
Cold-Tolerance Candidate Gene Identification in Maize Germination Using BSA, Transcriptome and Metabolome Profiling


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Construction of Segregating Pools
2.3. BSA-Seq and QTL-Seq Analysis
2.4. Bulked Segregant RNA-Seq Analysis
2.5. Metabolomics Analysis
3. Results
3.1. Statistical Analysis of Phenotypes of Maize’s SGRL
3.2. Sequencing Data Analysis of Four DNA Bulks
3.3. QTL-Seq Analysis
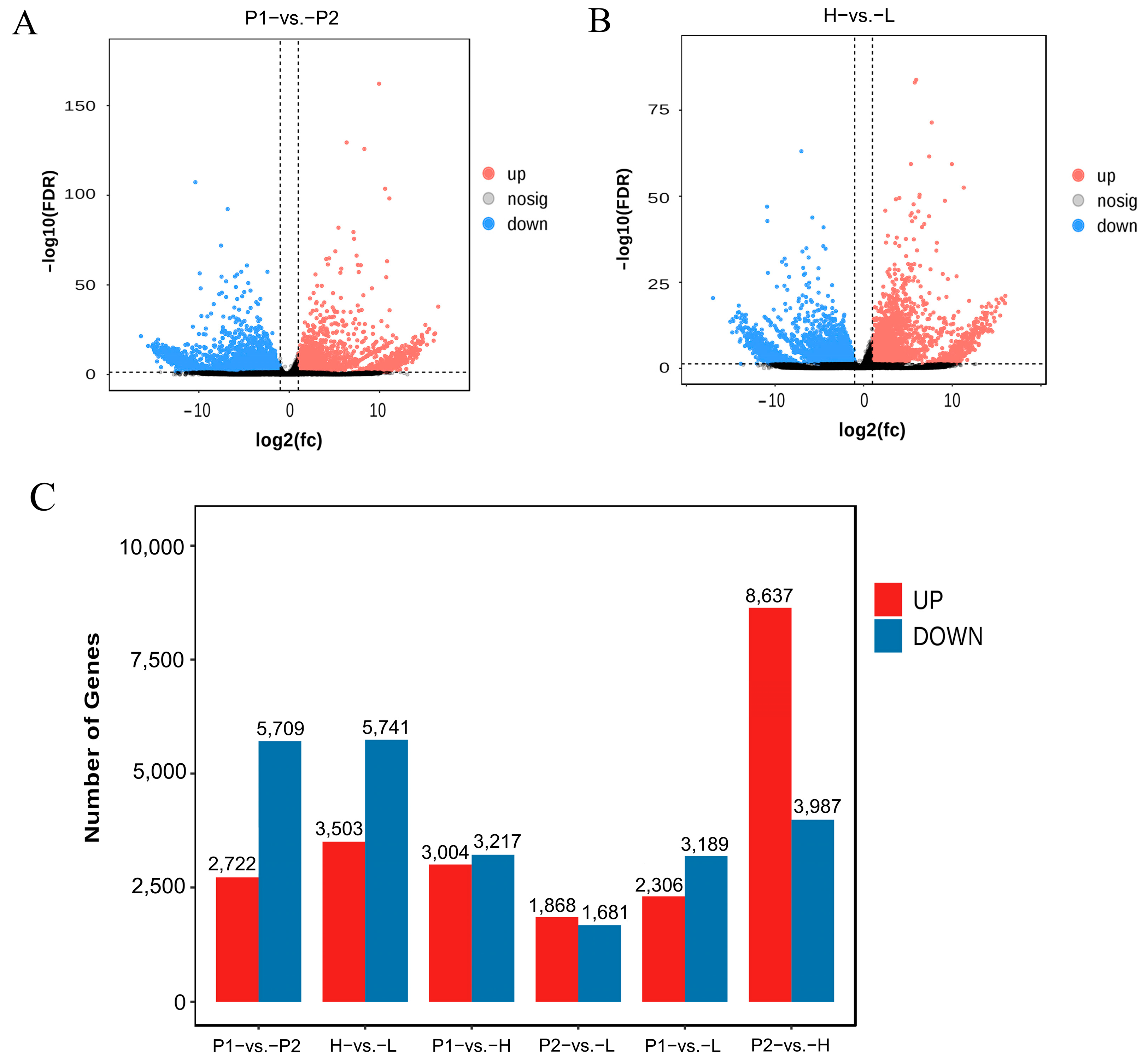
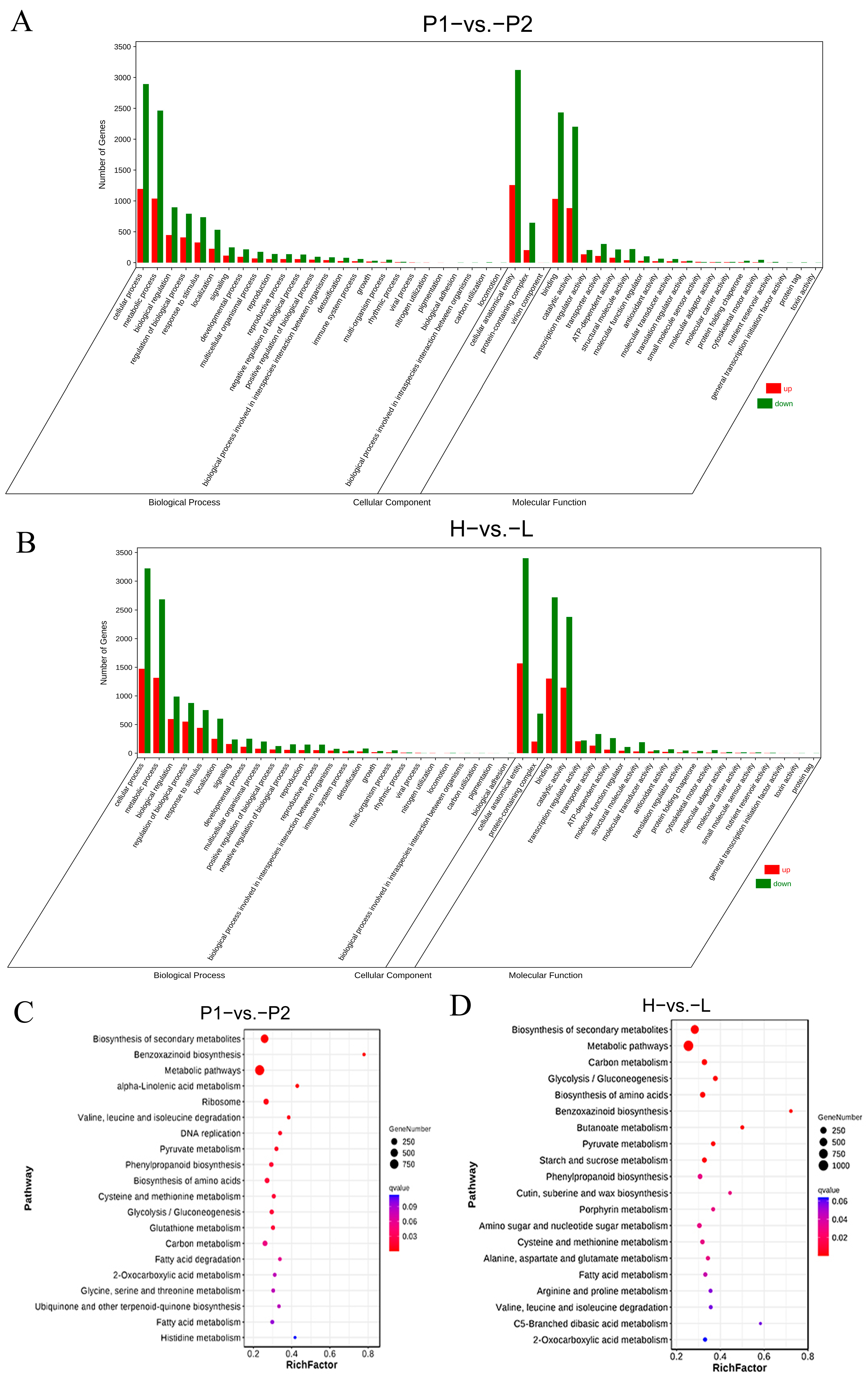
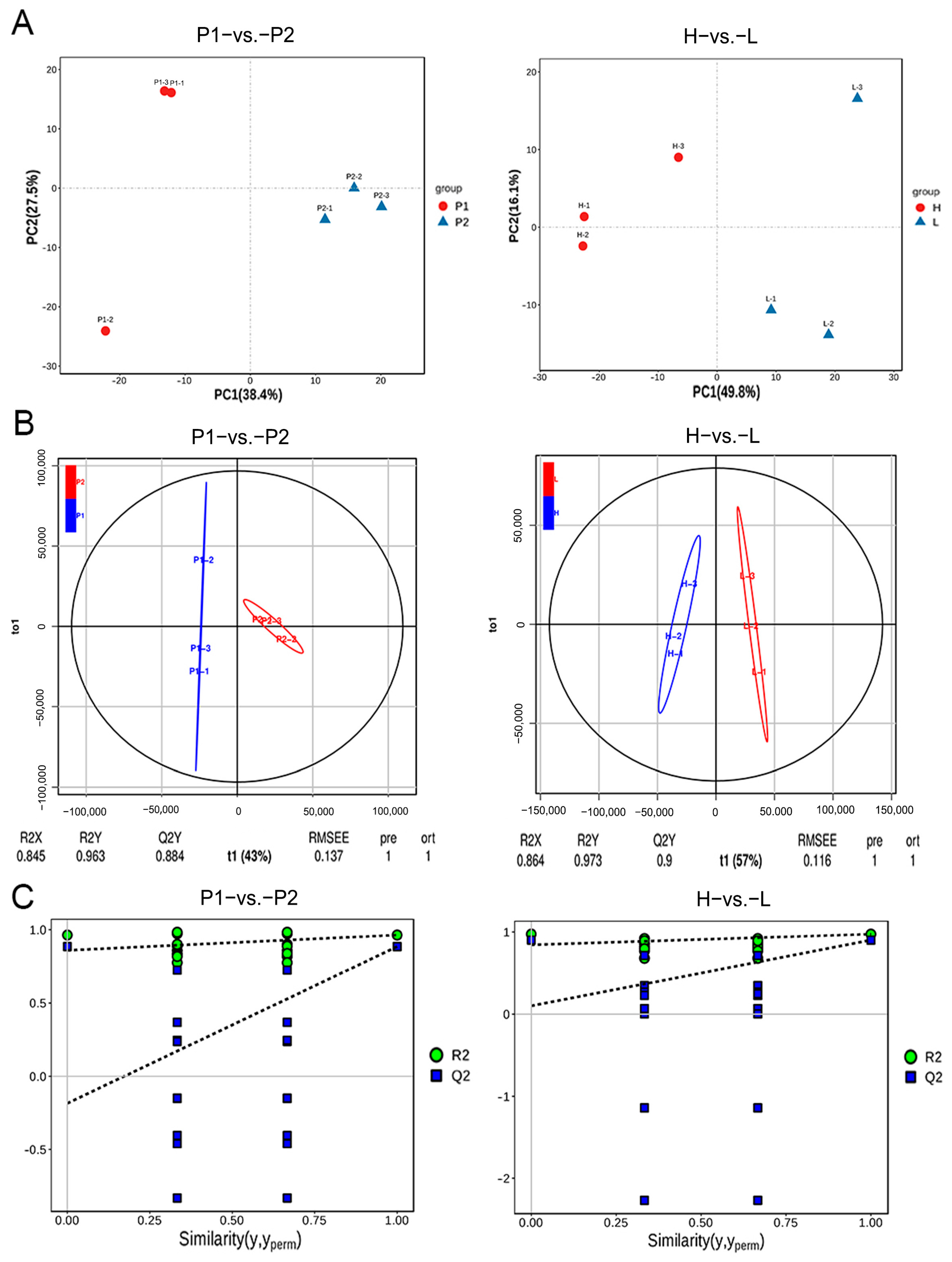
3.4. Transcriptome Analysis
3.5. Association Analysis of QTL-Seq and Transcriptome Data
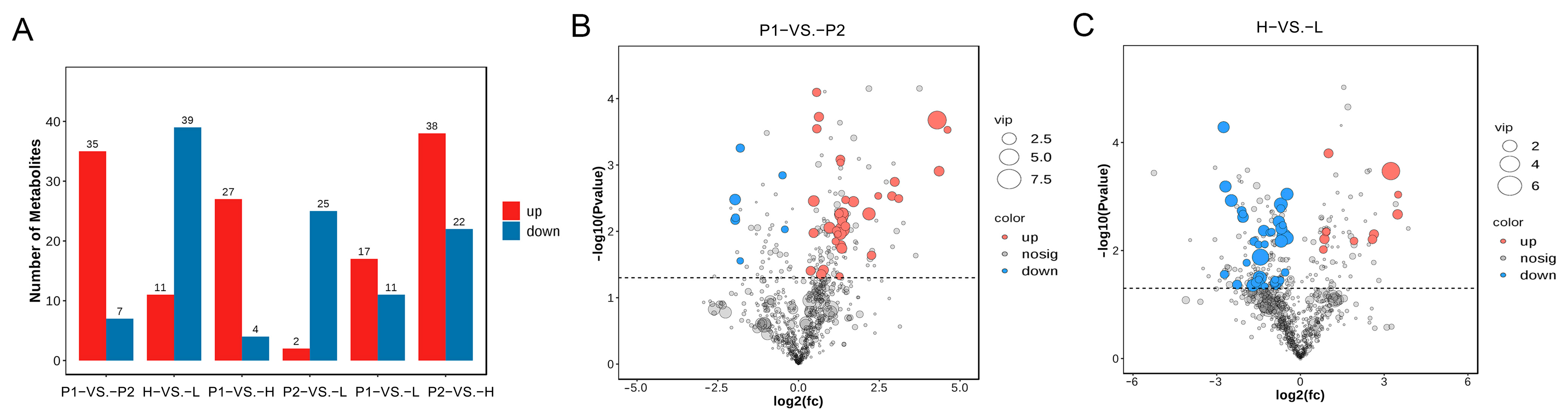
3.6. Metabolomic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Peter, R.; Eschholz, T.W.; Stamp, P. Swiss flint maize landraces-A rich pool of variability for early vigour in cool environments. Field Crop. Res. 2009, 110, 157–166. [Google Scholar] [CrossRef]
- Verheul, M.J.; Picatto, C.; Stamp, P. Growth and development of maize (Zea mays L.) seedlings under chilling conditions in the field. Eur. J. Agron. 1996, 5, 31–43. [Google Scholar] [CrossRef]
- Hund, A.; Richner, W.; Soldati, A.; Fracheboud, Y.; Stamp, P. Root morphology and photosynthetic performance of maize inbred lines at low temperature. Eur. J. Agron. 2007, 27, 52–61. [Google Scholar] [CrossRef]
- Muhammad, F.A.E.; Tariq, A.A.; Abdul, W.B.; Dong-Jin, L.C.; Kadambot, H.M. Chilling tolerance in maize: Agronomic and physiological approaches. Crop Pasture Sci. 2009, 60, 501–516. [Google Scholar]
- Guy, C.L. Cold acclimation and freezing stress tolerance: Role of protein metabolism. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1990, 41, 187–223. [Google Scholar] [CrossRef]
- Xiong, L.; Schumaker, K.S.; Zhu, J.K. Cell signaling during cold, drought, and salt stress. Plant Cell 2002, 14, S165–S183. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Y.; Wang, X. Physiological and molecular mechanisms of plant responses to cold stress. Front. Plant Sci. 2017, 8, 1844. [Google Scholar]
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef]
- Chengqun, L.; Baoling, H. Effects of low temperature stress on membrane lipid peroxidation and cell defense enzyme activity in leaves of E. grandis × E. urophylla seedlings. Guihaia 2004, 24, 64–68. [Google Scholar]
- Kim, J.S.; Jung, H.J.; Lee, H.J.; Kim, K.A.; Kang, H. Glycine-rich RNA-binding protein7 affects abiotic stress responses by regulating stomata opening and closing in Arabidopsis thaliana. Plant J. 2008, 55, 455–466. [Google Scholar] [CrossRef]
- Mastrangelo, A.M.; Marone, D.; Laidò, G.; Leonardis, A.M.D.; Vita, P.D. Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 2012, 185, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, G.; Sutoh, K.; Zhu, J.K.; Zhang, W. Identification of cold-inducible microRNAs in plants by transcriptome analysis. BBA-Gene Regul. Mech. 2008, 1779, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fu, J.; Gu, R.; Wang, J.; Chen, X.; Jia, J.; Zhang, J.; Wang, G. Isolation and analysis of cold stress inducible genes in Zea mays by suppression subtractive hybridization and cDNA macroarray. Plant Mol. Biol. Rep. 2009, 27, 38–49. [Google Scholar] [CrossRef]
- Lv, D.K.; Bai, X.; Li, Y.; Ding, X.D.; Ge, Y.; Cai, H.; Ji, W.; Wu, N.; Zhu, Y.M. Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene 2010, 459, 39–47. [Google Scholar] [CrossRef]
- Sadanandom, A.; Bailey, M.; Ewan, R.; Lee, J.; Nelis, S. The ubiquitin–proteasome system: Central modifier of plant signalling. New Phytol. 2012, 196, 13–28. [Google Scholar] [CrossRef]
- Jung, J.H.; Seo, P.J.; Park, C.M. The E3 ubiquitin ligase HOS1 regulates Arabidopsis flowering by mediating CONSTANS degradation under cold stress. J. Biol. Chem. 2012, 287, 43277–43287. [Google Scholar] [CrossRef]
- Han, Q.; Zhu, Q.; Shen, Y.; Lee, M.; Lübberstedt, T.; Zhao, G. QTL Mapping Low-Temperature Germination Ability in the Maize IBM Syn10 DH Population. Plants 2022, 11, 214. [Google Scholar] [CrossRef]
- Du, L.; Peng, X.; Zhang, H.; Xin, W.; Ma, K.; Liu, Y.; Hu, G. Transcriptome Analysis and QTL Mapping Identify Candidate Genes and Regulatory Mechanisms Related to Low-Temperature Germination Ability in Maize. Genes 2023, 14, 1917. [Google Scholar] [CrossRef]
- Jatimliansky, J.R.; García, M.D.; Molina, M.C. Response to chilling of Zea mays, Tripsacum dactyloides and their hybrid. Biol. Plant. 2004, 48, 561–567. [Google Scholar] [CrossRef]
- He, R.Y.; Zheng, J.J.; Chen, Y.; Pan, Z.Y.; Yang, T.; Zhou, Y.; Tang, Q.L. QTL-seq and transcriptomic integrative analyses reveal two positively regulated genes that control the low-temperature germination ability of MTP–maize introgression lines. Theor. Appl. Genet. 2023, 136, 116. [Google Scholar] [CrossRef]
- Li, X.; Wang, G.; Fu, J.; Li, L.; Jia, G.; Ren, L.; Lubberstedt, T.; Wang, G.; Wang, J.; Gu, R. QTL mapping in three connected populations reveals a set of consensus genomic regions for low temperature germination ability in Zea mays L. Front. Plant Sci. 2018, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Lübberstedt, T.; Zhao, G.; Lee, M. QTL mapping of low-temperature germination ability in the maize IBM Syn4 RIL population. PLoS ONE 2016, 11, e0152795. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, J.; Li, W.; Lee, M. Genome-wide Association Analysis of Ten Chilling Tolerance Indices at the Germination and Seedling Stages in Maize. J. Integr. Plant Biol. 2013, 55, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, J.; Xu, Q.; Wang, D.; Di, H.; Huang, J.; Yang, X.; Wang, Z.; Zhang, L.; Dong, L.; et al. Identification of candidate tolerance genes to low-temperature during maize germination by GWAS and RNA-seq approaches. BMC Plant Biol. 2020, 20, 333. [Google Scholar] [CrossRef]
- Sobkowiak, A.; Jończyk, M.; Jarochowska, E.; Biecek, P.; Trzcinska-Danielewicz, J.; Leipner, J.; Fronk, J.; Sowiński, P. Genome-wide transcriptomic analysis of response to low temperature reveals candidate genes determining divergent cold-sensitivity of maize inbred lines. Plant Mol. Biol. 2014, 85, 317–331. [Google Scholar] [CrossRef]
- Li, X.; Hu, H.; Hu, X.; Wang, G.; Du, X.; Li, L.; Wang, F.; Fu, J.; Wang, G.; Wang, J.; et al. Transcriptome analysis of near-isogenic lines provides novel insights into genes associated with seed low-temperature germination ability in Maize (Zea mays L.). Plants 2022, 11, 887. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Li, W.; Gao, X.; Xu, X.; Zhang, C.; Yu, S.; Dou, Y.; Luo, W.; Yu, L. Transcriptome Analysis Reveals POD as an Important Indicator for Assessing Low-Temperature Tolerance in Maize Radicles during Germination. Plants 2024, 13, 1362. [Google Scholar] [CrossRef]
- Yu, T.; Zhang, J.; Cao, J.; Cai, Q.; Li, X.; Sun, Y.; Li, S.; Li, Y.; Hu, G.; Cao, S.; et al. Leaf transcriptomic response mediated by cold stress in two maize inbred lines with contrasting tolerance levels. Genomics 2021, 113, 782–794. [Google Scholar] [CrossRef]
- Yang, G.; Zou, H.; Wu, Y.; Liu, H.; Yuan, Y. Identification and characterisation of candidate genes involved in chilling responses in maize (Zea Mays L.). Plant Cell 2011, 106, 127–141. [Google Scholar] [CrossRef]
- Zeng, R.; Li, Z.; Shi, Y.; Fu, D.; Yin, P.; Cheng, J.; Jiang, C.; Yang, S. Natural variation in a type-A response regulator confers maize chilling tolerance. Nat. Commun. 2021, 12, 4713. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zhang, J.; Ma, X.; Cao, S.; Li, W.; Yang, G. A Multi-Omics View of Maize’s (Zea mays L.) Response to Low Temperatures During the Seedling Stage. Int. J. Mol. Sci. 2024, 25, 12273. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Zuo, Y.; Gu, W.; Wei, S.; Li, J. Exogenous Proline Optimizes Osmotic Adjustment Substances and Active Oxygen Metabolism of Maize Embryo under Low-Temperature Stress and Metabolomic Analysis. Processes 2022, 10, 1388. [Google Scholar] [CrossRef]
- Jiang, F.; Lv, S.; Zhang, Z.; Chen, Q.; Mai, J.; Wan, X.; Liu, P. Integrated metabolomics and transcriptomics analysis during seed germination of waxy corn under low temperature stress. BMC Plant Biol. 2023, 23, 190. [Google Scholar] [CrossRef]
- Li, H.; Richard, D. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 754–1760. [Google Scholar] [CrossRef]
- Wang, K.; Mingyao, L.; Hakon, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Magwene, P.M.; Willis, J.H.; Kelly, J.K. The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput. Biol. 2011, 7, e1002255. [Google Scholar] [CrossRef]
- Mansfeld, B.N.; Grumet, R. QTLseqr: An R package for bulk segregant analysis with next-generation sequencing. Plant Genome 2018, 11, 180006. [Google Scholar] [CrossRef]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Su, Y.C.; Yost, H.J. MMAPPR: Mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef]
- Fisher, R.A. On the interpretation of $2 from contingency tables, and the calculation of P. J. R. Stat. Soc. 1922, 85, 87–94. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. bioRxiv 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNAseq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yoon, D.; Kim, Y.J.; Lee, W.K.; Choi, B.R.; Oh, S.M.; Lee, Y.S.; Kim, J.K.; Lee, D.Y. Metabolic Changes in Serum Metabolome of Beagle Dogs Fed Black Ginseng. Metabolites 2020, 10, 517. [Google Scholar] [CrossRef]
- Chen, Q.; Geng, T.; Hou, W.J.; Chen, C.Q. Impacts of Climate Warming on Growth and Yield of Spring Maize in Recent 20 Years in Northeast China. Sci. Agric. Sin. 2014, 47, 1904–1916. [Google Scholar]
- Shin, K.C.; Kim, S.; Le, A.Q.; Lee, H.S.; Adeva, C.; Jeon, Y.A.; Luong, N.H.; Kim, W.J.; Akhtamov, M.; Ahn, S.N. Fine mapping of a low-temperature germinability QTL qLTG1 using introgression lines derived from Oryza rufipogon. Plant Breed. Biotechnol. 2019, 7, 141–150. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Hagihara, S.; Hirai, D. Field assessment of a major QTL associated with tolerance to cold-induced seed coat discoloration in soybean. Breed. Sci. 2019, 69, 521–528. [Google Scholar] [CrossRef]
- Yang, L.M.; Liu, H.L.; Lei, L.; Zhao, H.W.; Wang, J.G.; Li, N.; Sun, J.; Zheng, H.L.; Zou, D.T. Identification of QTLs controlling low-temperature germinability and cold tolerance at the seedling stage in rice (Oryza Sativa L.). Euphytica 2018, 214, 1–4. [Google Scholar] [CrossRef]
- Livaja, M.; Wang, Y.; Wieckhorst, S.; Haseneyer, G.; Seidel, M.; Hahn, V.; Knapp, S.J.; Taudien, S.; Schön, C.-C.; Bauer, E. BSTA: A targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genom. 2013, 14, 628. [Google Scholar] [CrossRef]
- Liu, S.; Yeh, C.-T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [PubMed]
- Lara-Núñez, A.; Guerrero-Molina, E.D.; Vargas-Cortez, T.; Vázquez-Ramos, J.M. Interplay of CDKs and cyclins with glycolytic regulatory enzymes PFK and PK. J. Plant Physiol. 2024, 303, 154378. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wei, Y.; Chen, Y.; Ding, P.; Xu, F.; Shao, X. Gene co-expression network analysis of the transcriptome identifies cold-resistant hub genes related to carbohydrate metabolism in peach fruit. Postharvest Biol. Technol. 2025, 220, 113300. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, X.; Wang, Y.; Wang, Q.; Ma, L.; Wu, C.E.; Wang, R.; Shi, Y.; Watkins, C.B.; Zuo, J.; et al. Storage temperature affects metabolism of sweet corn. Postharvest Biol. Technol. 2025, 219, 113232. [Google Scholar] [CrossRef]
- Sproule, K.A. Microtubule Involvement in the Plant Low Temperature Response. Ph.D. Thesis, University of Saskatchewan, Saskatoon, SK, Canada, 2008. [Google Scholar]
- Xu, G.; Li, L.; Zhou, J.; He, M.; Lyu, D.; Zhao, D.; Qin, S. Integrated transcriptomics and metabolomics analyses reveal key genes and essential metabolic pathways for the acquisition of cold tolerance during dormancy in apple. Environ. Exp. Bot. 2023, 213, 105413. [Google Scholar] [CrossRef]
- Xu, J.; Huang, Z.; Du, H.; Tang, M.; Fan, P.; Yu, J.; Zhou, Y. SEC1-C3H39 module fine-tunes cold tolerance by mediating its target mRNA degradation in tomato. New Phytol. 2023, 237, 870–884. [Google Scholar] [CrossRef]
- Schuler, M.A. Plant cytochrome P450 monooxygenases. Crit. Rev. Plant Sci. 1996, 15, 235–284. [Google Scholar] [CrossRef]
- Pandian, B.A.; Sathishraj, R.; Djanaguiraman, M.; Prasad, P.V.V.; Jugulam, M. Role of cytochrome P450 enzymes in plant stress response. Antioxidants 2020, 9, 454. [Google Scholar] [CrossRef]
- Leonard, A.; Holloway, B.; Guo, M.; Rupe, M.; Yu, G.; Beatty, M.; Zastrow-Hayes, G.; Meeley, R.; Llaca, V.; Butler, K.; et al. tassel-less1 encodes a boron channel protein required for inflorescence development in maize. Plant Cell Physiol. 2014, 55, 1044–1054. [Google Scholar] [CrossRef]
- Matthes, M.S.; Darnell, Z.; Best, N.B.; Guthrie, K.; Robil, J.M.; Amstutz, J.; Durbak, A.; McSteen, P. Defects in meristem maintenance, cell division, and cytokinin signaling are early responses in the boron deficient maize mutant tassel-less1. Physiol. Plant. 2022, 174, e13670. [Google Scholar] [CrossRef]
- Ostrowski, M.; Jakubowska, A. UDP-glycosyltransferases of plant hormones. Adv. Cell Biol. 2014, 4, 43–60. [Google Scholar] [CrossRef]
- Gharabli, H.; Della Gala, V.; Welner, D.H. The function of UDP-glycosyltransferases in plants and their possible use in crop protection. Biotechnol. Adv. 2023, 67, 108182. [Google Scholar] [CrossRef]
- Jia, Y.; Zou, D.; Wang, J.; Sha, H.; Liu, H.; Inayat, M.A.; Sun, J.; Zheng, H.; Xia, N.; Zhao, H. Effects of γ-aminobutyric acid, glutamic acid, and calcium chloride on rice (Oryza sativa L.) under cold stress during the early vegetative stage. J. Plant Growth Regul. 2017, 36, 240–253. [Google Scholar] [CrossRef]
- Lager, I.; Yilmaz, J.L.; Zhou, X.R.; Jasieniecka, K.; Kazachkov, M.; Wang, P.; Zou, J.; Weselake, R.; Smith, M.A.; Bayon, S. Plant acyl-CoA: Lysophosphatidylcholine acyltransferases (LPCATs) have different specificities in their forward and reverse reactions. J. Biol. Chem. 2013, 288, 36902–36914. [Google Scholar] [CrossRef]
- Nair, P.; Kandasamy, S.; Zhang, J.; Ji, X.; Kirby, C.; Benkel, B.; Hodges, M.D.; Critchley, A.T.; Hiltz, D.; Prithiviraj, B. Transcriptional and metabolomic analysis of Ascophyllum nodosum mediated freezing tolerance in Arabidopsis thaliana. BMC Genom. 2012, 13, 643. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment | Liao2386 | Liao6082 | F1 | RIL | |
|---|---|---|---|---|---|
| Mean ± SD (%) | Mean ± SD (%) | Mean ± SD (%) | Mean ± SD (%) | Range (%) | |
| E1 | 84.67 ± 2.49 | 15.33 ± 1.89 | 50.00 ± 4.32 | 58.41 ± 23.13 | 0–98 |
| E2 | 85.67 ± 1.70 | 13.67 ± 1.25 | 48.67 ± 10.53 | 56.27 ± 24.90 | 2–100 |
| E3 | 85.33 ± 1.25 | 14.33 ± 1.70 | 50.33 ± 6.02 | 56.00 ± 23.29 | 1–97 |
| average | 85.22 ± 1.93 | 14.44 ± 1.77 | 49.66 ± 7.46 | 56.89 ± 23.81 | 0–100 |
| Bulk | Clean Reads | Date Generated (Gb) | Q30 (%) | Genome Coverage 10× (%) | Average Depth (×) | GC (%) | Total Mapped Efficiency (%) | Perfect Mapped Efficiency (%) |
|---|---|---|---|---|---|---|---|---|
| P1 | 4.96 × 108 | 74.37 | 93.05 | 91.80 | 34.11 | 46.68 | 99.18 | 88.11 |
| P2 | 4.96 × 108 | 74.41 | 89.80 | 83.13 | 34.13 | 48.08 | 99.04 | 86.02 |
| H | 5.44 × 108 | 81.61 | 92.97 | 88.13 | 37.44 | 46.29 | 98.76 | 83.83 |
| L | 4.73 × 108 | 70.89 | 92.82 | 86.69 | 32.52 | 46.86 | 98.76 | 83.51 |
| Calculation Models | CI | QTL Number | Genes Number | Chr |
|---|---|---|---|---|
| Δ(SNP-index) | 95% | 44 | 3460 | 1, 2, 3, 4, 5, 6, 7, 9, 10 |
| 99% | 13 | 739 | 1, 2, 3, 7, 9, 10 | |
| G′ value | 95% | 43 | 2922 | 1, 2, 3, 6, 7, 9, 10 |
| 99% | 8 | 528 | 1, 2, 3, 7, 9, 10 | |
| Euclidean distance | 95% | 40 | 2781 | 1, 2, 3, 6, 7, 9, 10 |
| 99% | 5 | 434 | 1, 2, 9, 10 | |
| Fisher’s exact test | 95% | 14 | 789 | 1, 2, 3, 7, 9, 10 |
| 99% | 4 | 109 | 1, 10 |
| Gene_id | P1-vs.-P2 | H-vs.-L | Annotation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Average Readcount of P1 | Average Readcount of P2 | Log2 Fold Change | p Value | Average Readcount of H | Average Readcount of L | Log2 Fold Change | p Value | ||
| Zm00001eb043000 | 8479 | 21377 | 1.35 | 0.001456 | 7275 | 16087 | 1.24 | 2.61 × 10−6 | Phosphofructose kinase2 |
| Zm00001eb043400 | 45 | 219 | 2.34 | 3.68 × 10−18 | 16 | 50 | 1.78 | 0.000237 | ATP hydrolysis activity |
| Zm00001eb043490 | 17 | 0 | −10.32 | 0.000124 | 18 | 0 | −10.36 | 6.47 × 10−5 | UDP-forming activity |
| Zm00001eb043620 | 1765 | 155 | −3.43 | 4.18 × 10−9 | 1957 | 242 | −3.03 | 0.000187 | Cytochrome P450 |
| Zm00001eb043650 | 293 | 108 | −1.38 | 0.00052 | 1060 | 197 | −2.40 | 2.22 × 10−7 | Tasselless1 |
| Zm00001eb043680 | 285 | 52 | −2.39 | 4.01 × 10−5 | 716 | 129 | −2.47 | 0.00037 | Kinesin-like protein |
| Zm00001eb043720 | 755 | 2213 | 1.58 | 0.000413 | 644 | 1803 | 1.60 | 3.37 × 10−7 | Zinc finger CCCH domain-containing protein |
| Comparison | R2X | R2Y | Q2 |
|---|---|---|---|
| P1-vs.-P2 | 0.845 | 0.963 | 0.884 |
| H-vs.-L | 0.864 | 0.973 | 0.9 |
| Class | P1-vs.-P2 | H-vs.-L | ||
|---|---|---|---|---|
| Up | Down | Up | Down | |
| Amino acid and derivatives | 15 | 4 | 6 | 14 |
| Amines | 1 | 0 | 0 | 1 |
| Phenols and its derivatives | 1 | 0 | 0 | 0 |
| Phenolic acids | 2 | 0 | 1 | 1 |
| Nucleotide and its derivates | 1 | 0 | 0 | 0 |
| Flavonoids | 1 | 0 | 0 | 1 |
| Alkaloids and derivatives | 2 | 0 | 0 | 1 |
| Organic acid and its derivatives | 4 | 1 | 3 | 2 |
| Lipids | 6 | 2 | 0 | 4 |
| Phytohormones | 2 | 0 | 0 | 2 |
| Carbohydrates and its derivatives | 0 | 0 | 1 | 13 |
| Total | 35 | 7 | 11 | 39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Hao, N.; Li, Y.; Sun, N.; Wang, L.; Ye, Y. Cold-Tolerance Candidate Gene Identification in Maize Germination Using BSA, Transcriptome and Metabolome Profiling. Agronomy 2025, 15, 1067. https://doi.org/10.3390/agronomy15051067
Wang C, Hao N, Li Y, Sun N, Wang L, Ye Y. Cold-Tolerance Candidate Gene Identification in Maize Germination Using BSA, Transcriptome and Metabolome Profiling. Agronomy. 2025; 15(5):1067. https://doi.org/10.3390/agronomy15051067
Chicago/Turabian StyleWang, Cheng, Nan Hao, Yueming Li, Nan Sun, Liwei Wang, and Yusheng Ye. 2025. "Cold-Tolerance Candidate Gene Identification in Maize Germination Using BSA, Transcriptome and Metabolome Profiling" Agronomy 15, no. 5: 1067. https://doi.org/10.3390/agronomy15051067
APA StyleWang, C., Hao, N., Li, Y., Sun, N., Wang, L., & Ye, Y. (2025). Cold-Tolerance Candidate Gene Identification in Maize Germination Using BSA, Transcriptome and Metabolome Profiling. Agronomy, 15(5), 1067. https://doi.org/10.3390/agronomy15051067