
Joint Transcriptome and Metabolome-Based Analysis Reveals Key Modules and Candidate Genes for Drought Tolerance in Wheat (Triticum aestivum L.) Seedlings

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant and Treatments
2.2. Transcriptome Sequencing and Data Analysis
2.3. Metabolomics Measurements and Data Analysis
2.4. Metabolite Module and Gene Module Construction
2.5. Gene–Metabolite Correlation Network Construction
2.6. Screening of Hub Genes
2.7. qRT-PCR
3. Results
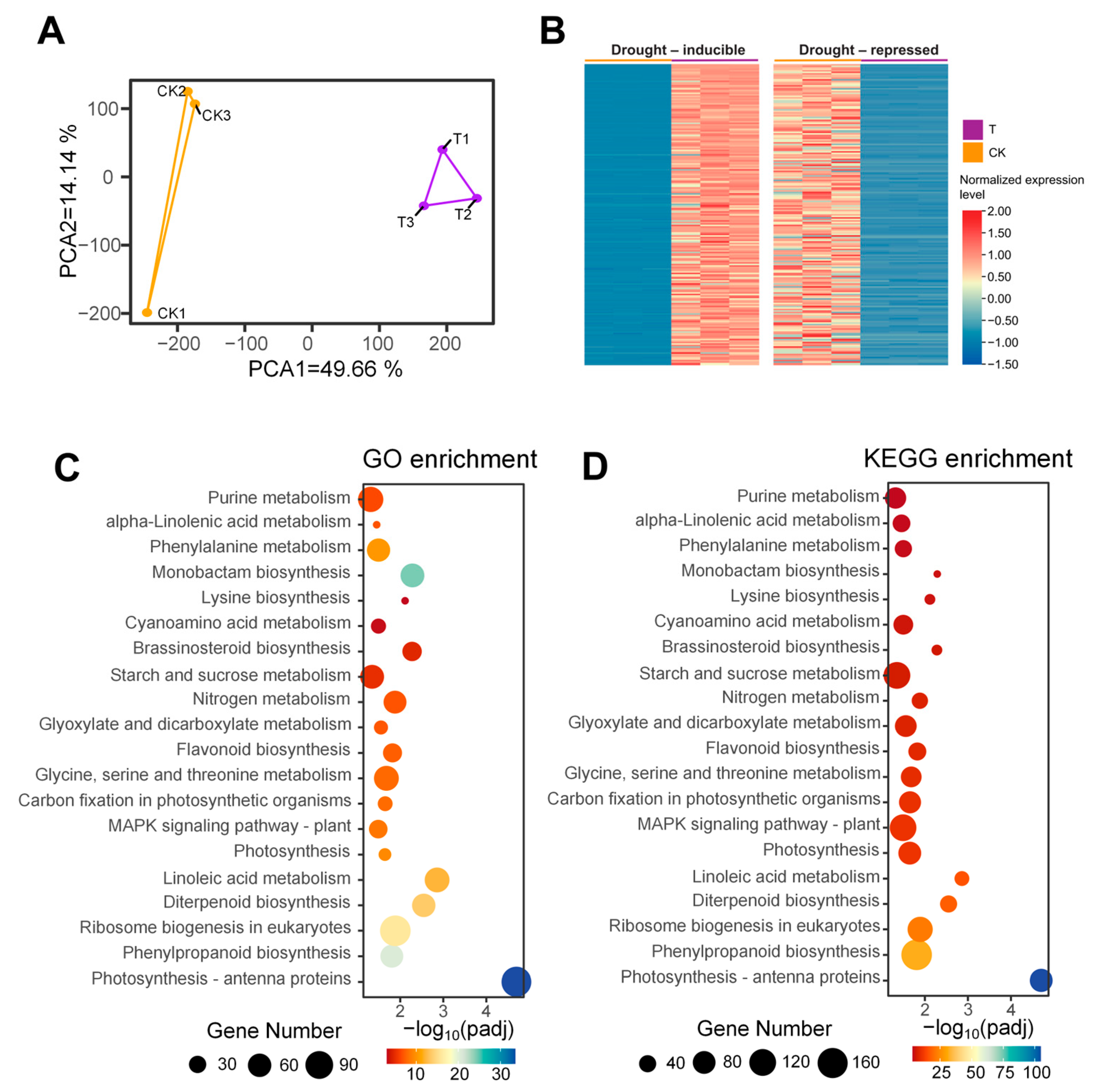
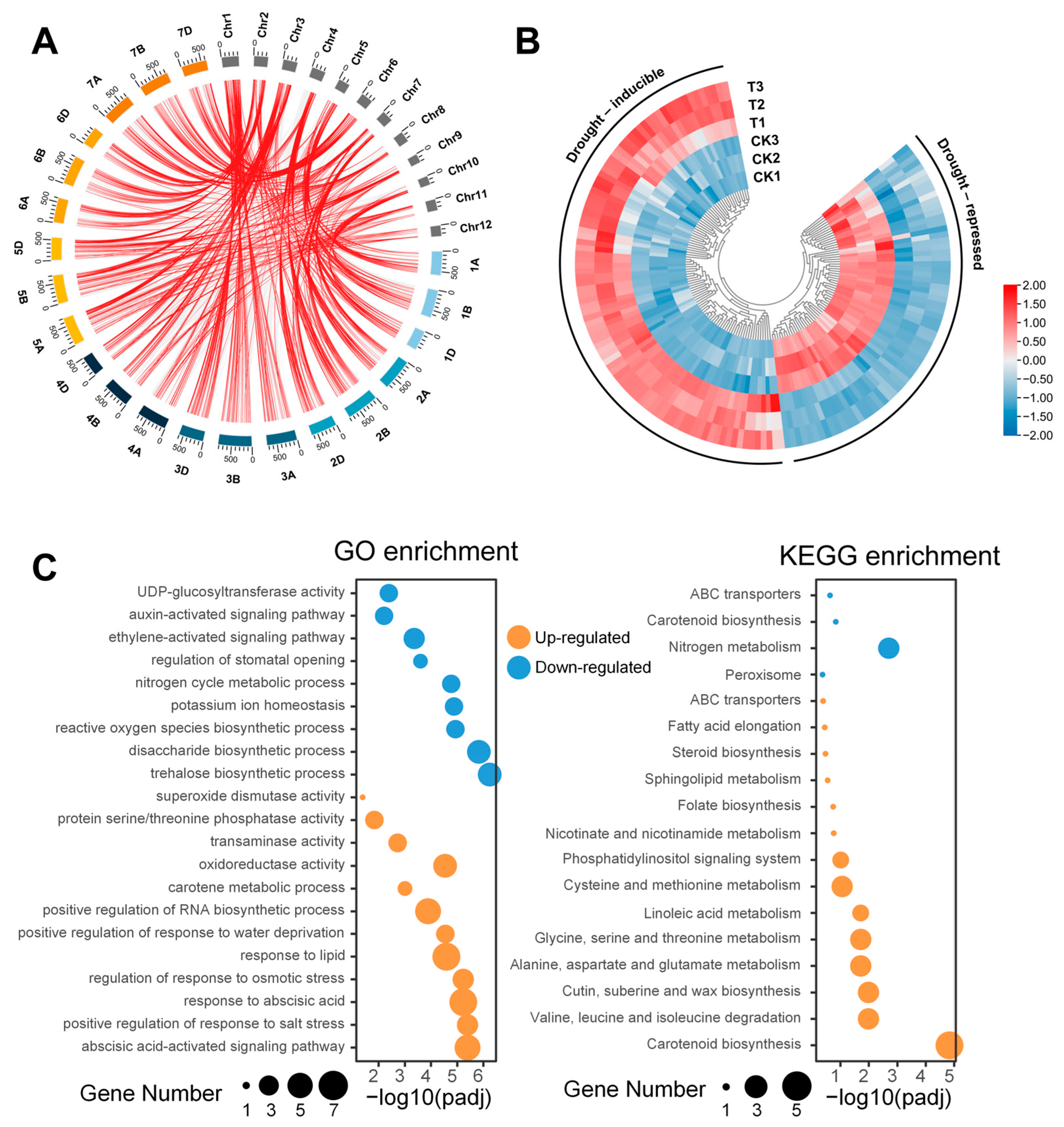
3.1. Transcriptome Analysis
3.2. Candidate Gene Mining and Functional Analysis Based on Homology Comparison
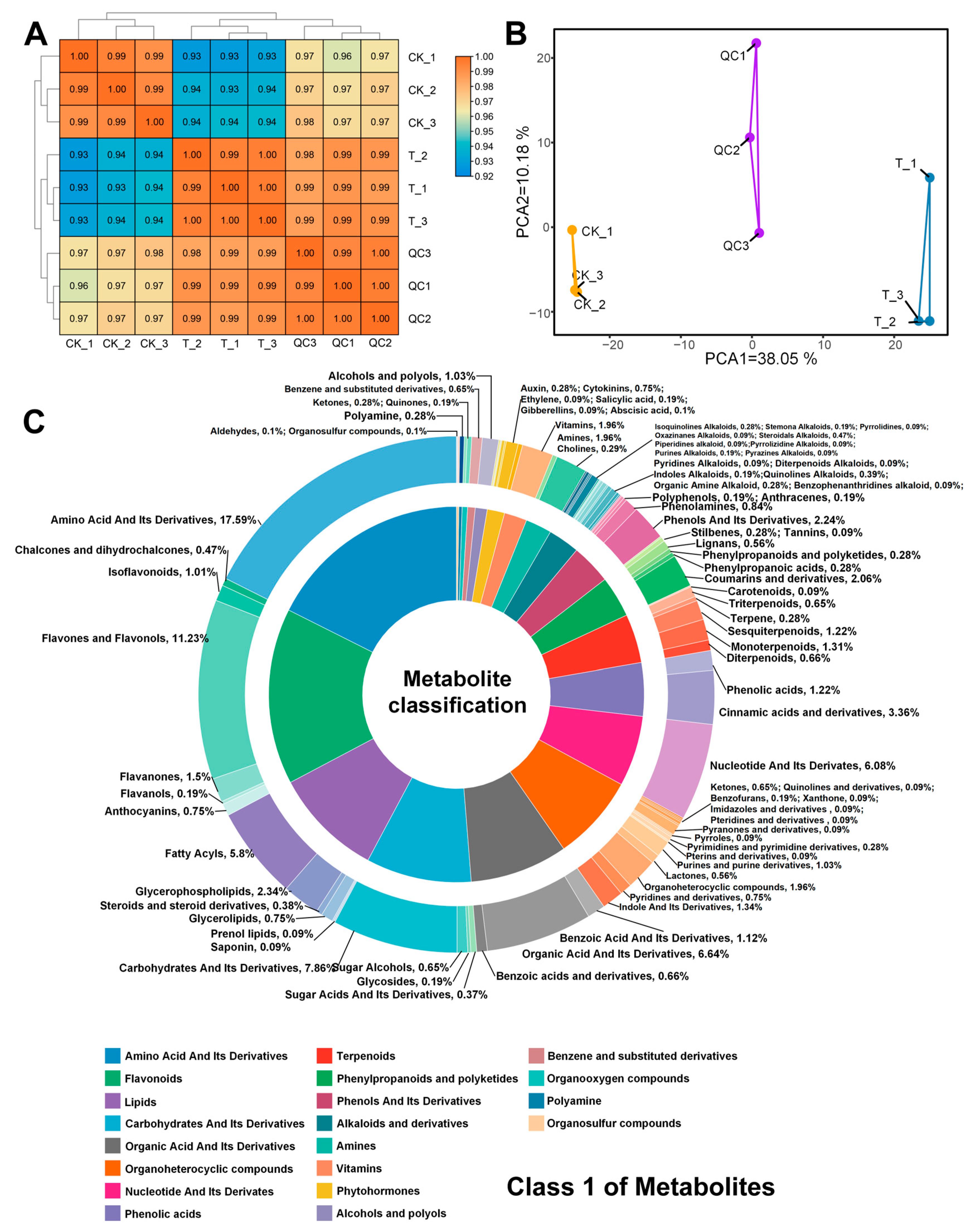
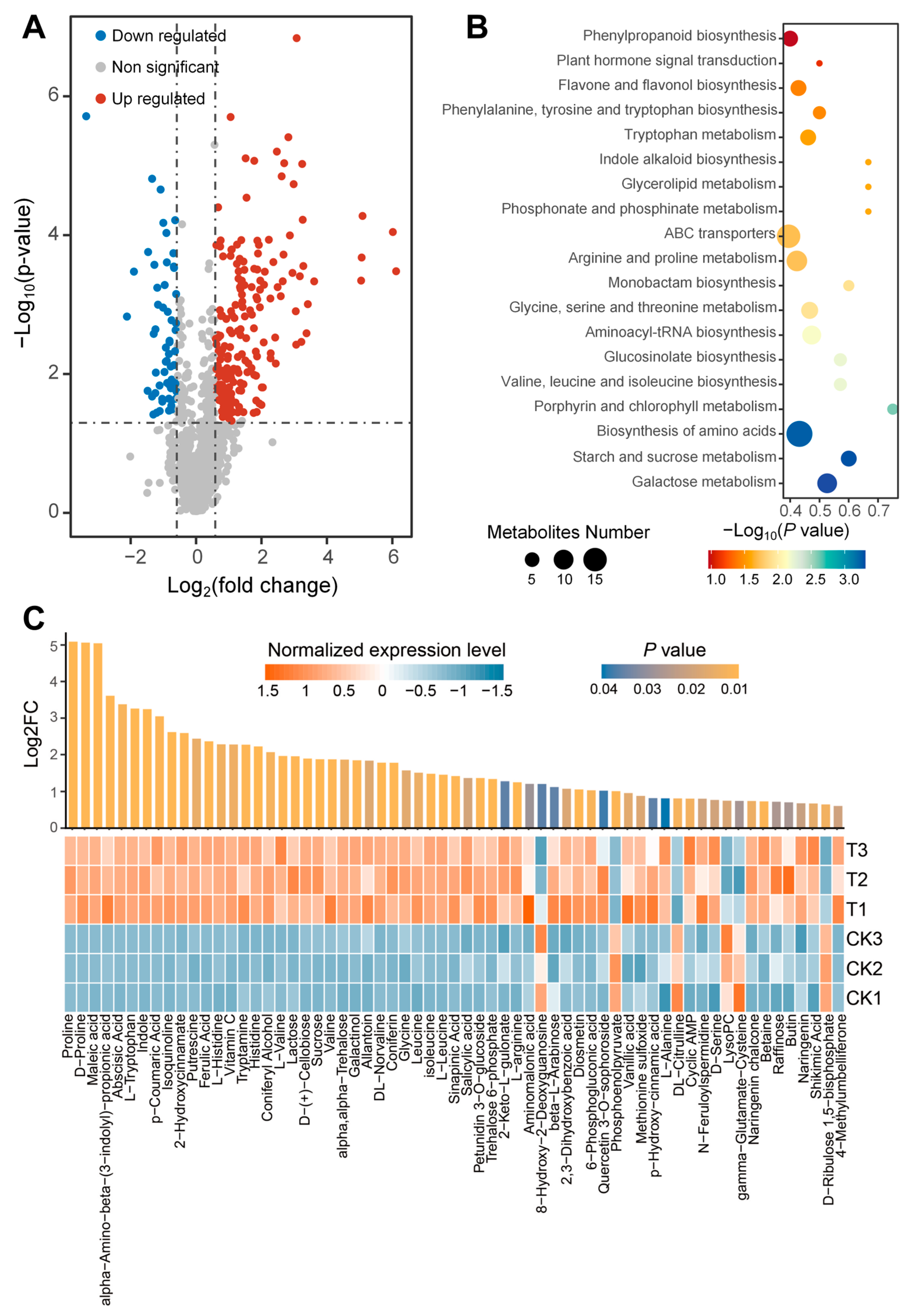
3.3. Metabolomics Analysis
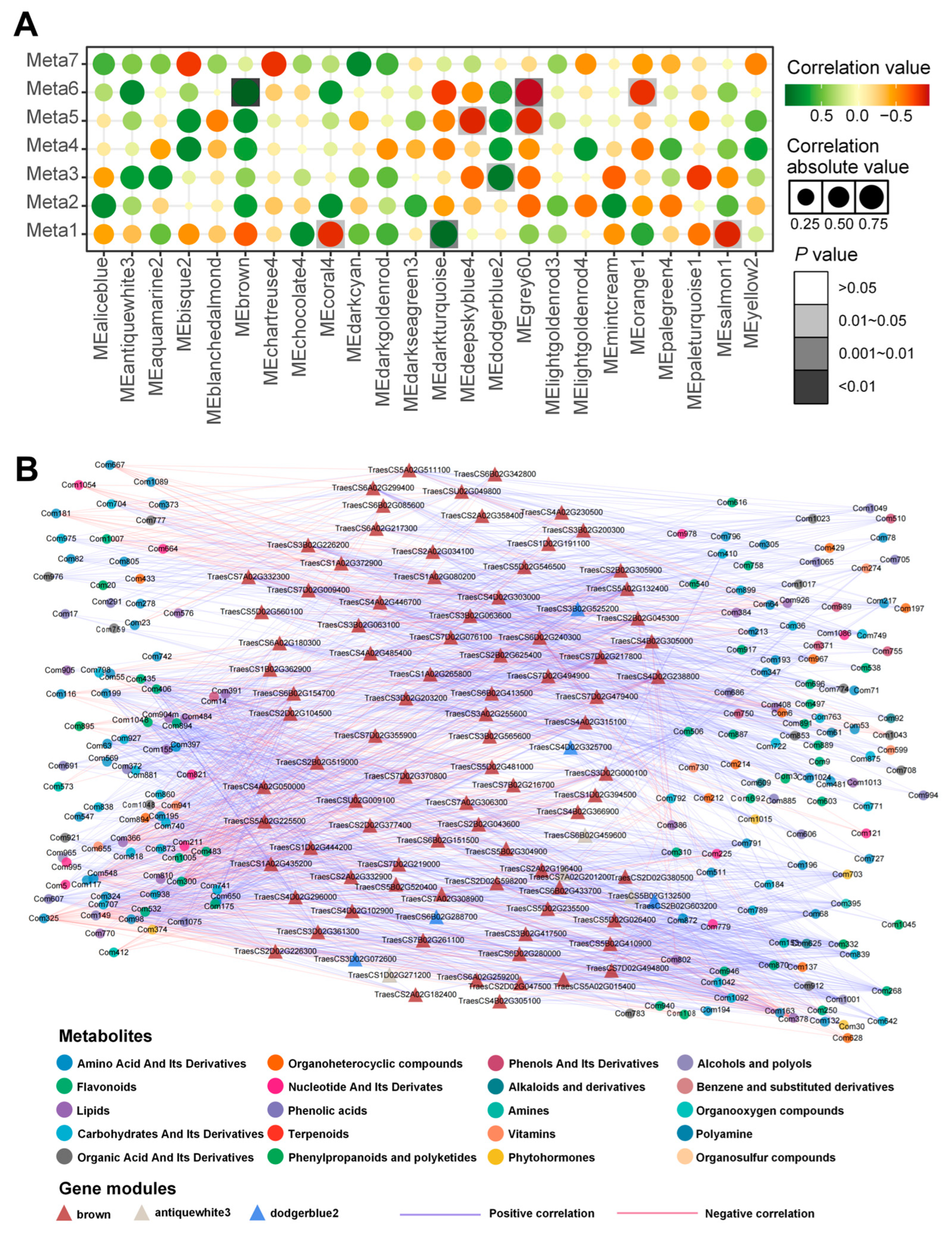
3.4. Metabolite Module Construction and Screening of Important Gene Modules
3.5. Combined Transcription-Metabolism Analysis
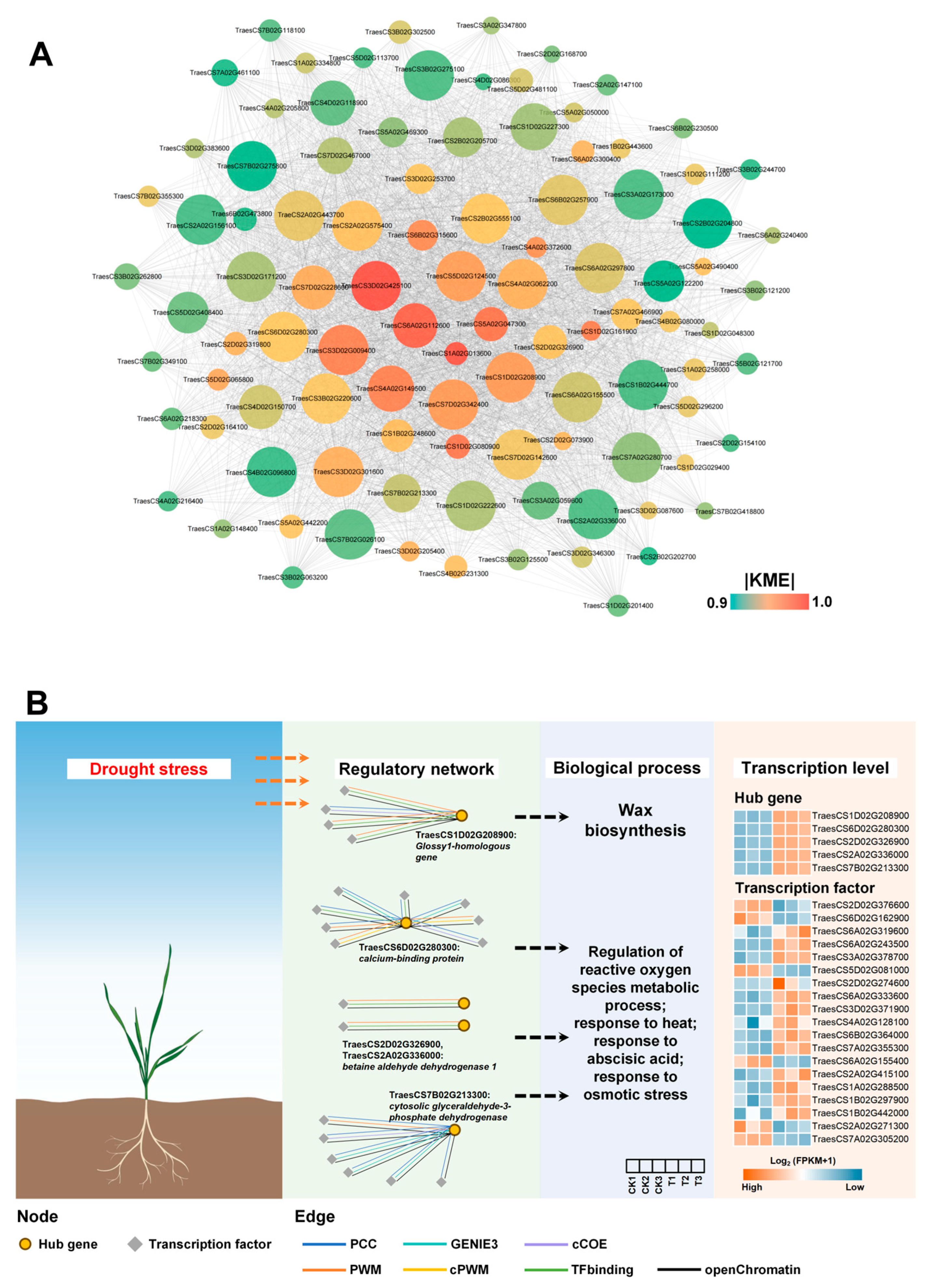
3.6. Mining and Analysis of Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tadesse, W.; Sanchez-Garcia, M.; Assefa, S.G.; Amri, A.; Bishaw, Z.; Ogbonnaya, F.; Baum, M. Genetic gains in wheat breeding and its role in feeding the world. Crop Breed. Genet. Genom. 2019, 1, e190005. [Google Scholar] [CrossRef]
- Piao, S.L.; Ciais, P.; Huang, Y.; Shen, Z.H.; Peng, S.S.; Li, J.S.; Zhou, L.P.; Liu, H.Y.; Ma, Y.C.; Ding, Y.H.; et al. The impacts of climate change on water resources and agriculture in China. Nature 2010, 467, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.; Halford, N.G. Food security: The challenge of increasing wheat yield and the importance of not compromising food safety. Ann. Appl. Biol. 2014, 164, 354–372. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Nouraei, S.; Mia, M.S.; Liu, H.; Turner, N.C.; Yan, G. Transcriptome Analyses of Near Isogenic Lines Reveal Putative Drought Tolerance Controlling Genes in Wheat. Front. Plant Sci. 2022, 13, 857829. [Google Scholar] [CrossRef]
- Xi, W.; Hao, C.; Li, T.; Wang, H.; Zhang, X. Transcriptome Analysis of Roots from Wheat (Triticum aestivum L.) Varieties in Response to Drought Stress. Int. J. Mol. Sci. 2023, 24, 7245. [Google Scholar] [CrossRef]
- Ma, J.; Li, R.; Wang, H.; Li, D.; Wang, X.; Zhang, Y.; Zhen, W.; Duan, H.; Yan, G.; Li, Y. Transcriptomics Analyses Reveal Wheat Responses to Drought Stress during Reproductive Stages under Field Conditions. Front. Plant Sci. 2017, 8, 592. [Google Scholar] [CrossRef]
- Niu, Y.; Li, J.; Sun, F.; Song, T.; Han, B.; Liu, Z.; Su, P. Comparative transcriptome analysis reveals the key genes and pathways involved in drought stress response of two wheat (Triticum aestivum L.) varieties. Genomics 2023, 115, 110688. [Google Scholar] [CrossRef]
- Hall, R.D. Plant metabolomics in a nutshell: Potential and future challenges. In Biology of Plant Metabolomics; Blackwell: Oxford, UK, 2011; Volume 43, pp. 1–24. [Google Scholar] [CrossRef]
- Meena, K.K.; Sorty, A.M.; Bitla, U.M.; Choudhary, K.; Gupta, P.; Pareek, A.; Prabha, R.; Sahu, P.K.; Gupta, V.K. Abiotic stress responses and microbe-mediated mitigation in plants: The omics strategies. Front. Plant Sci. 2017, 8, 172. [Google Scholar] [CrossRef]
- Guo, X.; Xin, Z.; Yang, T.; Ma, X.; Zhang, Y.; Wang, Z.; Ren, Y.; Lin, T. Metabolomics Response for Drought Stress Tolerance in Chinese Wheat Genotypes (Triticum aestivum). Plants 2020, 9, 520. [Google Scholar] [CrossRef] [PubMed]
- Ullah, N.; Yüce, M.; Gökçe, Z.N.O.; Budak, H. Comparative metabolite profiling of drought stress in roots and leaves of seven Triticeae species. BMC Genom. 2017, 18, 969. [Google Scholar] [CrossRef]
- Guo, R.; Shi, L.; Jiao, Y.; Li, M.; Zhong, X.; Gu, F.; Liu, Q.; Xia, X.; Li, H. Metabolic responses to drought stress in the tissues of drought-tolerant and drought-sensitive wheat genotype seedlings. AoB Plants 2018, 10, ply016. [Google Scholar] [CrossRef]
- Kang, Z.; Babar, M.A.; Khan, N.; Guo, J.; Khan, J.; Islam, S.; Shrestha, S.; Shahi, D. Comparative metabolomic profiling in the roots and leaves in contrasting genotypes reveals complex mechanisms involved in post-anthesis drought tolerance in wheat. PLoS ONE 2019, 14, e0213502. [Google Scholar] [CrossRef]
- Bouhaddani, S.E.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.W. Evaluation of O2PLS in Omics data integration. BMC Bioinform. 2016, 17 (Suppl. S2), 11. [Google Scholar] [CrossRef]
- Bylesjö, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data integration in plant biology: The O2PLS method for combined modeling of transcript and metabolite data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef]
- Szymanski, J.; Brotman, Y.; Willmitzer, L.; Cuadros-Inostroza, Á. Linking gene expression and membrane lipid composition of Arabidopsis. Plant Cell 2014, 26, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, P.; Sun, J.; Li, B.; Jia, J.; Yuan, J.; Li, X.; Gu, S.; Pang, X. CsMYC2 is involved in the regulation of phenylpropanoid biosynthesis induced by trypsin in cucumber (Cucumis sativus) during storage. Plant Physiol. Biochem. 2023, 196, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ebeed, H.T.; Stevenson, S.R.; Cuming, A.C.; Baker, A. Conserved and differential transcriptional responses of peroxisome associated pathways to drought, dehydration and ABA. J. Exp. Bot. 2018, 69, 4971–4985. [Google Scholar] [CrossRef]
- Nxele, X.; Klein, A.; Ndimba, B.K. Drought and salinity stress alters ROS accumulation, water retention, and osmolyte content in sorghum plants. S. Afr. J. Bot. 2017, 108, 261–266. [Google Scholar] [CrossRef]
- Varoquaux, N.; Cole, B.; Gao, C.; Pierroz, G.; Baker, C.R.; Patel, D.; Madera, M.; Jeffers, T.; Hollingsworth, J.; Sievert, J.; et al. Transcriptomic analysis of field-droughted sorghum from seedling to maturity reveals biotic and metabolic responses. Proc. Natl. Acad. Sci. USA 2019, 116, 27124–27132. [Google Scholar] [CrossRef]
- Xiong, H.; Yu, J.; Miao, J.; Li, J.; Zhang, H.; Wang, X.; Liu, P.; Zhao, Y.; Jiang, C.; Yin, Z.; et al. Natural variation in OsLG3 increases drought tolerance in rice by inducing ROS scavenging. Plant Physiol. 2018, 178, 451–467. [Google Scholar] [CrossRef]
- Ijaz, R.; Ejaz, J.; Gao, S.; Liu, T.; Imtiaz, M.; Ye, Z.; Wang, T. Overexpression of Annexin gene AnnSp2, enhances drought and salt tolerance through modulation of ABA synthesis and scavenging ROS in tomato. Sci. Rep. 2017, 7, 12087. [Google Scholar] [CrossRef]
- Anjum, S.A.; Ashraf, U.; Tanveer, M.; Khan, I.; Hussain, S.; Shahzad, B.; Zohaib, A.; Abbas, F.; Saleem, M.F.; Ali, I.; et al. Drought induced changes in growth, osmolyte accumulation and antioxidant metabolism of three maize hybrids. Front. Plant Sci. 2017, 8, 69. [Google Scholar] [CrossRef]
- Qiao, B.; Zhang, Q.; Liu, D.; Wang, H.; Yin, J.; Wang, R.; He, M.; Cui, M.; Shang, Z.; Wang, D.; et al. A calcium-binding protein, rice annexin OsANN1, enhances heat stress tolerance by modulating the production of H2O2. J. Exp. Bot. 2015, 66, 5853–5866. [Google Scholar] [CrossRef]
- Jia, Y.; Gu, X.; Chai, J.; Yao, X.; Cheng, S.; Liu, L.; He, S.; Peng, Y.; Zhang, Q.; Zhu, Z. Rice OsANN9 enhances drought tolerance through modulating ROS scavenging systems. Int. J. Mol. Sci. 2023, 24, 17495. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Pan, G.; Zhou, N.; Zhai, Z.; Yang, L.; Zhu, H.; Cui, X.; Zhao, P.; Zhang, H.; Li, S.; et al. Calcium-dependent protein kinase 5 (CPK5) positively modulates drought tolerance through phosphorylating ABA-responsive element binding factors in oilseed rape (Brassica napus L.). Plant Sci. 2022, 315, 111125. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.; Lim, C.W.; Lee, S.C. Roles of pepper bZIP transcription factor CaATBZ1 and its interacting partner RING-type E3 ligase CaASRF1 in modulation of ABA signalling and drought tolerance. Plant J. 2019, 100, 399–410. [Google Scholar] [CrossRef]
- Pan, Y.; Hu, X.; Li, C.; Xu, X.; Su, C.; Li, J.; Song, H.; Zhang, X.; Pan, Y. SlbZIP38, a tomato bZIP family gene downregulated by abscisic acid, is a negative regulator of drought and salt stress tolerance. Genes 2017, 8, 402. [Google Scholar] [CrossRef]
- Gulzar, F.; Fu, J.; Zhu, C.; Yan, J.; Li, X.; Meraj, T.A.; Shen, Q.; Hassan, B.; Wang, Q. Maize WRKY transcription factor ZmWRKY79 positively regulates drought tolerance through elevating ABA biosynthesis. Int. J. Mol. Sci. 2021, 22, 10080. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; He, L.; Wu, Y. Wheat WRKY transcription factor TaWRKY24 confers drought and salt tolerance in transgenic plants. Plant Physiol. Biochem. 2023, 205, 108137. [Google Scholar] [CrossRef]
- Liang, Y.; Ma, F.; Li, B.; Guo, C.; Hu, T.; Zhang, M.; Liang, Y.; Zhu, J.; Zhan, X. A bHLH transcription factor, SlbHLH96, promotes drought tolerance in tomato. Hortic. Res. 2022, 9, uhac198. [Google Scholar] [CrossRef]
- Li, C.; Yan, C.; Sun, Q.; Wang, J.; Yuan, C.; Mou, Y.; Shan, S.; Zhao, X. The bHLH transcription factor AhbHLH112 improves the drought tolerance of peanut. BMC Plant Biol. 2021, 21, 540. [Google Scholar] [CrossRef]
- Gao, S.S.; Wang, Y.L.; Yu, S.; Huang, Y.Q.; Liu, H.C.; Chen, W.; He, X.Y. Effects of drought stress on growth, physiology and secondary metabolites of two Adonis species in Northeast China. Sci. Hortic. 2020, 259, 108855. [Google Scholar] [CrossRef]
- Weretilnyk, E.A.; Hanson, A.D. Molecular cloning of a plant betaine-aldehyde dehydrogenase, an enzyme implicated in adaptation to salinity and drought. Proc. Natl. Acad. Sci. USA 1990, 87, 2745–2749. [Google Scholar] [CrossRef]
- Tang, W.; Sun, J.; Liu, J.; Liu, F.; Yan, J.; Gou, X.; Lu, B.R.; Liu, Y. RNAi-directed downregulation of betaine aldehyde dehydrogenase 1 (OsBADH1) results in decreased stress tolerance and increased oxidative markers without affecting glycine betaine biosynthesis in rice (Oryza sativa). Plant Mol. Biol. 2014, 86, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.J.; Zhang, M.; Zhang, H.X.; Zhang, P. Improved tolerance to various abiotic stresses in transgenic sweet potato (Ipomoea batatas) expressing spinach betaine aldehyde dehydrogenase. PLoS ONE 2012, 7, e37344. [Google Scholar] [CrossRef]
- Yang, C.L.; Zhou, Y.; Fan, J.; Fu, Y.H.; Shen, L.B.; Yao, Y.; Li, R.; Fu, S.; Duan, R.; Hu, X.; et al. SpBADH of the halophyte Sesuvium portulacastrum strongly confers drought tolerance through ROS scavenging in transgenic Arabidopsis. Plant Physiol. Biochem. 2015, 96, 377–387. [Google Scholar] [CrossRef]
- Yu, M.; Yu, Y.; Guo, S.; Zhang, M.; Li, N.; Zhang, S.; Zhou, H.; Wei, F.; Song, T.; Cheng, J.; et al. Identification of TaBADH-A1 allele for improving drought resistance and salt tolerance in wheat (Triticum aestivum L.). Front. Plant Sci. 2022, 13, 942359. [Google Scholar] [CrossRef]
- Ranjbar, M.; Khakdan, F.; Ghorbani, A.; Zargar, M.; Chen, M. The variations in gene expression of GAPDH in Ocimum basilicum cultivars under drought-induced stress conditions. Environ. Sci. Pollut. Res. 2023, 30, 119187–119203. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Yang, S. Cytosolic TaGAPC2 enhances tolerance to drought stress in transgenic Arabidopsis plants. Int. J. Mol. Sci. 2020, 21, 7499. [Google Scholar] [CrossRef]
- Zeng, L.; Deng, R.; Guo, Z.; Yang, S.; Deng, X. Genome-wide identification and characterization of glyceraldehyde-3-phosphate dehydrogenase genes family in wheat (Triticum aestivum). BMC Genom. 2016, 17, 240. [Google Scholar] [CrossRef]
- Geng, S.; Li, S.; Zhao, J.; Gao, W.; Chen, Q.; Zheng, K.; Wang, Y.; Jiao, Y.; Long, Y.; Liu, P.; et al. Glyceraldehyde-3-phosphate dehydrogenase Gh_GAPDH9 is associated with drought resistance in Gossypium hirsutum. PeerJ 2023, 11, e16445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Rao, X.L.; Shi, H.T.; Li, R.J.; Lu, Y.T. Overexpression of a cytosolic glyceraldehyde-3-phosphate dehydrogenase gene OsGAPC3 confers salt tolerance in rice. Plant Cell Tissue Organ Cult. 2011, 107, 1–11. [Google Scholar] [CrossRef]
- Sun, M.; Li, Y.; Zheng, J.; Wu, D.; Li, C.; Li, Z.; Zang, Z.; Zhang, Y.; Fang, Q.; Li, W.; et al. A Nuclear Factor Y-B Transcription Factor, GmNFYB17, Regulates Resistance to Drought Stress in Soybean. Int. J. Mol. Sci. 2022, 23, 7242. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Zhang, X.; Lu, X.; Chen, G.; Chen, Z.H. Molecular and evolutionary mechanisms of cuticular wax for plant drought tolerance. Front. Plant Sci. 2017, 8, 621. [Google Scholar] [CrossRef]
- Patwari, P.; Salewski, V.; Gutbrod, K.; Kreszies, T.; Dresen-Scholz, B.; Peisker, H.; Steiner, U.; Meyer, A.J.; Schreiber, L.; Dörmann, P. Surface wax esters contribute to drought tolerance in Arabidopsis. Plant J. 2019, 98, 727–744. [Google Scholar] [CrossRef]
- Seo, P.J.; Lee, S.B.; Suh, M.C.; Park, M.J.; Go, Y.S.; Park, C.M. The MYB96 transcription factor regulates cuticular wax biosynthesis under drought conditions in Arabidopsis. Plant Cell 2011, 23, 1138–1152. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, H.U.; Suh, M.C. MYB94 and MYB96 additively activate cuticular wax biosynthesis in Arabidopsis. Plant Cell Physiol. 2016, 57, 2300–2311. [Google Scholar] [CrossRef]
- Kim, H.; Chung, M.S.; Yoo, S. DEWAX2 transcription factor negatively regulates cuticular wax biosynthesis in Arabidopsis leaves. Plant Cell Physiol. 2018, 59, 966–977. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Li, C.; Hu, N.; Zhu, Y.; He, Z.; Sun, Y.; Wang, Z.; Wang, Y. ECERIFERUM1-6A is required for the synthesis of cuticular wax alkanes and promotes drought tolerance in wheat. Plant Physiol. 2022, 190, 1640–1657. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; He, S.; Zhai, H.; Li, R.; Zhao, N.; Liu, Q. A sweet potato auxin response factor gene (IbARF5) is involved in carotenoid biosynthesis and salt and drought tolerance in transgenic Arabidopsis. Front. Plant Sci. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, X.; Liu, X.; Wu, C.; Yu, C.; Hu, G.; Chen, L.; Chen, R.; Bouzayen, M.; Zouine, M.; et al. Knockout of auxin response factor SlARF4 improves tomato resistance to water deficit. Int. J. Mol. Sci. 2021, 22, 3347. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Li, L.; Wei, A.; Zhou, L.; Wang, B.; Liu, M.; Lei, Y.; Xie, Y.; Li, X. TaMYB44-5A reduces drought tolerance by repressing transcription of TaRD22-3A in the abscisic acid signaling pathway. Planta 2024, 260, 52. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Zeng, C.; Men, Y.; Li, N.; Zhao, Y.; Chen, Z.; Huang, Y.; Hu, Y.; Zotova, L.; Dauren, S.; et al. Joint Transcriptome and Metabolome-Based Analysis Reveals Key Modules and Candidate Genes for Drought Tolerance in Wheat (Triticum aestivum L.) Seedlings. Agronomy 2025, 15, 922. https://doi.org/10.3390/agronomy15040922
Li L, Zeng C, Men Y, Li N, Zhao Y, Chen Z, Huang Y, Hu Y, Zotova L, Dauren S, et al. Joint Transcriptome and Metabolome-Based Analysis Reveals Key Modules and Candidate Genes for Drought Tolerance in Wheat (Triticum aestivum L.) Seedlings. Agronomy. 2025; 15(4):922. https://doi.org/10.3390/agronomy15040922
Chicago/Turabian StyleLi, Ling, Chaowu Zeng, Yihan Men, Na Li, Yujiao Zhao, Zeyu Chen, Yanju Huang, Yingang Hu, Lyudmila Zotova, Serikbay Dauren, and et al. 2025. "Joint Transcriptome and Metabolome-Based Analysis Reveals Key Modules and Candidate Genes for Drought Tolerance in Wheat (Triticum aestivum L.) Seedlings" Agronomy 15, no. 4: 922. https://doi.org/10.3390/agronomy15040922
APA StyleLi, L., Zeng, C., Men, Y., Li, N., Zhao, Y., Chen, Z., Huang, Y., Hu, Y., Zotova, L., Dauren, S., Song, Q., Li, J., & Chen, L. (2025). Joint Transcriptome and Metabolome-Based Analysis Reveals Key Modules and Candidate Genes for Drought Tolerance in Wheat (Triticum aestivum L.) Seedlings. Agronomy, 15(4), 922. https://doi.org/10.3390/agronomy15040922