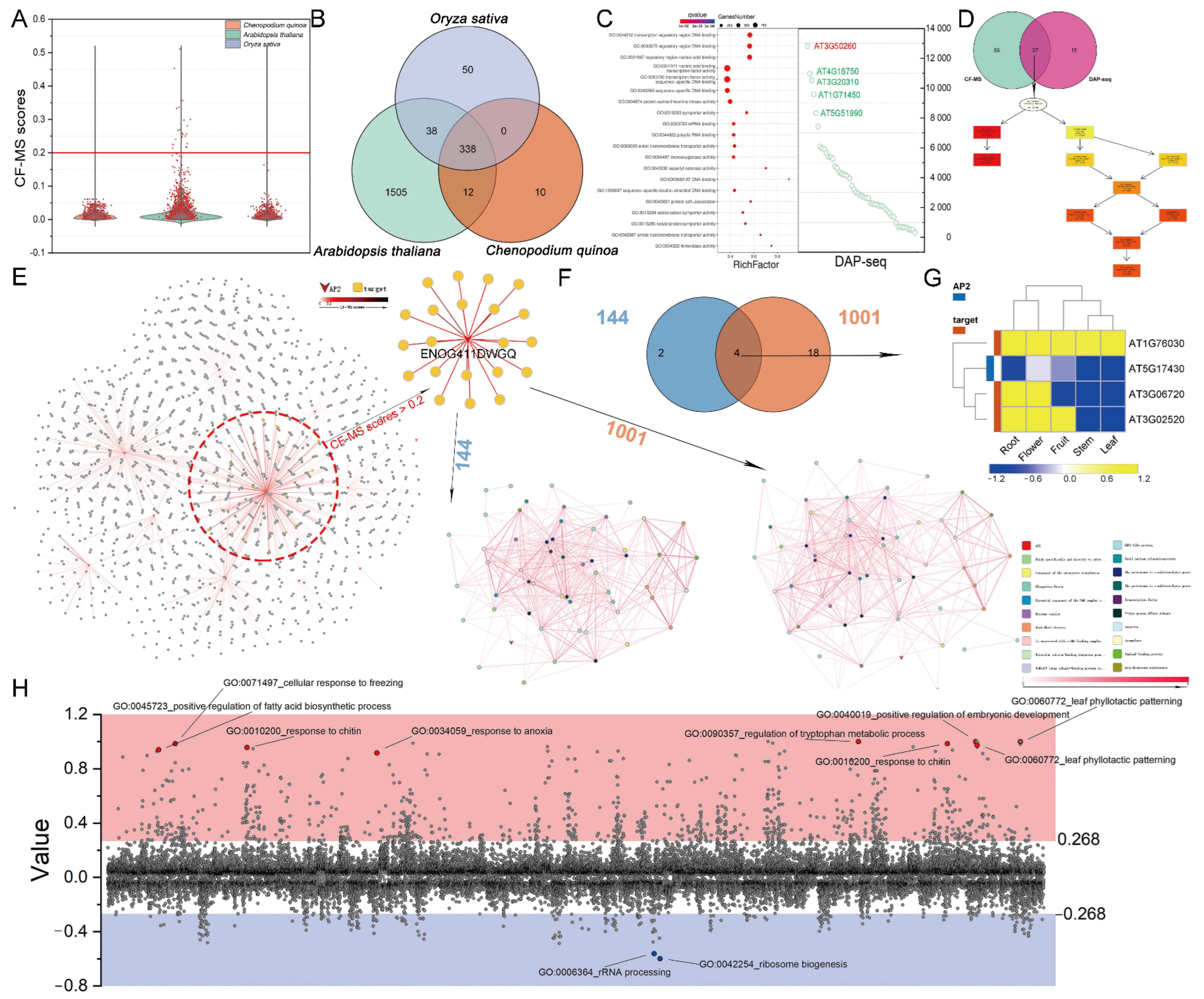
3.5. CF-MS and DNA Affinity Purification Sequencing (DAP-Seq) Determine the Functions of AP2/ERFs
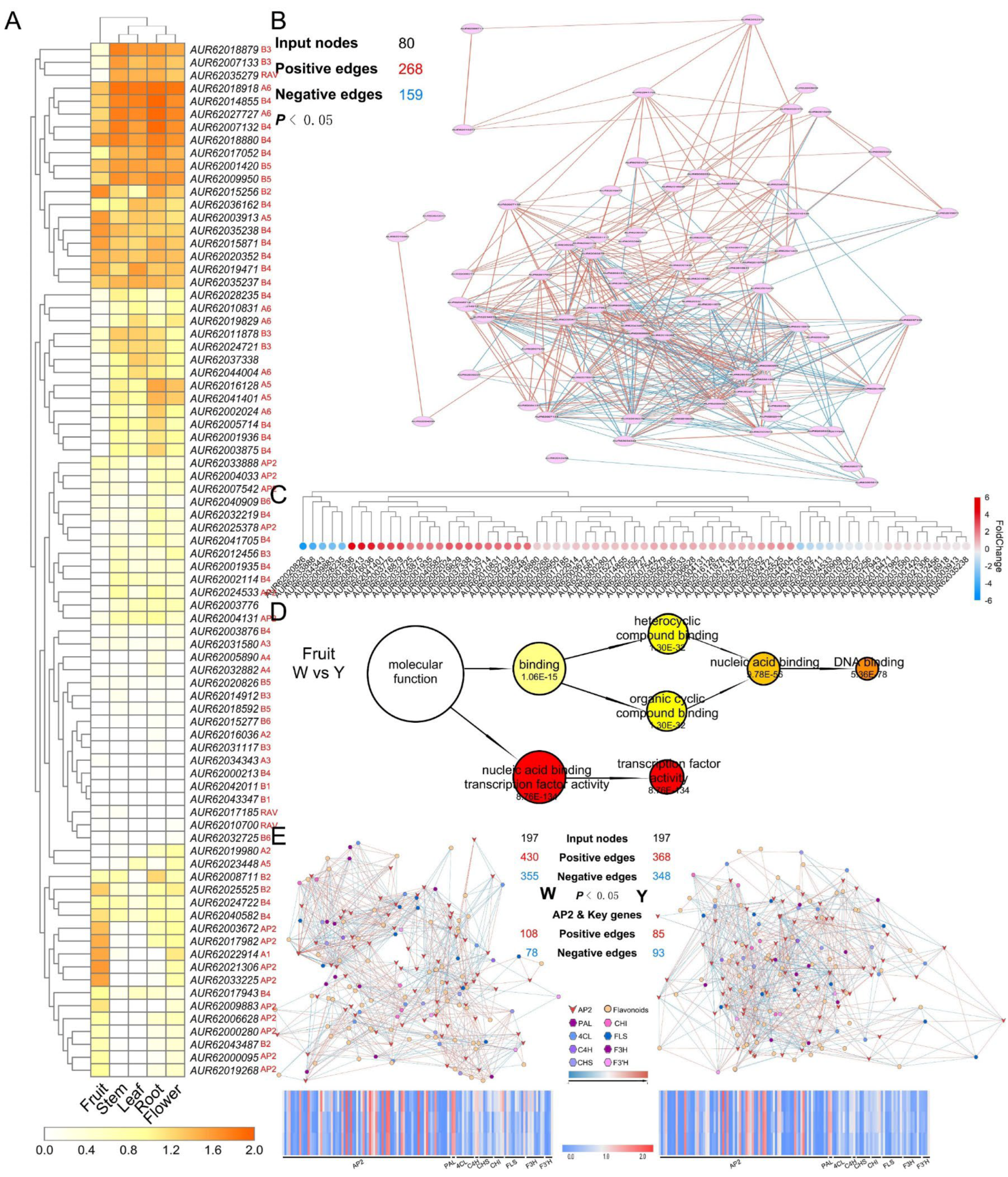
3.6. Polyploidy-Related CqAP2/ERFs May Promote Environmental Adaptability by Regulating the Fatty Acid Synthesis Pathway
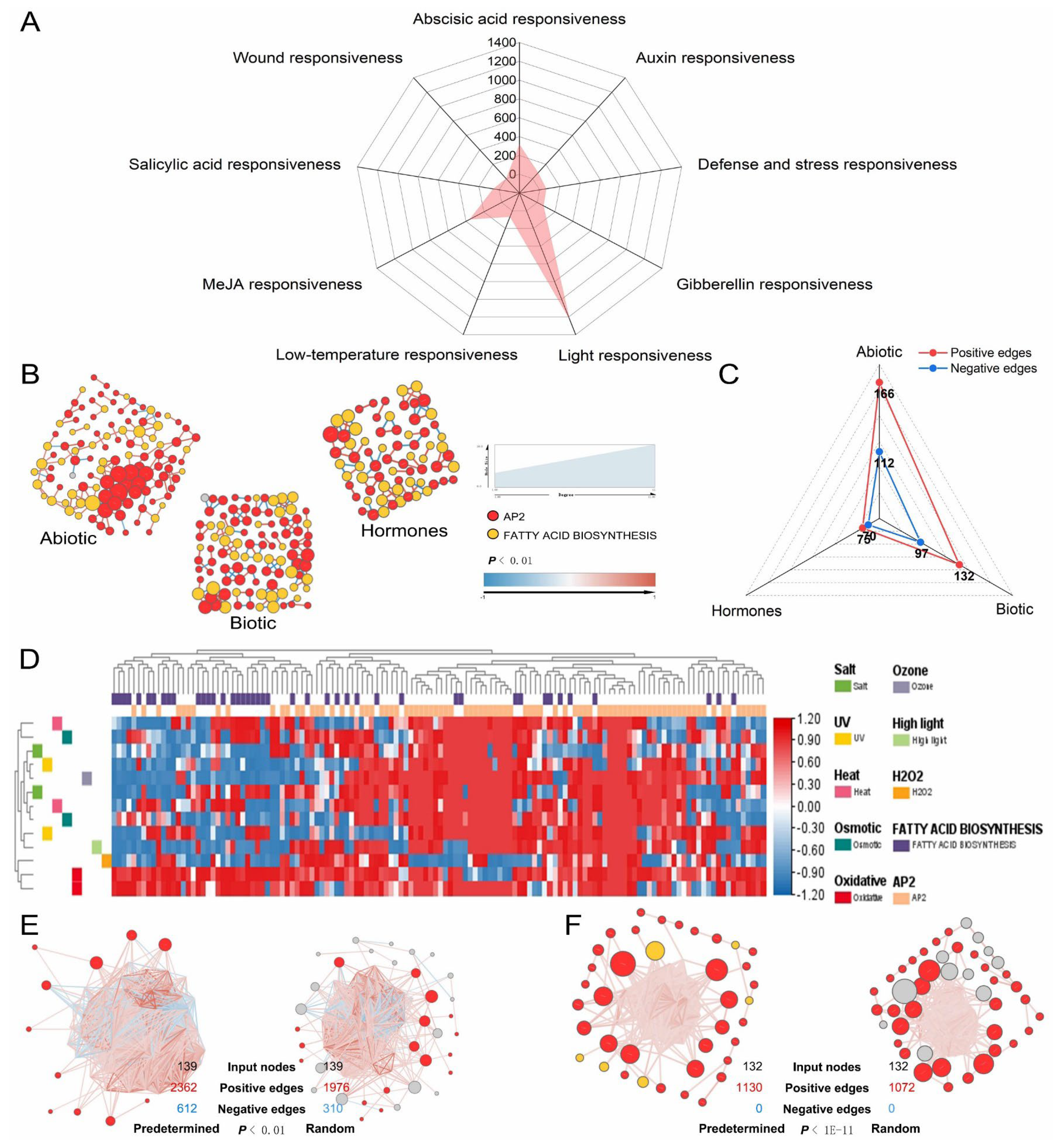
3.7. General Applicability Network of AP2/ERFs and Fatty Acid Pathway Genes
4. Discussion
4.1. The Evolution of Auxiliary Motifs in AP2/ERFs and Their Neofunctionalization
4.2. AP2/ERFs Generated by Amplification May Facilitate Environmental Adaptation
4.3. Comparative Perspectives and Breeding Prospects of AP2/ERF Expansion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guyomarc’h, S.; Boutté, Y.; Laplaze, L. AP2/ERF transcription factors orchestrate very long chain fatty acid biosynthesis during Arabidopsis lateral root development. Mol. Plant 2021, 14, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Xue, L.; Cheng, J.; Yang, X.; Xie, H.; Song, X.; Qiang, S. Polyploidization-driven differentiation of freezing tolerance in Solidago canadensis. Plant Cell Environ. 2020, 43, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, K.; Dai, X.; Qin, G.; Lu, D.; Gao, Z.; Li, X.; Song, B.; Bian, J.; Ren, D.; et al. A telomere-to-telomere genome assembly coupled with multi-omic data provides insights into the evolution of hexaploid bread wheat. Nat. Genet. 2025, 57, 1008–1020. [Google Scholar] [CrossRef] [PubMed]
- Soltis, P.S.; Soltis, D.E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef]
- Liu, M.; Yu, J.; Yang, M.; Cao, L.; Chen, C. Adaptive evolution of chloroplast division mechanisms during plant terrestrialization. Cell Rep. 2024, 43, 113950. [Google Scholar] [CrossRef]
- Xu, L.; Yang, L.; Li, A.; Guo, J.; Wang, H.; Qi, H.; Li, M.; Yang, P.; Song, S. An AP2/ERF transcription factor confers chilling tolerance in rice. Sci. Adv. 2024, 10, eado4788. [Google Scholar] [CrossRef]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Xie, Z.; Nolan, T.M.; Jiang, H.; Yin, Y. AP2/ERF Transcription Factor Regulatory Networks in Hormone and Abiotic Stress Responses in Arabidopsis. Front. Plant Sci. 2019, 10, 228. [Google Scholar] [CrossRef]
- Wang, Z.; Miao, L.; Chen, Y.; Peng, H.; Ni, Z.; Sun, Q.; Guo, W. Deciphering the evolution and complexity of wheat germplasm from a genomic perspective. J. Genet. Genom. 2023, 50, 846–860. [Google Scholar] [CrossRef]
- Yu, H.; Liao, J.; Jiang, Y.; Zhong, M.; Tao, S.; Chai, S.; Wang, L.; Lin, L.; Yang, R.; Deng, X.; et al. Ecotype-specific phenolic acid accumulation and root softness in Salvia miltiorrhiza are driven by environmental and genetic factors. Plant Biotechnol. J. 2025. [Google Scholar] [CrossRef]
- Münzbergová, Z.; Haisel, D. Effects of polyploidization on the contents of photosynthetic pigments are largely population-specific. Photosynth. Res. 2019, 140, 289–299. [Google Scholar] [CrossRef]
- Chen, Y.; Cui, Q.; Xu, Y.; Yang, S.; Gao, M.; Wang, Y. Effects of tung oilseed FAD2 and DGAT2 genes on unsaturated fatty acid accumulation in Rhodotorula glutinis and Arabidopsis thaliana. Mol. Genet. Genom. 2015, 290, 1605–1613. [Google Scholar] [CrossRef]
- Zuo, S.; Guo, X.; Mandáková, T.; Edginton, M.; Al-Shehbaz, I.A.; Lysak, M.A. Genome diploidization associates with cladogenesis, trait disparity, and plastid gene evolution. Plant Physiol. 2022, 190, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Magar, M.M.; Liu, H.; Yan, G. Genome-Wide Analysis of AP2/ERF Superfamily Genes in Contrasting Wheat Genotypes Reveals Heat Stress-Related Candidate Genes. Front. Plant Sci. 2022, 13, 853086. [Google Scholar] [CrossRef]
- Xiao, L.; Ren, J.-Z.; Li, Q.; Yang, B.; Liu, Z.-J.; Chen, R.-B.; Zhang, L. Genome-wide analysis of AP2/ERF superfamily in Isatis indigotica. J. Integr. Med. 2023, 21, 77–88. [Google Scholar] [CrossRef]
- Ghorbani, R.; Zakipour, Z.; Alemzadeh, A.; Razi, H. Genome-wide analysis of AP2/ERF transcription factors family in Brassica napus. Physiol. Mol. Biol. Plants 2020, 26, 1463–1476. [Google Scholar] [CrossRef]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Genome-Wide Analysis of the ERF Gene Family in Arabidopsis and Rice. Plant Physiol. 2006, 140, 411–432. [Google Scholar] [CrossRef]
- Zhao, M.; Haxim, Y.; Liang, Y.; Qiao, S.; Gao, B.; Zhang, D.; Li, X. Genome-wide investigation of AP2/ERF gene family in the desert legume Eremosparton songoricum: Identification, classification, evolution, and expression profiling under drought stress. Front. Plant Sci. 2022, 13, 885694. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Y.; Song, Y.; Fan, D.; Li, J.; Zhang, Z.; Wang, L.; He, J.; Chen, C.; Ma, C. Hierarchical Regulatory Networks Reveal Conserved Drivers of Plant Drought Response at the Cell-Type Level. Adv. Sci. 2025, e2415106. [Google Scholar] [CrossRef]
- Aanniz, T.; El Baaboua, A.; Aboulaghras, S.; Bouyahya, A.; Benali, T.; Balahbib, A.; El Omari, N.; Butnariu, M.; Muzammil, K.; Yadav, K.K.; et al. Impact of water stress to plant epigenetic mechanisms in stress and adaptation. Physiol. Plant. 2025, 177, e70058. [Google Scholar] [CrossRef]
- Landis, J.B.; Soltis, D.E.; Li, Z.; Marx, H.E.; Barker, M.S.; Tank, D.C.; Soltis, P.S. Impact of whole-genome duplication events on diversification rates in angiosperms. Am. J. Bot. 2018, 105, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, M.; Zhao, Y.; Chen, Y.; Wu, L.; Yin, H.; Xiong, S.; Wang, S.; Wang, J.; Yang, Y.; et al. LcERF19, an AP2/ERF transcription factor from Litsea cubeba, positively regulates geranial and neral biosynthesis. Hortic. Res. 2022, 9, uhac093. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Ding, C.; Hu, H.; Dong, G.; Zhang, G.; Qian, Q.; Ren, D. Molecular Events of Rice AP2/ERF Transcription Factors. Int. J. Mol. Sci. 2022, 23, 12013. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Singh, S.K.; Patra, B.; Liu, X.; Pattanaik, S.; Yuan, L. Mutually Regulated AP2/ERF Gene Clusters Modulate Biosynthesis of Specialized Metabolites in Plants. Plant Physiol. 2020, 182, 840–856. [Google Scholar] [CrossRef]
- Kandhol, N.; Jain, M.; Tripathi, D.K. Nanoparticles as potential hallmarks of drought stress tolerance in plants. Physiol. Plant. 2022, 174, e13665. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, Y.S.; Redillas, M.C.F.R.; Jang, G.; Jung, H.; Bang, S.W.; Choi, Y.D.; Ha, S.-H.; Reuzeau, C.; Kim, J.-K. OsNAC5 overexpression enlarges root diameter in rice plants leading to enhanced drought tolerance and increased grain yield in the field. Plant Biotechnol. J. 2013, 11, 101–114. [Google Scholar] [CrossRef]
- Parks, M.B.; Nakov, T.; Ruck, E.C.; Wickett, N.J.; Alverson, A.J. Phylogenomics reveals an extensive history of genome duplication in diatoms (Bacillariophyta). Am. J. Bot. 2018, 105, 330–347. [Google Scholar] [CrossRef]
- Gong, Z.; Xiong, L.; Shi, H.; Yang, S.; Herrera-Estrella, L.R.; Xu, G.; Chao, D.-Y.; Li, J.; Wang, P.-Y.; Qin, F.; et al. Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 2020, 63, 635–674. [Google Scholar] [CrossRef]
- Ma, N.; Sun, P.; Li, Z.-Y.; Zhang, F.-J.; Wang, X.-F.; You, C.-X.; Zhang, C.-L.; Zhang, Z. Plant disease resistance outputs regulated by AP2/ERF transcription factor family. Stress Biol. 2024, 4, 2. [Google Scholar] [CrossRef]
- He, M.; Qin, C.-X.; Wang, X.; Ding, N.-Z. Plant Unsaturated Fatty Acids: Biosynthesis and Regulation. Front. Plant Sci. 2020, 11, 390. [Google Scholar] [CrossRef]
- He, M.; Ding, N.-Z. Plant Unsaturated Fatty Acids: Multiple Roles in Stress Response. Front. Plant Sci. 2020, 11, 562785. [Google Scholar] [CrossRef]
- Feng, D.; Cheng, J.; Yang, X.; Tian, Z.; Liu, Y.; Zhang, Y.; Qiang, S. Polyploidization-enhanced effective clonal reproduction endows the successful invasion of Solidago canadensis. Ecol. Appl. 2024, 34, e2738. [Google Scholar] [CrossRef] [PubMed]
- Clo, J. Polyploidization: Consequences of genome doubling on the evolutionary potential of populations. Am. J. Bot. 2022, 109, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Alix, K.; Gérard, P.R.; Schwarzacher, T.; Heslop-Harrison, J.S.P. Polyploidy and interspecific hybridization: Partners for adaptation, speciation and evolution in plants. Ann. Bot. 2017, 120, 183–194. [Google Scholar] [CrossRef]
- Liu, M.; Yang, M.; Liang, H.; Luo, B.; Deng, J.; Cao, L.; Zheng, D.; Chen, C. Polyploidy drives autophagy to participate in plant-specific functions. iMeta 2024, 3, e252. [Google Scholar] [CrossRef]
- Heslop-Harrison, J.S.P.; Schwarzacher, T.; Liu, Q. Polyploidy: Its consequences and enabling role in plant diversification and evolution. Ann. Bot. 2023, 131, 1–10. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Su, Y.; Shen, H. Rice Responses to Abiotic Stress: Key Proteins and Molecular Mechanisms. Int. J. Mol. Sci. 2025, 26, 896. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Li, R.; Ge, Y.; Li, Y.; Li, R. Plants’ Response to Abiotic Stress: Mechanisms and Strategies. Int. J. Mol. Sci. 2023, 24, 10915. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, X.; Wei, L.; Wansee, S.; Rabbani Nasab, H.; Chen, L.; Kang, Z.; Wang, J. TaAP2-10, an AP2/ERF transcription factor, contributes to wheat resistance against stripe rust. J. Plant Physiol. 2023, 288, 154078. [Google Scholar] [CrossRef]
- Zhang, Y.; Ji, A.; Xu, Z.; Luo, H.; Song, J. The AP2/ERF transcription factor SmERF128 positively regulates diterpenoid biosynthesis in Salvia miltiorrhiza. Plant Mol. Biol. 2019, 100, 83–93. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, R.; Xiang, C.; Zhang, R.; Wang, Q.; Wang, T.; Li, X.; Lu, X.; Gao, S.; Liu, Z.; et al. Transcriptomic and Physiological Analysis Reveal That α-Linolenic Acid Biosynthesis Responds to Early Chilling Tolerance in Pumpkin Rootstock Varieties. Front. Plant Sci. 2021, 12, 669565. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, S.; Chang, X.; Wang, X.; Zhao, Y.; Xia, Y.; Trigiano, R.N.; Jiao, Y.; Chen, F. The ancient wave of polyploidization events in flowering plants and their facilitated adaptation to environmental stress. Plant Cell Environ. 2020, 43, 2847–2856. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, N.W.K.; Chung, F.Y.; Yamaguchi, N.; Gan, E.-S.; Ito, T. Transcriptional Regulators of Plant Adaptation to Heat Stress. Int. J. Mol. Sci. 2023, 24, 13297. [Google Scholar] [CrossRef]
- Ren, R.; Wang, H.; Guo, C.; Zhang, N.; Zeng, L.; Chen, Y.; Ma, H.; Qi, J. Widespread Whole Genome Duplications Contribute to Genome Complexity and Species Diversity in Angiosperms. Mol. Plant 2018, 11, 414–428. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, J.; Yang, M.; Liang, H.; Zheng, D.; Zhu, G.; Ye, Z.; Lai, X.; Liu, M. A Comprehensive Analysis of the Multiple AP2/ERF Regulatory Network Unveils Putative Components of the Fatty Acid Pathway for Environmental Adaptation. Agronomy 2025, 15, 1112. https://doi.org/10.3390/agronomy15051112
Deng J, Yang M, Liang H, Zheng D, Zhu G, Ye Z, Lai X, Liu M. A Comprehensive Analysis of the Multiple AP2/ERF Regulatory Network Unveils Putative Components of the Fatty Acid Pathway for Environmental Adaptation. Agronomy. 2025; 15(5):1112. https://doi.org/10.3390/agronomy15051112
Chicago/Turabian StyleDeng, Junjie, Ming Yang, Heng Liang, Daojun Zheng, Guangshun Zhu, Zhenpei Ye, Xinjie Lai, and Moyang Liu. 2025. "A Comprehensive Analysis of the Multiple AP2/ERF Regulatory Network Unveils Putative Components of the Fatty Acid Pathway for Environmental Adaptation" Agronomy 15, no. 5: 1112. https://doi.org/10.3390/agronomy15051112
APA StyleDeng, J., Yang, M., Liang, H., Zheng, D., Zhu, G., Ye, Z., Lai, X., & Liu, M. (2025). A Comprehensive Analysis of the Multiple AP2/ERF Regulatory Network Unveils Putative Components of the Fatty Acid Pathway for Environmental Adaptation. Agronomy, 15(5), 1112. https://doi.org/10.3390/agronomy15051112