
A Genome-Wide Analysis of the VuR2R3-MYB Gene Family in Cowpea and Its Expression in Anthocyanin Accumulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Genome-Wide Identification of VuR2R3-MYB TFs in Cowpea
2.2. Phylogenetic Analysis and Conservation Analysis of VuR2R3-MYB Proteins
2.3. Analysis of Gene Structure and Promoter Characteristics of VuR2R2-MYBs
2.4. VuR2R3-MYB Physical Localization, Collinearity Analysis, and Ka/Ks Calculation of Duplicated VuR2R3-MYB TFs
2.5. Transcriptome Date Analysis
2.6. Verification of RNA-Seq Data by qRT-PCR
2.7. Metabolome Detection and Analysis
3. Results
3.1. Identification and Characteristics of R2R3-MYB TF Family Members in Cowpea
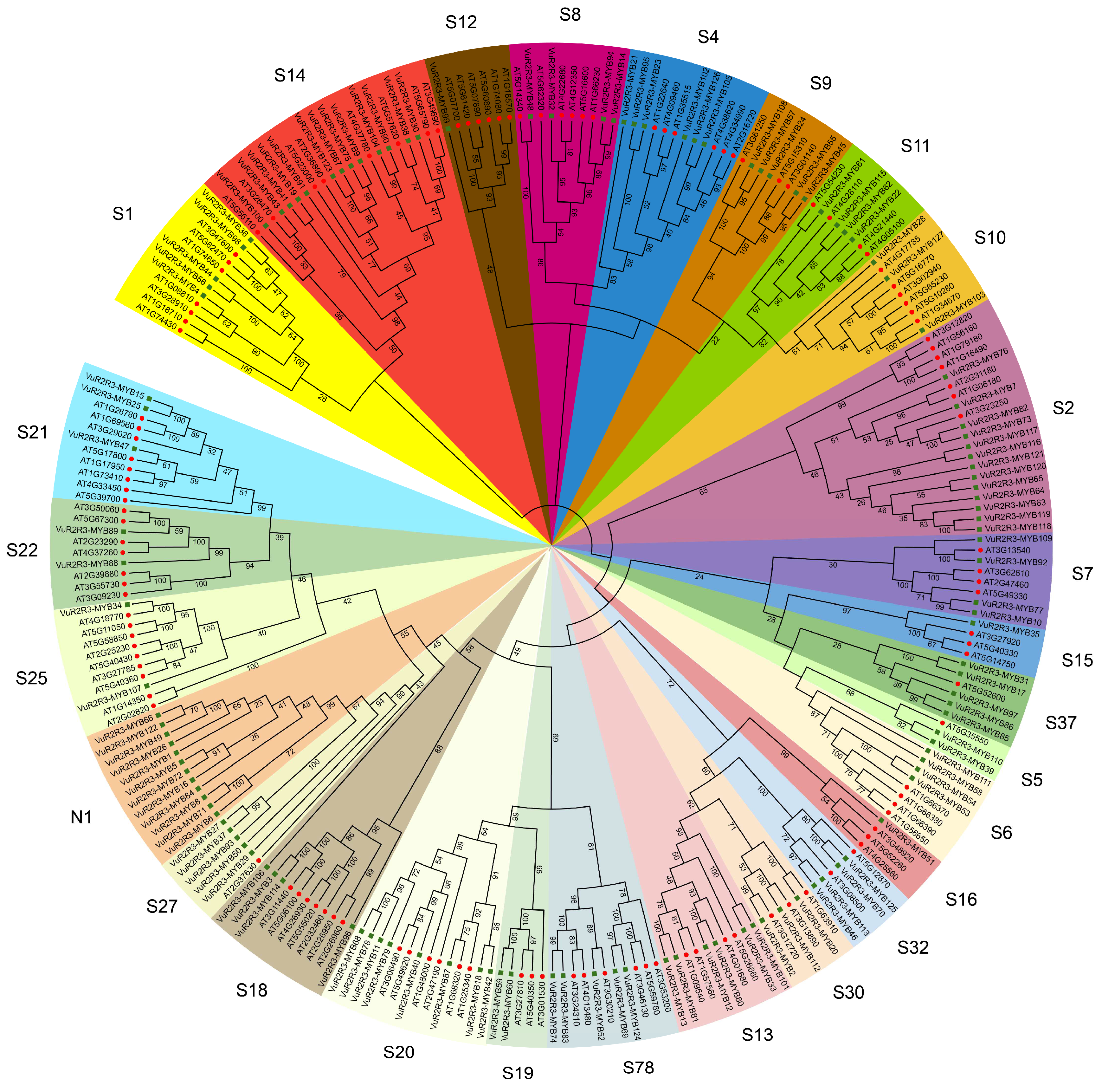
3.2. Phylogenetic Analysis of VuR2R3-MYB TFs in Cowpea
3.3. Analysis of Gene Structure, Conserved Motifs, and Protein Sequence Conservation in VuR2R3-MYB TFs
3.4. Cis-Acting Element Analysis of VuR2R3-MYB TF Promoters in Cowpea
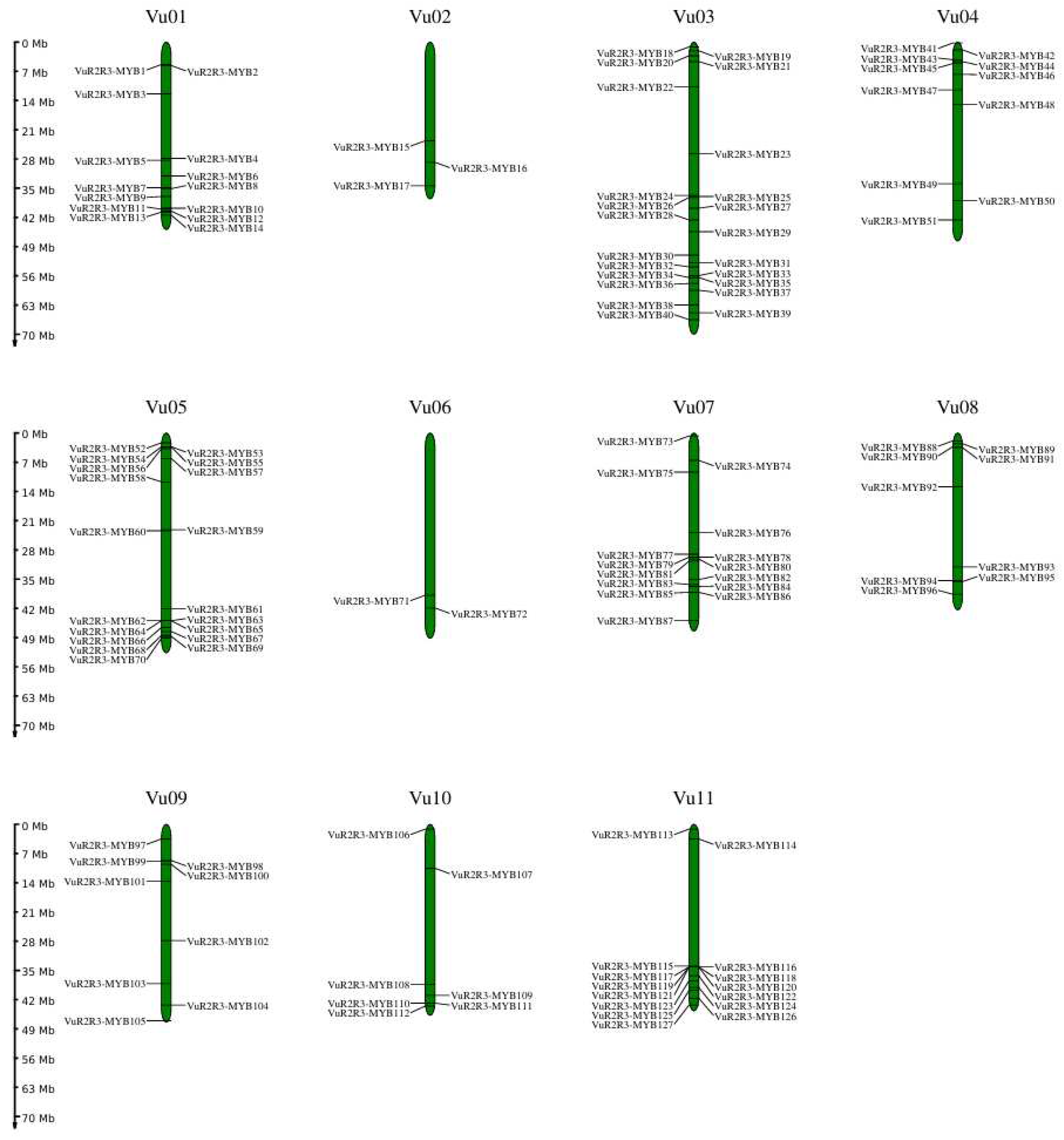
3.5. Chromosomal Distribution, Tandem and Segmental Duplications Analysis, and Ka/Ks Analysis of VuR2R3-MYB TFs
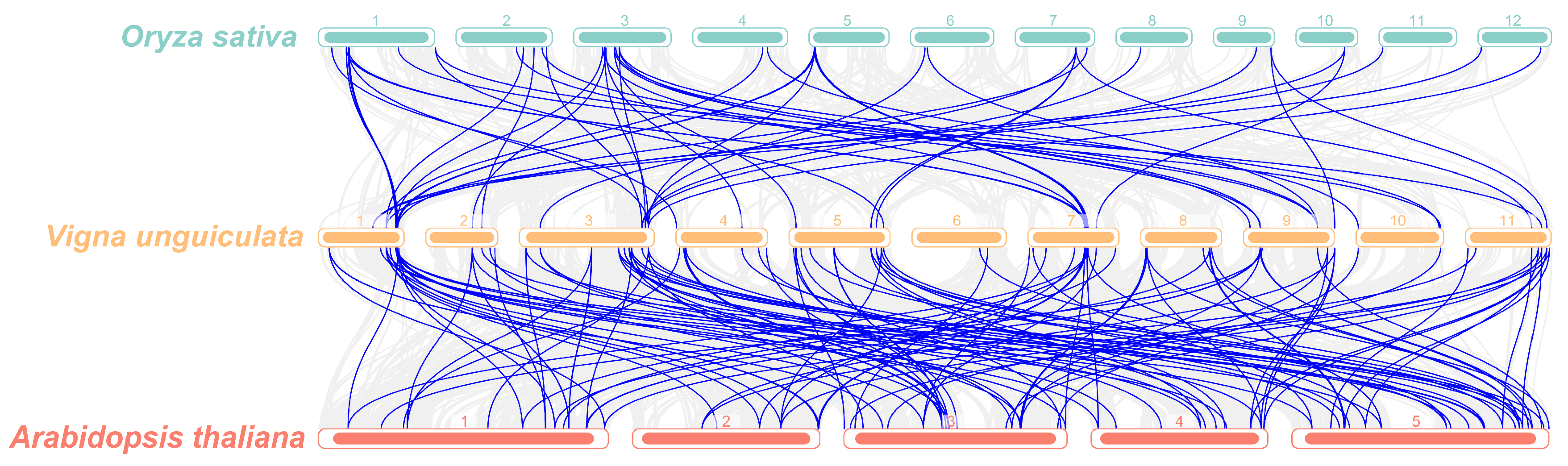
3.6. Evolutionary Analysis of R2R3-MYB TFs Across Multiple Species
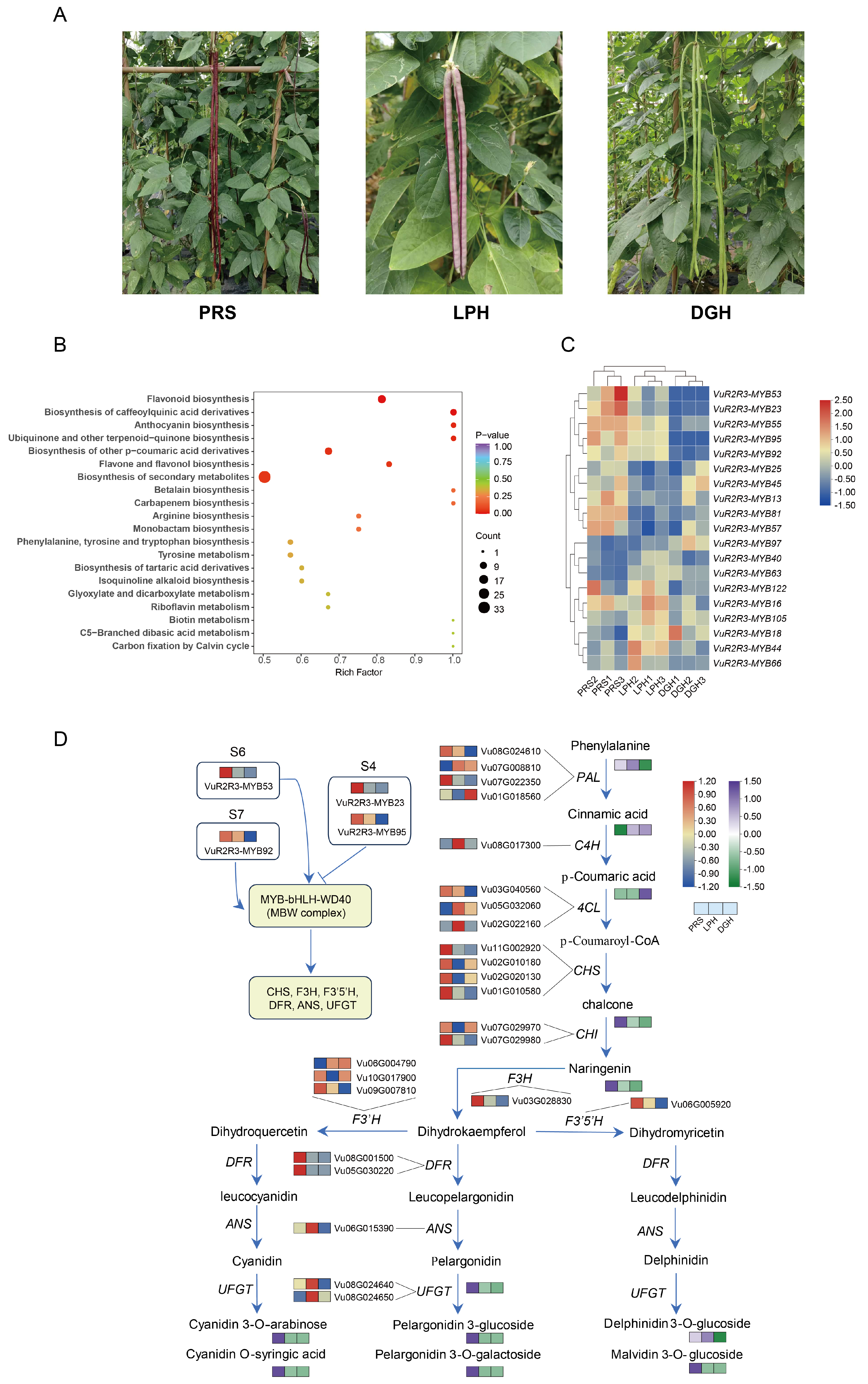
3.7. Integrated Transcriptomic and Metabolomic Analysis with Anthocyanin Biosynthesis Candidate TF Screening
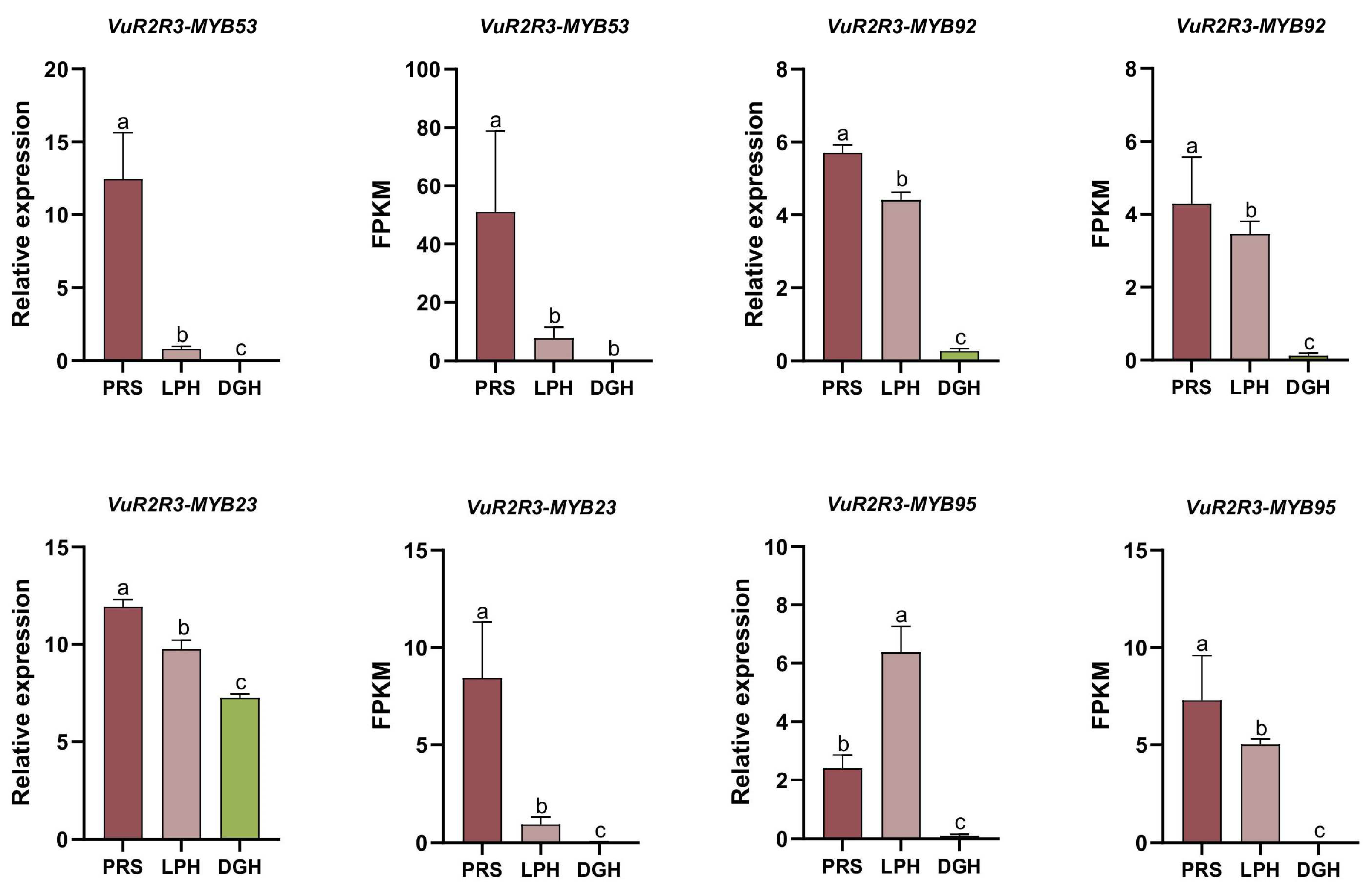
3.8. Expression Analysis of VuR2R3-MYBs Through qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xia, Q.; Pan, L.; Zhang, R.; Ni, X.; Wang, Y.; Dong, X.; Gao, Y.; Zhang, Z.; Kui, L.; Li, Y.; et al. The Genome Assembly of Asparagus Bean, Vigna unguiculata Ssp. Sesquipedialis. Sci. Data 2019, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Moshelion, M.; Wu, X.; Halperin, O.; Wang, B.; Luo, J.; Wallach, R.; Wu, X.; Lu, Z.; Li, G. Natural Variation and Gene Regulatory Basis for the Responses of Asparagus Beans to Soil Drought. Front. Plant Sci. 2015, 6, 891. [Google Scholar] [CrossRef] [PubMed]
- Kongjaimun, A.; Kaga, A.; Tomooka, N.; Somta, P.; Vaughan, D.A.; Srinives, P. The Genetics of Domestication of Yardlong Bean, Vigna unguiculata (L.) Walp. Ssp. unguiculata Cv.-Gr. Sesquipedalis. Ann. Bot. 2012, 109, 1185–1200. [Google Scholar] [CrossRef]
- Wu, X.; Cortés, A.J.; Blair, M.W. Genetic Differentiation of Grain, Fodder and Pod Vegetable Type Cowpeas (Vigna unguiculata L.) Identified through Single Nucleotide Polymorphisms from Genotyping-by-Sequencing. Mol. Hortic. 2022, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Wu, X.; Muñoz-Amatriaín, M.; Wang, B.; Wu, X.; Hu, Y.; Huynh, B.; Close, T.J.; Roberts, P.A.; Zhou, W.; et al. Genomic Regions, Cellular Components and Gene Regulatory Basis Underlying Pod Length Variations in Cowpea (V. unguiculata L. Walp). Plant Biotechnol. J. 2017, 15, 547–557. [Google Scholar] [CrossRef]
- Pan, L.; Liu, M.; Kang, Y.; Mei, X.; Hu, G.; Bao, C.; Zheng, Y.; Zhao, H.; Chen, C.; Wang, N. Comprehensive Genomic Analyses of Vigna unguiculata Provide Insights into Population Differentiation and the Genetic Basis of Key Agricultural Traits. Plant Biotechnol. J. 2023, 21, 1426–1439. [Google Scholar] [CrossRef]
- Wu, X.; Hu, Z.; Zhang, Y.; Li, M.; Liao, N.; Dong, J.; Wang, B.; Wu, J.; Wu, X.; Wang, Y.; et al. Differential Selection of Yield and Quality Traits Has Shaped Genomic Signatures of Cowpea Domestication and Improvement. Nat. Genet. 2024, 56, 992–1005. [Google Scholar] [CrossRef]
- Herniter, I.A.; Muñoz-Amatriaín, M.; Lo, S.; Guo, Y.-N.; Close, T.J. Identification of Candidate Genes Controlling Black Seed Coat and Pod Tip Color in Cowpea (Vigna unguiculata [L.] Walp). G3 2018, 8, 3347–3355. [Google Scholar] [CrossRef]
- Jaeger, S.R.; Antúnez, L.; Ares, G.; Swaney-Stueve, M.; Jin, D.; Harker, F.R. Quality Perceptions Regarding External Appearance of Apples: Insights from Experts and Consumers in Four Countries. Postharvest Biol. Technol. 2018, 146, 99–107. [Google Scholar] [CrossRef]
- Naing, A.H.; Kim, C.K. Abiotic Stress-induced Anthocyanins in Plants: Their Role in Tolerance to Abiotic Stresses. Physiol. Plant. 2021, 172, 1711–1723. [Google Scholar] [CrossRef]
- Speer, H.; D’Cunha, N.M.; Alexopoulos, N.I.; McKune, A.J.; Naumovski, N. Anthocyanins and Human Health—A Focus on Oxidative Stress, Inflammation and Disease. Antioxidants 2020, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Isayenkov, S.V. The Role of Anthocyanins in Plant Tolerance to Drought and Salt Stresses. Plants 2023, 12, 2558. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liu, T.; Nan, W.; Jeewani, D.C.; Niu, Y.; Li, C.; Wang, Y.; Shi, X.; Wang, C.; Wang, J.; et al. Two Transcription Factors TaPpm1 and TaPpb1 Co-Regulate Anthocyanin Biosynthesis in Purple Pericarps of Wheat. J. Exp. Bot. 2018, 69, 2555–2567. [Google Scholar] [CrossRef]
- Rahim, M.A.; Resentini, F.; Dalla Vecchia, F.; Trainotti, L. Effects on Plant Growth and Reproduction of a Peach R2R3-MYB Transcription Factor Overexpressed in Tobacco. Front. Plant Sci. 2019, 10, 1143. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Wang, X.; Zhou, M.; Zhou, X.; Ye, X.; Wei, X. An R2R3 MYB Transcription Factor in Wheat, Ta PIMP 1, Mediates Host Resistance to Bipolaris sorokiniana and Drought Stresses through Regulation of Defense- and Stress-related Genes. New Phytol. 2012, 196, 1155–1170. [Google Scholar] [CrossRef]
- Zhou, Z.; Wei, X.; Lan, H. CgMYB1, an R2R3-MYB Transcription Factor, Can Alleviate Abiotic Stress in an Annual Halophyte Chenopodium glaucum. Plant Physiol. Biochem. 2023, 196, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Teng, Y.; Cen, Q.; Fang, Y.; Tian, Q.; Zhang, X.; Wang, H.; Zhang, X.; Xue, D. Genome-Wide Identification of R2R3-MYB Transcription Factor and Expression Analysis under Abiotic Stress in Rice. Plants 2022, 11, 1928. [Google Scholar] [CrossRef]
- Wu, Y.; Wen, J.; Xia, Y.; Zhang, L.; Du, H. Evolution and Functional Diversification of R2R3-MYB Transcription Factors in Plants. Hortic. Res. 2022, 9, uhac058. [Google Scholar] [CrossRef]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the Anthocyanin Biosynthetic Pathway by the TTG1/bHLH/Myb Transcriptional Complex in Arabidopsis Seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Li, C.; Yu, W.; Xu, J.; Lu, X.; Liu, Y. Anthocyanin Biosynthesis Induced by MYB Transcription Factors in Plants. Int. J. Mol. Sci. 2022, 23, 11701. [Google Scholar] [CrossRef]
- Chen, L.; Cui, Y.; Yao, Y.; An, L.; Bai, Y.; Li, X.; Yao, X.; Wu, K. Genome-Wide Identification of WD40 Transcription Factors and Their Regulation of the MYB-bHLH-WD40 (MBW) Complex Related to Anthocyanin Synthesis in Qingke (Hordeum vulgare L. Var. nudum Hook. f.). BMC Genomics 2023, 24, 166. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Z.; Li, J.; Zhang, H.; Peng, Y.; Li, Z. Uncovering Hierarchical Regulation among MYB-bHLH-WD40 Proteins and Manipulating Anthocyanin Pigmentation in Rice. Int. J. Mol. Sci. 2022, 23, 8203. [Google Scholar] [CrossRef]
- Ma, R.; Huang, W.; Hu, Q.; Tian, G.; An, J.; Fang, T.; Liu, J.; Hou, J.; Zhao, M.; Sun, L. Tandemly Duplicated MYB Genes Are Functionally Diverged in the Regulation of Anthocyanin Biosynthesis in Soybean. Plant Physiol. 2024, 194, 2549–2563. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Lv, A.; Wen, W.; Fan, N.; You, X.; Gao, L.; Zhou, P.; Shi, F.; An, Y. MsMYB206–MsMYB450–MsHY5 Complex Regulates Alfalfa Tolerance to Salt Stress via Regulating Flavonoid Biosynthesis during the Day and Night Cycles. Plant J. 2025, 121, e17216. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hought, J.; Baseggio, M.; Hart, J.P.; Gore, M.A.; Ilut, D.C. Genomic Characterization of the Native Seeds/SEARCH Common Bean (Phaseolus vulgaris L.) Collection and Its Seed Coat Patterns. Genet. Resour. Crop Evol. 2019, 66, 1469–1482. [Google Scholar] [CrossRef]
- Li, H.; Yao, Y.; An, L.; Li, X.; Cui, Y.; Bai, Y.; Yao, X.; Wu, K. Isolation and Expression Analysis of the HvnAnt2 Gene in Qingke Barley (Hordeum Vulgare L. Var. Nudum Hook. f.) Varieties with Different Grain Colours. Czech J. Genet. Plant Breed. 2024, 60, 107–118. [Google Scholar] [CrossRef]
- Wang, L.; Ni, D.; Yang, F.; Lin, L.; Yang, Y.; Sun, C.; Ye, X.; Cao, J.; Kong, X. Transcriptome Profiling of Sorghum bicolor Reveals Cultivar-Specific Molecular Signatures Associated with Starch and Phenolic Compounds Biosyntheses and Accumulation during Sorghum Grain Development. Czech J. Genet. Plant Breed. 2023, 59, 235–252. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Z.; Wu, Z.; Li, T.; Shen, Z.; Zhou, X.; Wu, X.; Li, G.; Zhang, Y. A Near-complete Assembly of Asparagus Bean Provides Insights into Anthocyanin Accumulation in Pods. Plant Biotechnol. J. 2023, 21, 2473–2489. [Google Scholar] [CrossRef]
- Jiang, C.K.; Rao, G.Y. Insights into the Diversification and Evolution of R2R3-MYB Transcription Factors in Plants. Plant Physiol. 2020, 183, 637–655. [Google Scholar] [CrossRef]
- Zheng, J.; Wu, H.; Zhao, M.; Yang, Z.; Zhou, Z.; Guo, Y.; Lin, Y.; Chen, H. OsMYB3 Is a R2R3-MYB Gene Responsible for Anthocyanin Biosynthesis in Black Rice. Mol. Breed. 2021, 41, 51. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Q.; Xie, X.; Cai, Y.; Li, J.; Feng, Y.; Zhang, Y. Integrated Metabolomics and Transcriptomics Analyses Reveal the Molecular Mechanisms Underlying the Accumulation of Anthocyanins and Other Flavonoids in Cowpea Pod (Vigna unguiculata L.). J. Agric. Food Chem. 2020, 68, 9260–9275. [Google Scholar] [CrossRef]
- Krylova, E.A.; Mikhailova, A.S.; Zinchenko, Y.N.; Perchuk, I.N.; Razgonova, M.P.; Khlestkina, E.K.; Burlyaeva, M.O. The Content of Anthocyanins in Cowpea (Vigna unguiculata (L.) Walp.) Seeds and Contribution of the MYB Gene Cluster to Their Coloration Pattern. Plants 2023, 12, 3624. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Chen, Y.; Ravelombola, W.; Mou, B.; Sun, X.; Zhang, Q.; Xiao, Y.; Tian, Y.; Luo, Q.; Alatawi, I.; et al. Genetic Dissection of Diverse Seed Coat Patterns in Cowpea through a Comprehensive GWAS Approach. Plants 2024, 13, 1275. [Google Scholar] [CrossRef] [PubMed]
- Lay, L.; Khan, W.; Jo, H.; Kim, S.-H.; Kim, Y. Genome-Wide Association Study on Cowpea Seed Coat Color Using RGB Images. Mol. Breed. 2024, 44, 80. [Google Scholar] [CrossRef]
- Pal, L.; Dwivedi, V.; Gupta, S.K.; Saxena, S.; Pandey, A.; Chattopadhyay, D. Biochemical Analysis of Anthocyanin and Proanthocyanidin and Their Regulation in Determining Chickpea Flower and Seed Coat Colour. J. Exp. Bot. 2023, 74, 130–148. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pal, L.; Rajput, R.; Chhatwal, H.; Singh, N.; Chattopadhyay, D.; Pandey, A. CaLAP1 and CaLAP2 Orchestrate Anthocyanin Biosynthesis in the Seed Coat of Cicer arietinum. Planta 2024, 260, 38. [Google Scholar] [CrossRef]
- Ma, C.; Feng, Y.; Zhou, S.; Zhang, J.; Guo, B.; Xiong, Y.; Wu, S.; Li, Y.; Li, Y.; Li, C. Metabolomics and Transcriptomics Provide Insights into the Molecular Mechanisms of Anthocyanin Accumulation in the Seed Coat of Differently Colored Mung Bean (Vigna radiata L.). Plant Physiol. Biochem. 2023, 200, 107739. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A Biologist-Centric Software for Evolutionary Analysis of DNA and Protein Sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for Motif Discovery and Searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, İ.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “One for All, All for One” Bioinformatics Platform for Biological Big-Data Mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching during Cell Differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, W.; Liu, J.; Liu, H.; Lv, Z.; Zhang, C.; Chen, D.; Jiao, Z. Postharvest UV-C Irradiation Increased the Flavonoids and Anthocyanins Accumulation, Phenylpropanoid Pathway Gene Expression, and Antioxidant Activity in Sweet Cherries (Prunus avium L.). Postharvest Biol. Technol. 2021, 175, 111490. [Google Scholar] [CrossRef]
- Chong, J.; Xia, J. MetaboAnalystR: An R Package for Flexible and Reproducible Analysis of Metabolomics Data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB Transcription Factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, H.; Zhang, Y.; Zhao, Y.; Zhang, Y.; Feng, X.; Lin, H. Diverse Roles of MYB Transcription Factors in Plants. J. Integr. Plant Biol. 2025, 67, 539–562. [Google Scholar] [CrossRef]
- Du, H.; Yang, S.-S.; Liang, Z.; Feng, B.-R.; Liu, L.; Huang, Y.-B.; Tang, Y.-X. Genome-Wide Analysis of the MYB Transcription Factor Superfamily in Soybean. BMC Plant Biol. 2012, 12, 106. [Google Scholar] [CrossRef]
- Wang, S.; Xu, Z.; Yang, Y.; Ren, W.; Fang, J.; Wan, L. Genome-Wide Analysis of R2R3-MYB Genes in Cultivated Peanut (Arachis hypogaea L.): Gene Duplications, Functional Conservation, and Diversification. Front. Plant Sci. 2023, 14, 1102174. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, J.; Qin, L.; Shi, W.; Xia, G.; Liu, S. TaCYP81D5, One Member in a Wheat Cytochrome P450 Gene Cluster, Confers Salinity Tolerance via Reactive Oxygen Species Scavenging. Plant Biotechnol. J. 2020, 18, 791–804. [Google Scholar] [CrossRef]
- Chen, L.; Hu, B.; Qin, Y.; Hu, G.; Zhao, J. Advance of the Negative Regulation of Anthocyanin Biosynthesis by MYB Transcription Factors. Plant Physiol. Biochem. 2019, 136, 178–187. [Google Scholar] [CrossRef]
- Jin, H. Transcriptional Repression by AtMYB4 Controls Production of UV-Protecting Sunscreens in Arabidopsis. EMBO J. 2000, 19, 6150–6161. [Google Scholar] [CrossRef]
- Preston, J.; Wheeler, J.; Heazlewood, J.; Li, S.F.; Parish, R.W. AtMYB32 is Required for Normal Pollen Development in Arabidopsis thaliana. Plant J. 2004, 40, 979–995. [Google Scholar] [CrossRef]
- Stracke, R.; Ishihara, H.; Huep, G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential Regulation of Closely Related R2R3-MYB Transcription Factors Controls Flavonol Accumulation in Different Parts of the Arabidopsis thaliana Seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef]
- Agarwal, M.; Hao, Y.; Kapoor, A.; Dong, C.H.; Fujii, H.; Zheng, X.; Zhu, J.K. A R2R3 Type MYB Transcription Factor Is Involved in the Cold Regulation of CBF Genes and in Acquired Freezing Tolerance. J. Biol. Chem. 2006, 281, 37636–37645. [Google Scholar] [CrossRef]
- Dias, A.P.; Braun, E.L.; McMullen, M.D.; Grotewold, E. Recently Duplicated Maize R2R3 Myb Genes Provide Evidence for Distinct Mechanisms of Evolutionary Divergence after Duplication. Plant Physiol. 2003, 131, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Denekamp, M.; Smeekens, S.C. Integration of Wounding and Osmotic Stress Signals Determines the Expression of the AtMYB102 Transcription Factor Gene. Plant Physiol. 2003, 132, 1415–1423. [Google Scholar] [CrossRef]
- Ballester, A.-R.; Molthoff, J.; De Vos, R.; Hekkert, B.T.L.; Orzaez, D.; Fernaݩndez-Moreno, J.-P.; Tripodi, P.; Grandillo, S.; Martin, C.; Heldens, J.; et al. Biochemical and Molecular Analysis of Pink Tomatoes: Deregulated Expression of the Gene Encoding Transcription Factor SlMYB12 Leads to Pink Tomato Fruit Color. Plant Physiol. 2009, 152, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.K.; Pandey, A.; Hamurcu, M.; Vyhnánek, T.; Zargar, S.M.; Kahraman, A.; Topal, A.; Gezgin, S. Exploring Strigolactones for Inducing Abiotic Stress Tolerance in Plants. Czech J. Genet. Plant Breed. 2024, 60, 55–69. [Google Scholar] [CrossRef]
- Xing, M.; Xin, P.; Wang, Y.; Han, C.; Lei, C.; Huang, W.; Zhang, Y.; Zhang, X.; Cheng, K.; Zhang, X. A Negative Feedback Regulatory Module Comprising R3-MYB Repressor MYBL2 and R2R3-MYB Activator PAP1 Fine-Tunes High Light-Induced Anthocyanin Biosynthesis in Arabidopsis. J. Exp. Bot. 2024, 75, 7381–7400. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Guan, M.; Zhao, C.; Geng, P.; Zhao, Q. Arabidopsis MYB4 Plays Dual Roles in Flavonoid Biosynthesis. Plant J. 2020, 101, 637–652. [Google Scholar] [CrossRef]
- Huang, W.; Khaldun, A.B.M.; Chen, J.; Zhang, C.; Lv, H.; Yuan, L.; Wang, Y. A R2R3-MYB Transcription Factor Regulates the Flavonol Biosynthetic Pathway in a Traditional Chinese Medicinal Plant, Epimedium Sagittatum. Front. Plant Sci. 2016, 7, 1089. [Google Scholar] [CrossRef]
- Jiang, L.; Yue, M.; Liu, Y.; Zhang, N.; Lin, Y.; Zhang, Y.; Wang, Y.; Li, M.; Luo, Y.; Zhang, Y.; et al. A Novel R2R3-MYB Transcription Factor FaMYB5 Positively Regulates Anthocyanin and Proanthocyanidin Biosynthesis in Cultivated Strawberries (Fragaria × Ananassa). Plant Biotechnol. J. 2023, 21, 1140–1158. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Deng, X.; Zhang, M.; Sun, H.; Gao, L.; Song, H.; Xin, J.; Ming, R.; Yang, D.; et al. Transcription Factor NnMYB5 Controls Petal Color by Regulating GLUTATHIONE S-TRANSFERASE2 in Nelumbo nucifera. Plant Physiol. 2023, 193, 1213–1226. [Google Scholar] [CrossRef]
- Yao, G.; Ming, M.; Allan, A.C.; Gu, C.; Li, L.; Wu, X.; Wang, R.; Chang, Y.; Qi, K.; Zhang, S.; et al. Map-based Cloning of the Pear Gene MYB114 Identifies an Interaction with Other Transcription Factors to Coordinately Regulate Fruit Anthocyanin Biosynthesis. Plant J. 2017, 92, 437–451. [Google Scholar] [CrossRef]
- Ni, J.; Bai, S.; Zhao, Y.; Qian, M.; Tao, R.; Yin, L.; Gao, L.; Teng, Y. Ethylene Response Factors Pp4ERF24 and Pp12ERF96 Regulate Blue Light-Induced Anthocyanin Biosynthesis in ‘Red Zaosu’ Pear Fruits by Interacting with MYB114. Plant Mol. Biol. 2019, 99, 67–78. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Yu, C.; Zhou, X.; Wu, Z.; Shen, Z.; Li, T.; Zhang, Y. A Genome-Wide Analysis of the VuR2R3-MYB Gene Family in Cowpea and Its Expression in Anthocyanin Accumulation. Agronomy 2025, 15, 1075. https://doi.org/10.3390/agronomy15051075
Yang Y, Yu C, Zhou X, Wu Z, Shen Z, Li T, Zhang Y. A Genome-Wide Analysis of the VuR2R3-MYB Gene Family in Cowpea and Its Expression in Anthocyanin Accumulation. Agronomy. 2025; 15(5):1075. https://doi.org/10.3390/agronomy15051075
Chicago/Turabian StyleYang, Yi, Canye Yu, Xuan Zhou, Zengxiang Wu, Zhuo Shen, Tinyao Li, and Yan Zhang. 2025. "A Genome-Wide Analysis of the VuR2R3-MYB Gene Family in Cowpea and Its Expression in Anthocyanin Accumulation" Agronomy 15, no. 5: 1075. https://doi.org/10.3390/agronomy15051075
APA StyleYang, Y., Yu, C., Zhou, X., Wu, Z., Shen, Z., Li, T., & Zhang, Y. (2025). A Genome-Wide Analysis of the VuR2R3-MYB Gene Family in Cowpea and Its Expression in Anthocyanin Accumulation. Agronomy, 15(5), 1075. https://doi.org/10.3390/agronomy15051075