

Comparative Transcriptomic Profiling Reveals Divergent Drought-Response Mechanisms Between Resistant and Susceptible Apple Genotype Roots


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Experimental Design of This Study
2.2. RNA Extraction and RNA-Seq Library Construction
2.3. The Bioinformatic Analysis of RNA-Seq Data
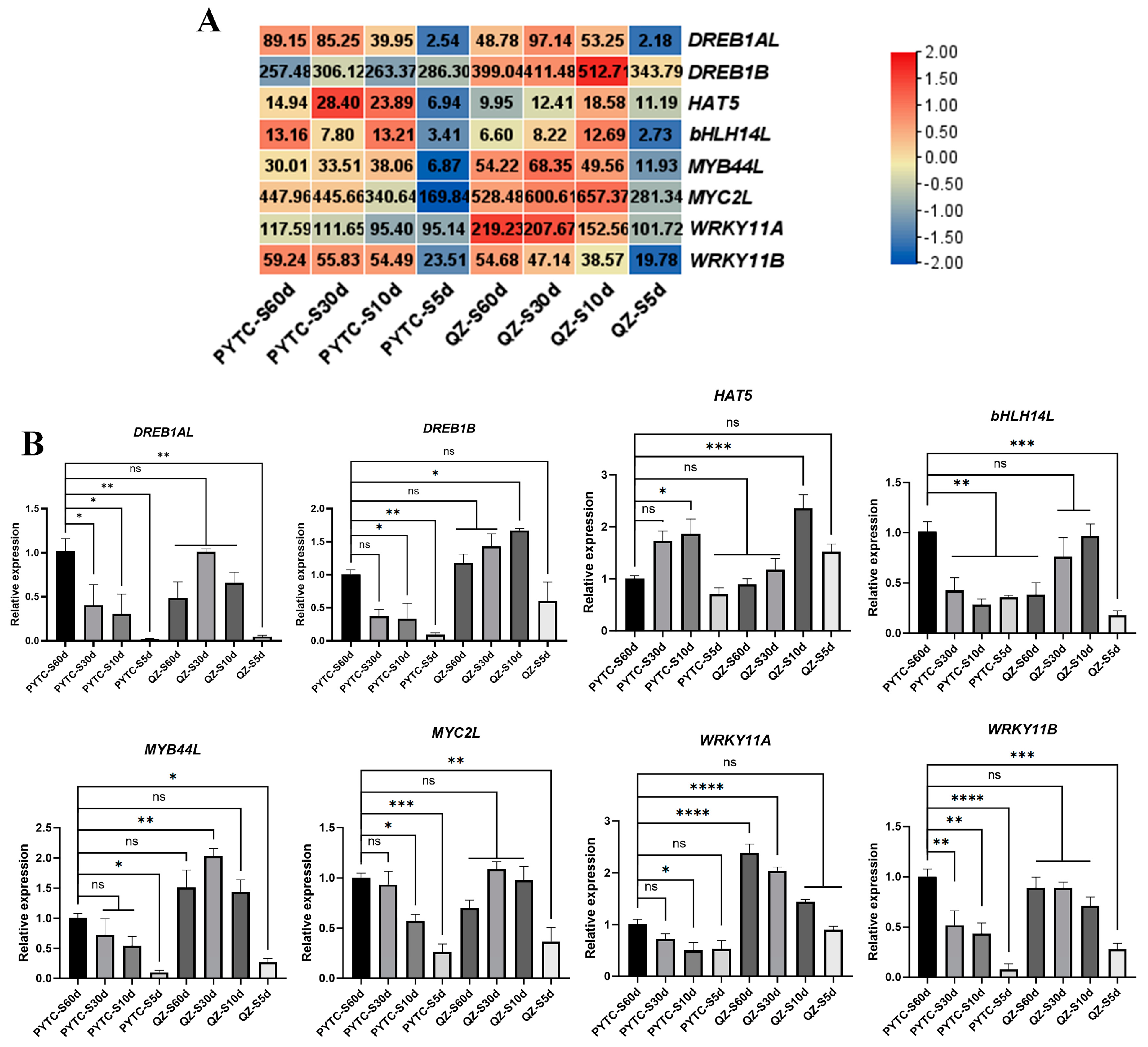
2.4. Quantitative RT-PCR Analysis
2.5. Co-Expression Network Construction and Trait Association Analysis of Modules
2.6. Statistical Analysis
3. Results
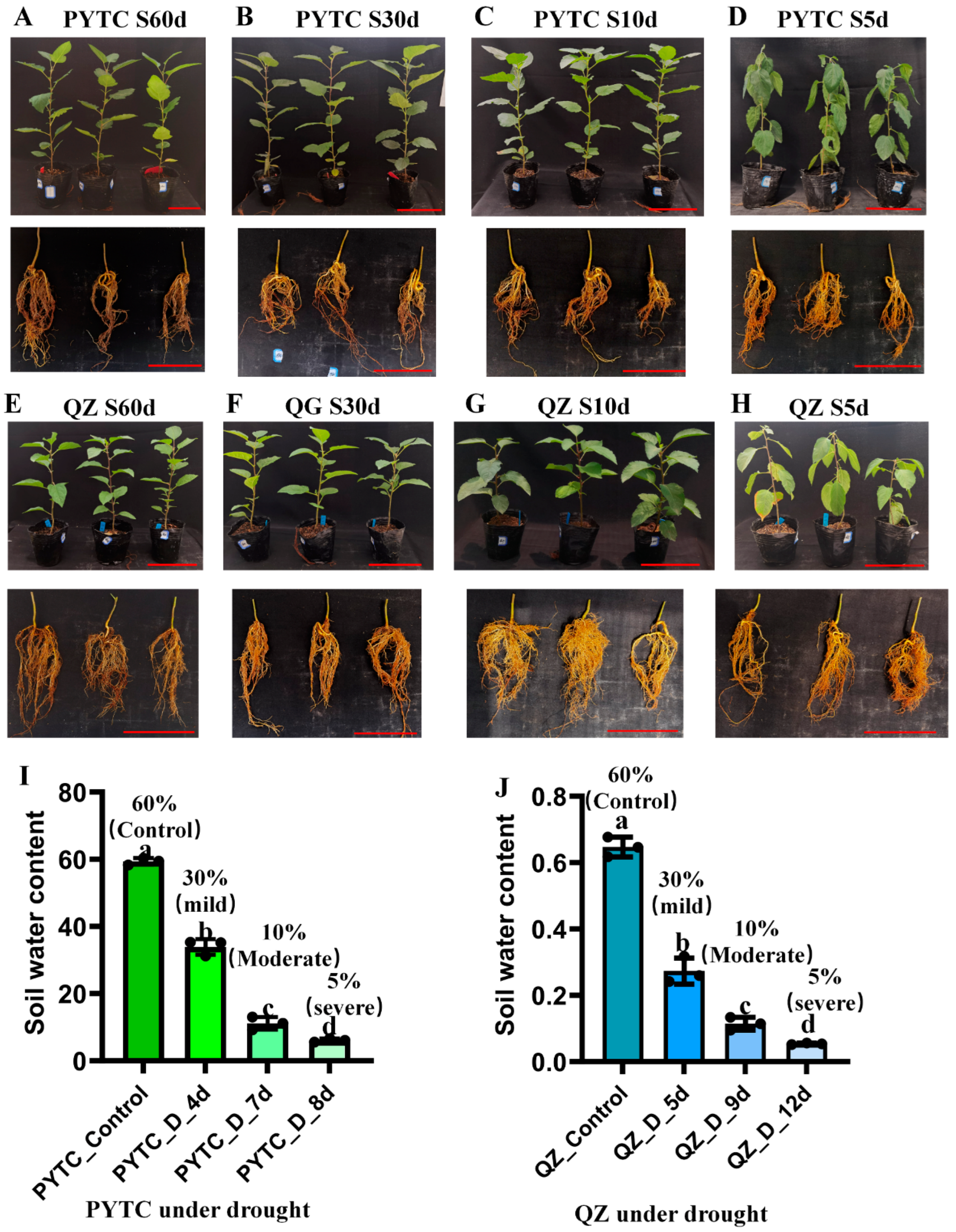
3.1. Drought Treatments on Drought Resistant (Malus prunifolia) and Susceptible (Malus hupehensis) Apple Genotype Roots
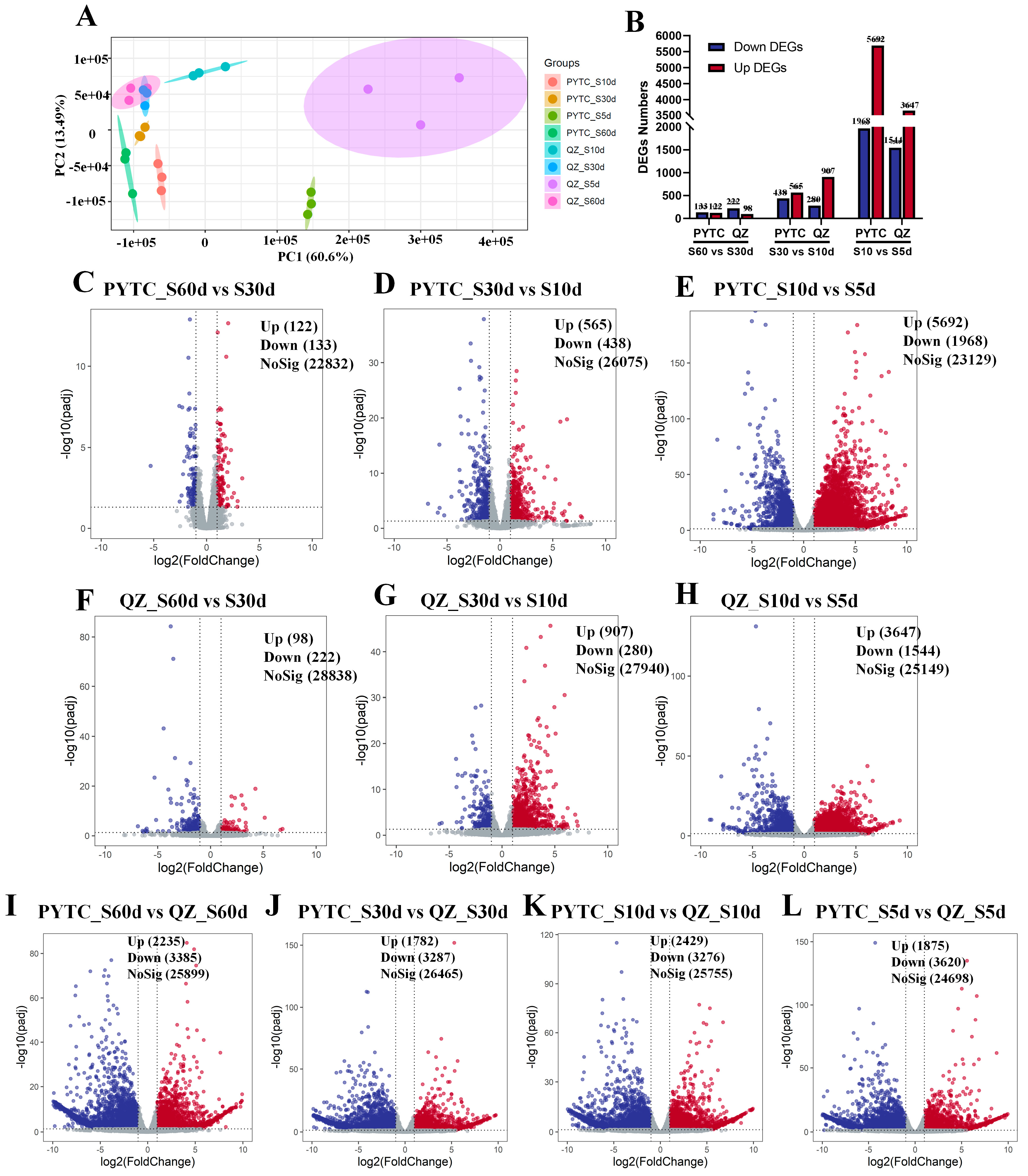
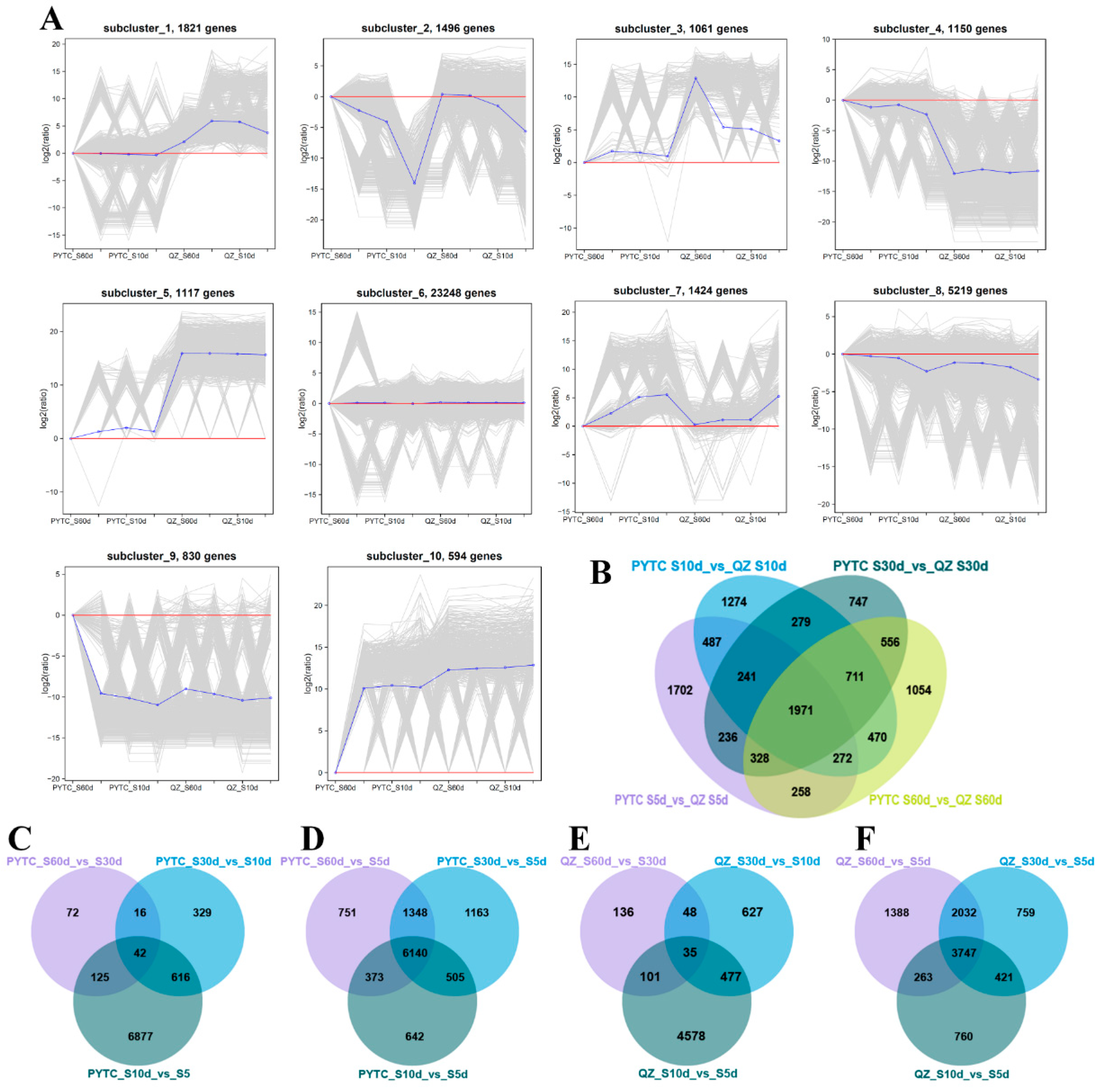
3.2. Transcriptomic Analysis of Malus prunifolia and Malus hupehensis Roots Under Drought
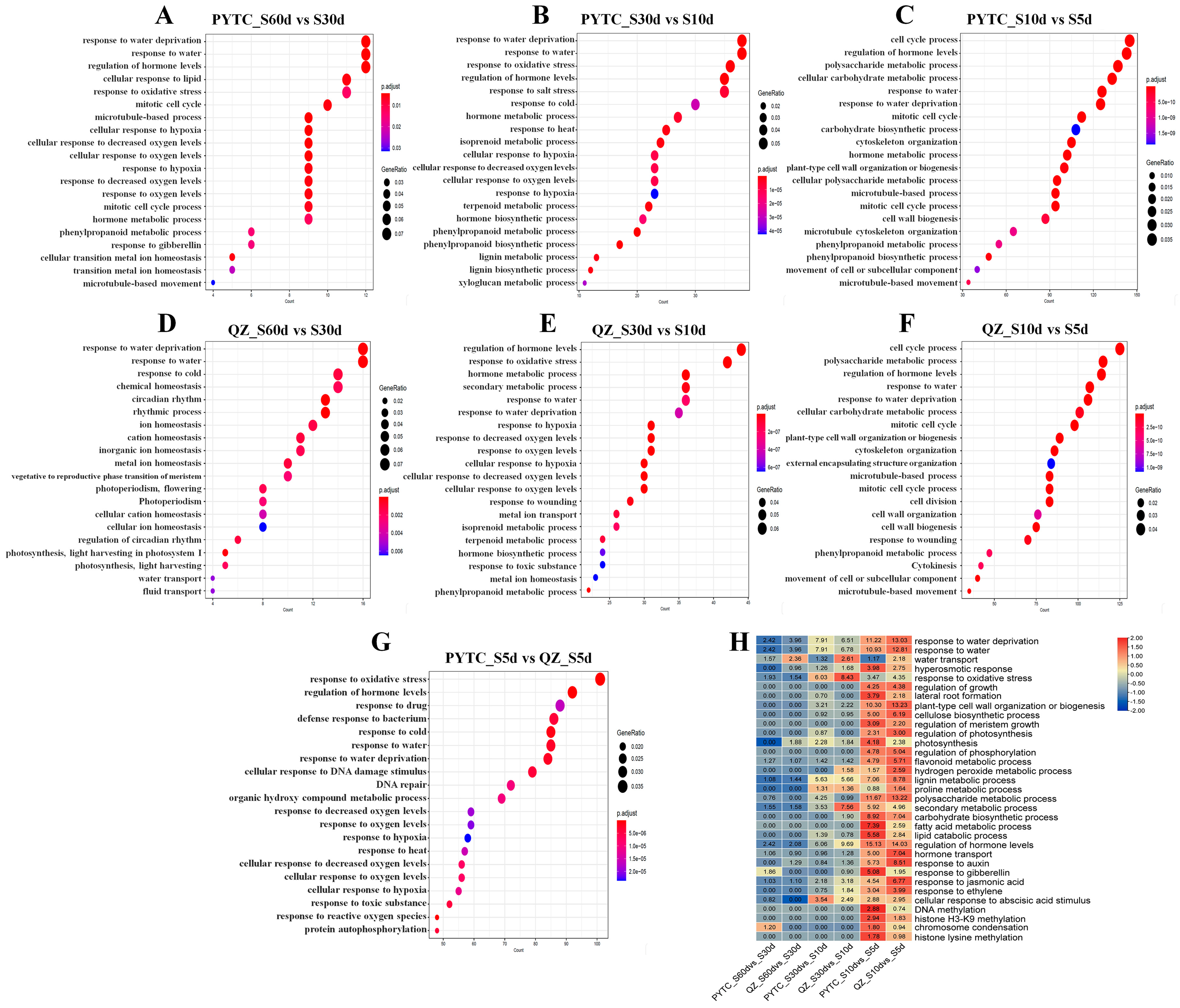
3.3. Gene Ontology (GO) Analysis Revealed the Gene Functions of DEGs Among Malus prunifolia and Malus hupehensis Roots Under Drought
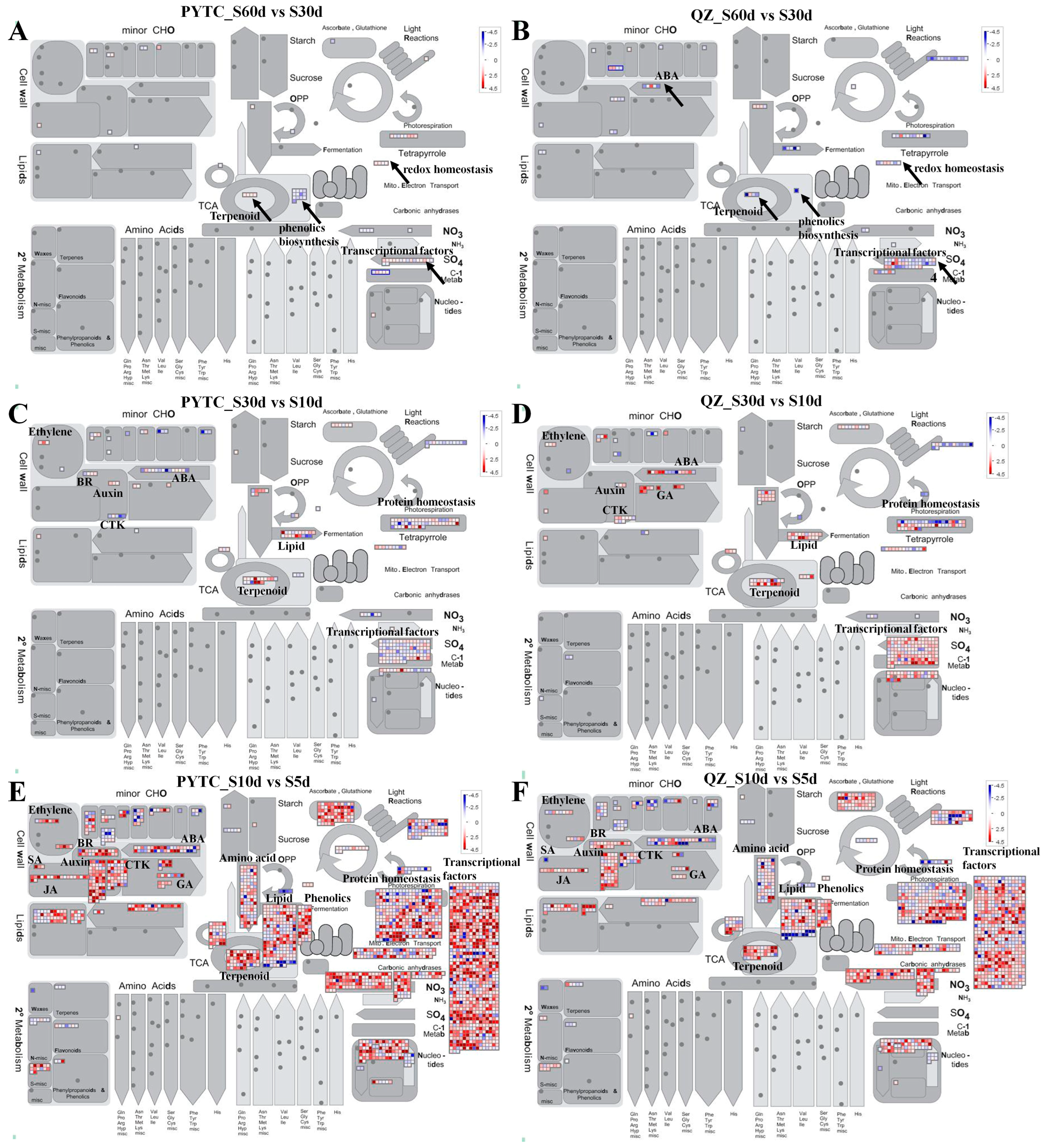
3.4. Differentially Expressed Genes Involved in Various Biological Regulatory Pathways
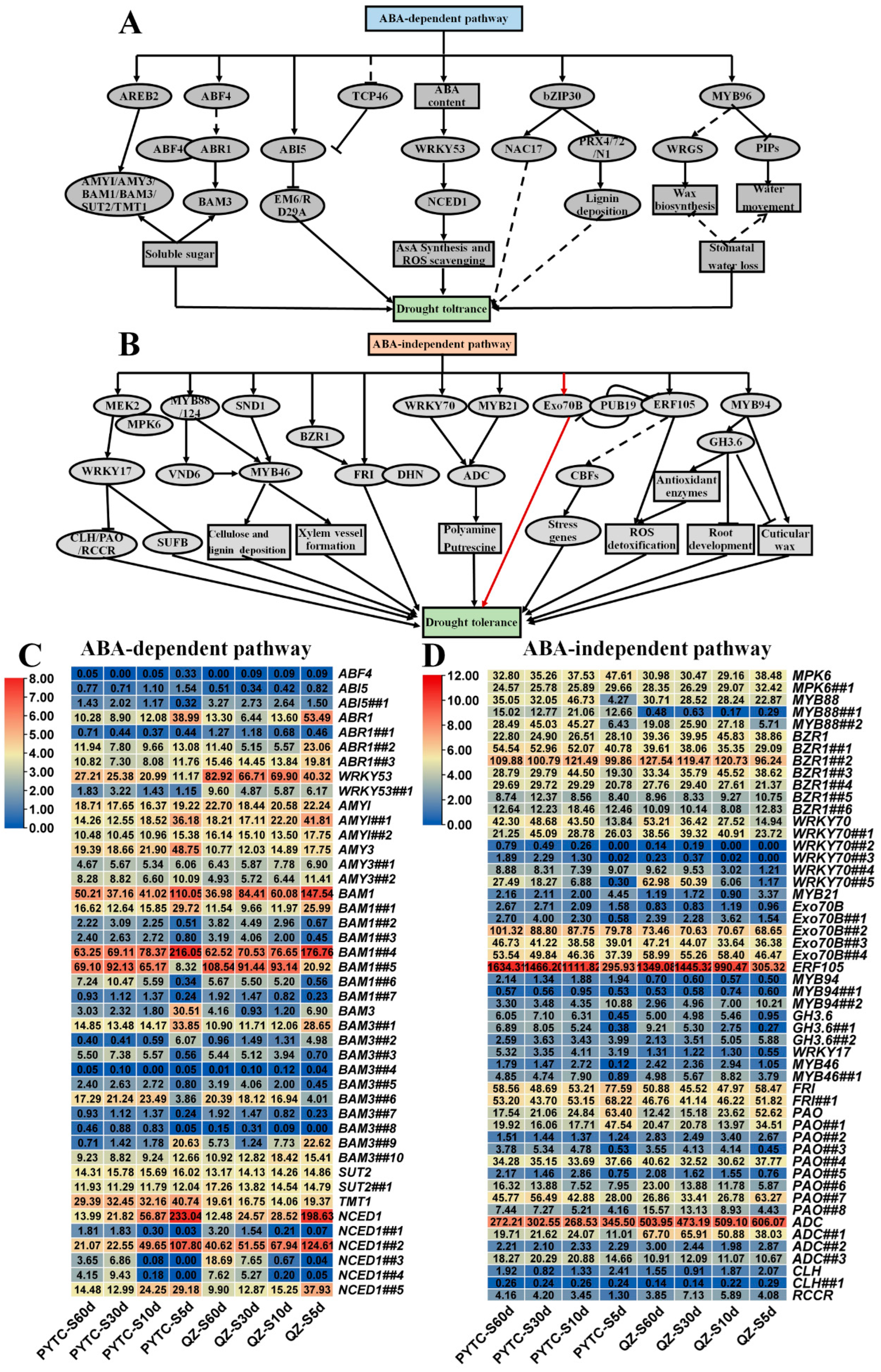
3.5. Gene Expression Alterations in Drought-Responsive Pathways
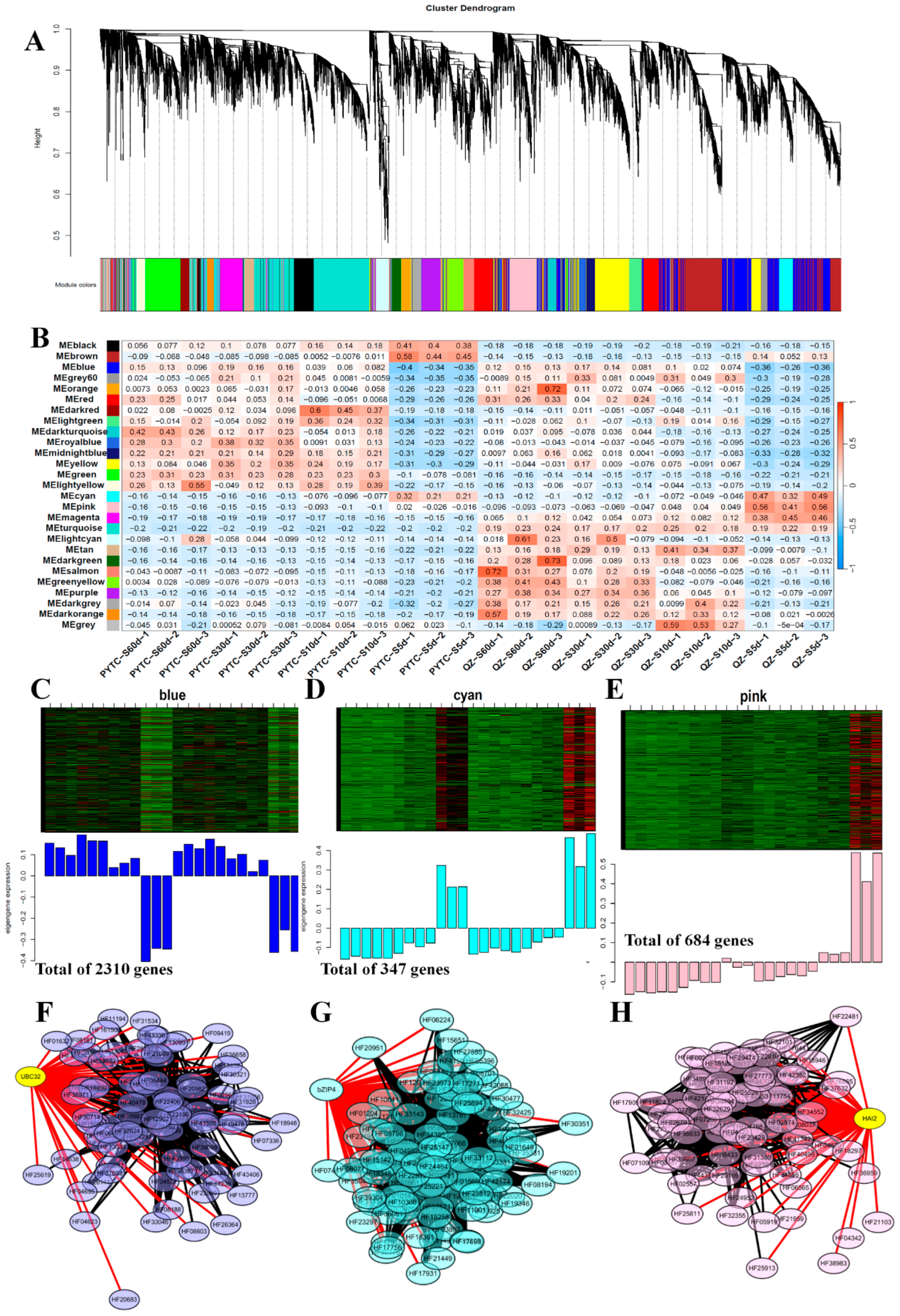
3.6. Weighted Gene Co-Expression Network Analysis (WGCNA) of DEGs Among Apple Root Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Ding, Y.L.; Yang, Y.Q.; Song, C.P.; Wang, B.S.; Yang, S.H.; Guo, Y.; Gong, Z.Z. Protein kinases in plant responses to drought, salt, and cold stress. J. Integr. Plant Biol. 2021, 63, 53–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, T.; Liu, C.; Mao, K.; Gong, X.; Li, C.; Ma, F. Fruit crops combating drought: Physiological responses and regulatory pathways. Plant Physiol. 2023, 192, 1768–1784. [Google Scholar] [CrossRef]
- Li, W.J.; Deng, M.T.; Wang, S.C.; Wang, C.X.; Guo, M.M.; Song, Y.; Guo, J.X.; Yan, J.J.; Ma, F.W.; Guan, Q.M.; et al. HISTONE DEACETYLASE 6 interaction with ABSCISIC ACID-INSENSITIVE 5 decreases apple drought tolerance. Plant Physiol. 2023, 193, 2711–2733. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sheng, L.; Li, F.; Zhou, T.; Guo, J.; Chang, Y.; Yang, J.; Jin, Y.; Chen, Y.; Lu, X. Seasonal drought promotes citrate accumulation in citrus fruit through the CsABF3-activated pathway. New Phytol. 2024, 242, 1131–1145. [Google Scholar] [CrossRef]
- Liu, Y.J.; An, J.P.; Gao, N.; Wang, X.; Chen, X.X.; Wang, X.F.; Zhang, S.; You, C.X. MdTCP46 interacts with MdABI5 to negatively regulate ABA signalling and drought response in apple. Plant Cell Environ. 2022, 45, 3233–3248. [Google Scholar] [CrossRef]
- Ma, Q.-J.; Sun, M.-H.; Lu, J.; Liu, Y.-J.; Hu, D.-G.; Hao, Y.-J. Transcription factor AREB2 is involved in soluble sugar accumulation by activating sugar transporter and amylase genes. Plant Physiol. 2017, 174, 2348–2362. [Google Scholar] [CrossRef]
- Zhang, T.T.; Lin, Y.J.; Liu, H.F.; Liu, Y.Q.; Zeng, Z.F.; Lu, X.Y.; Li, X.W.; Zhang, Z.L.; Zhang, S.; You, C.X.; et al. The AP2/ERF transcription factor MdDREB2A regulates nitrogen utilisation and sucrose transport under drought stress. Plant Cell Environ. 2024, 47, 1668–1684. [Google Scholar] [CrossRef]
- Mei, F.M.; Chen, B.; Du, L.Y.; Li, S.M.; Zhu, D.H.; Chen, N.; Zhang, Y.F.; Li, F.F.; Wang, Z.X.; Cheng, X.X.; et al. A gain-of-function allele of a DREB transcription factor gene ameliorates drought tolerance in wheat. Plant Cell 2022, 34, 4472–4494. [Google Scholar] [CrossRef]
- Kim, J.-S.; Kidokoro, S.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Regulatory networks in plant responses to drought and cold stress. Plant Physiol. 2024, 195, 170–189. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, X.; Shen, X. Regulatory network established by transcription factors transmits drought stress signals in plant. Stress. Biology 2022, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Geng, D.L.; Chen, P.X.; Shen, X.X.; Zhang, Y.; Li, X.W.; Jiang, L.J.; Xie, Y.P.; Niu, C.D.; Zhang, J.; Huang, X.H.; et al. MdMYB88 and MdMYB124 Enhance Drought Tolerance by Modulating Root Vessels and Cell Walls in Apple. Plant Physiol. 2018, 178, 1296–1309. [Google Scholar] [CrossRef]
- Chen, K.; Guo, Y.; Song, M.; Liu, L.; Xue, H.; Dai, H.; Zhang, Z. Dual role of MdSND1 in the biosynthesis of lignin and in signal transduction in response to salt and osmotic stress in apple. Hortic. Res. 2020, 7, 204. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, D.; Liu, C.; Shen, W.; He, J.; Yue, Q.; Niu, C.; Yang, F.; Li, X.; Shen, X. MdGH3. 6 is targeted by MdMYB94 and plays a negative role in apple water-deficit stress tolerance. Plant J. 2022, 109, 1271–1289. [Google Scholar] [CrossRef]
- Shan, D.; Wang, C.; Song, H.; Bai, Y.; Zhang, H.; Hu, Z.; Wang, L.; Shi, K.; Zheng, X.; Yan, T. The MdMEK2–MdMPK6–MdWRKY17 pathway stabilizes chlorophyll levels by directly regulating MdSUFB in apple under drought stress. Plant J. 2021, 108, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.K.; Ao, Q.Q.; Yang, L.Q.; Lu, F.X.; Cheng, H.K.; Fang, Q.X.; Li, C.; Chen, Q.Q.; Yan, J.L.; Wei, Y.S.; et al. Rapeseed PP2C37 Interacts with PYR/PYL Abscisic Acid Receptors and Negatively Regulates Drought Tolerance. J. Agr. Food Chem. 2024, 72, 12445–12458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Ma, C.N.; Gu, K.D.; Wang, J.H.; Yu, J.Q.; Liu, B.; Wang, Y.; He, J.X.; Hu, D.G.; Sun, Q. The BTB-BACK-TAZ domain protein MdBT2 reduces drought resistance by weakening the positive regulatory effect of MdHDZ27 on apple drought tolerance via ubiquitination. Plant J. 2024, 119, 283–299. [Google Scholar] [CrossRef]
- Hou, N.; Li, C.S.; He, J.Q.; Liu, Y.; Yu, S.S.; Malnoy, M.; Tahir, M.M.; Xu, L.F.; Ma, F.W.; Guan, Q.M. MdMTA-mediated m(6)A modification enhances drought tolerance by promoting mRNA stability and translation efficiency of genes involved in lignin deposition and oxidative stress. New Phytol. 2022, 234, 1294–1314. [Google Scholar] [CrossRef]
- Shen, X.X.; He, J.Q.; Ping, Y.K.; Guo, J.X.; Hou, N.; Cao, F.G.; Li, X.W.; Geng, D.L.; Wang, S.C.; Chen, P.X.; et al. The positive feedback regulatory loop of miR160-Auxin Response Factor 17-HYPONASTIC LEAVES 1 mediates drought tolerance in apple trees. Plant Physiol. 2022, 188, 1686–1708. [Google Scholar] [CrossRef]
- Li, Z.; Wang, L.; He, J.; Li, X.; Hou, N.; Guo, J.; Niu, C.; Li, C.; Liu, S.; Xu, J.; et al. Chromosome-scale reference genome provides insights into the genetic origin and grafting-mediated stress tolerance of Malus prunifolia. Plant Biotechnol. J. 2022, 20, 1015–1017. [Google Scholar] [CrossRef]
- Ye, N.H.; Zhai, H.; Du, Z.J.; Xu, H. Evaluation of drought resistance of ten apple rootstocks. J. Fruit Sci. 2004, 21, 395–398. [Google Scholar]
- Wang, S.; Liang, D.; Li, C.; Hao, Y.; Ma, F.; Shu, H. Influence of drought stress on the cellular ultrastructure and antioxidant system in leaves of drought-tolerant and drought-sensitive apple rootstocks. Plant Physiol. Biochem. 2012, 51, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; He, J.; Deng, M.; Wang, C.; Wang, R.; Yan, J.; Luo, M.; Ma, F.; Guan, Q.; Xu, J. Integrating ATAC-seq and RNA-seq Reveals the Dynamics of Chromatin Accessibility and Gene Expression in Apple Response to Drought. Int. J. Mol. Sci. 2022, 23, 11191. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Hu, J.; Han, X.L.; Li, J.J.; Gao, Y.; Richards, C.M.; Zhang, C.X.; Tian, Y.; Liu, G.M.; Gul, E.A.; et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 2019, 10, 1494. [Google Scholar] [CrossRef]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nature Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Lyu, F.; Han, F.; Ge, C.; Mao, W.; Chen, L.; Hu, H.; Chen, G.; Lang, Q.; Fang, C. OmicStudio: A composable bioinformatics cloud platform with real-time feedback that can generate high-quality graphs for publication. Imeta 2023, 2, e85. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef]
- Takahashi, F.; Suzuki, T.; Osakabe, Y.; Betsuyaku, S.; Kondo, Y.; Dohmae, N.; Fukuda, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K. A small peptide modulates stomatal control via abscisic acid in long-distance signalling. Nature 2018, 556, 235–238. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Qu, Z.; Guo, W.; van Nocker, S.; Zhang, C. Grapevine VviERF105 promotes tolerance to abiotic stress and is degraded by the E3 ubiquitin ligase VviPUB19. Environ. Exp. Bot. 2022, 201, 105001. [Google Scholar] [CrossRef]
- Xu, Y.-Y.; Zeng, R.-F.; Zhou, H.; Qiu, M.-Q.; Gan, Z.-M.; Yang, Y.-L.; Hu, S.-F.; Zhou, J.-J.; Hu, C.-G.; Zhang, J.-Z. Citrus FRIGIDA cooperates with its interaction partner dehydrin to regulate drought tolerance. Plant J. 2022, 111, 164–182. [Google Scholar] [CrossRef]
- Seleiman, M.F.; Al-Suhaibani, N.; Ali, N.; Akmal, M.; Alotaibi, M.; Refay, Y.; Dindaroglu, T.; Abdul-Wajid, H.H.; Battaglia, M.L. Drought stress impacts on plants and different approaches to alleviate its adverse effects. Plants 2021, 10, 259. [Google Scholar] [CrossRef]
- Mukherjee, A.; Dwivedi, S.; Bhagavatula, L.; Datta, S. Integration of light and ABA signaling pathways to combat drought stress in plants. Plant Cell Rep. 2023, 42, 829–841. [Google Scholar] [CrossRef]
- Muhammad Aslam, M.; Waseem, M.; Jakada, B.H.; Okal, E.J.; Lei, Z.; Saqib, H.S.A.; Yuan, W.; Xu, W.; Zhang, Q. Mechanisms of Abscisic Acid-Mediated Drought Stress Responses in Plants. Int. J. Mol. Sci. 2022, 23, 1084. [Google Scholar] [CrossRef]
- Antoni, R.; Gonzalez-Guzman, M.; Rodriguez, L.; Peirats-Llobet, M.; Pizzio, G.A.; Fernandez, M.A.; De Winne, N.; De Jaeger, G.; Dietrich, D.; Bennett, M.J.; et al. PYRABACTIN RESISTANCE1-LIKE8 Plays an Important Role for the Regulation of Abscisic Acid Signaling in Root. Plant Physiol. 2013, 161, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Wang, G.; Augstein, F.; de Vries, J.; Carlsbecker, A. Continuous root xylem formation and vascular acclimation to water deficit involves endodermal ABA signalling via miR165. Development 2018, 145, dev159202. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Complex plant responses to drought and heat stress under climate change. Plant J. 2024, 117, 1873–1892. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef]
- Wang, C.; Yan, J.; Hu, B.; Wang, R.; Miao, B.; Zeng, F.; Ma, F.; Guan, Q.; Xu, J. Genome-wide analysis of histone deacetylases in Apple and functional analysis of MdHDA6 in drought and salt stress responses. Sci. Hortic. 2023, 320, 112196. [Google Scholar] [CrossRef]
- Xu, J.; He, J.; Hu, B.; Hou, N.; Guo, J.; Wang, C.; Li, X.; Li, Z.; Zhai, J.; Zhang, T.; et al. Global hypermethylation of the N6-methyladenosine RNA modification associated with apple heterografting. Plant Physiol. 2023, 193, 2513–2537. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Liu, R.; Wu, Y.; Wei, S.; Wang, Q.; Zheng, Y.; Xia, R.; Shang, X.; Yu, F.; Yang, X.; et al. ERAD-related E2 and E3 enzymes modulate the drought response by regulating the stability of PIP2 aquaporins. Plant Cell 2021, 33, 2883–2898. [Google Scholar] [CrossRef]
- Feng, H.; Wang, S.; Dong, D.; Zhou, R.; Wang, H. Arabidopsis Ubiquitin-Conjugating Enzymes UBC7, UBC13, and UBC14 Are Required in Plant Responses to Multiple Stress Conditions. Plants 2020, 9, 723. [Google Scholar] [CrossRef]
- Fu, X.; Tang, X.; Zhang, N.; Si, H. StUBC13, a Ubiquitin-Conjugating Enzyme, Positively Regulates Salt and Osmotic Stresses in Potato. Int. J. Mol. Sci. 2024, 25, 13197. [Google Scholar] [CrossRef]
- Zhiguo, E.; Zhang, Y.; Li, T.; Wang, L.; Zhao, H. Characterization of the Ubiquitin-Conjugating Enzyme Gene Family in Rice and Evaluation of Expression Profiles under Abiotic Stresses and Hormone Treatments. PLoS ONE 2015, 10, e0122621. [Google Scholar]
- Wan, X.; Mo, A.; Liu, S.; Yang, L.; Li, L. Constitutive expression of a peanut ubiquitin-conjugating enzyme gene in Arabidopsis confers improved water-stress tolerance through regulation of stress-responsive gene expression. J. Biosci. Bioeng. 2011, 111, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-A.; Chang, R.-Z.; Qiu, L.-J. Overexpression of soybean ubiquitin-conjugating enzyme gene GmUBC2 confers enhanced drought and salt tolerance through modulating abiotic stress-responsive gene expression in Arabidopsis. Plant Mol. Biol. 2009, 72, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.; Huque, A.K.M.M.; Jung, K.W.; Kim, Y.Y.; Shin, J.S. A Stress-Responsive CaM-Binding Transcription Factor, bZIP4, Confers Abiotic Stress Resistance in Arabidopsis. J. Plant Biol. 2021, 64, 359–370. [Google Scholar] [CrossRef]
- Chen, Z.; Luan, Y.; Zhao, D.; Tao, J. Isolation and functional analysis of drought related PobZIP4 transcription factor in Paeonia ostii. Sci. Hortic. 2024, 332, 113187. [Google Scholar] [CrossRef]
- Bhaskara, G.B.; Nguyen, T.T.; Verslues, P.E. Unique Drought Resistance Functions of theHighly ABA-InducedClade A Protein Phosphatase 2Cs. Plant Physiol. 2012, 160, 379–395. [Google Scholar] [CrossRef]
- Shazadee, H.; Khan, N.; Wang, L.; Wang, X. GhHAI2, GhAHG3, and GhABI2 Negatively Regulate Osmotic Stress Tolerance via ABA-Dependent Pathway in Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2022, 13, 905181. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Wang, S.; Xiao, D.; Yang, M.; Miao, B.; Niu, B.; Wang, J.; Wei, J.; Ma, F.; Xu, J. Comparative Transcriptomic Profiling Reveals Divergent Drought-Response Mechanisms Between Resistant and Susceptible Apple Genotype Roots. Agronomy 2025, 15, 748. https://doi.org/10.3390/agronomy15030748
Yan J, Wang S, Xiao D, Yang M, Miao B, Niu B, Wang J, Wei J, Ma F, Xu J. Comparative Transcriptomic Profiling Reveals Divergent Drought-Response Mechanisms Between Resistant and Susceptible Apple Genotype Roots. Agronomy. 2025; 15(3):748. https://doi.org/10.3390/agronomy15030748
Chicago/Turabian StyleYan, Jinjiao, Shicong Wang, Dan Xiao, Mengyao Yang, Bingjie Miao, Bolin Niu, Jiangbo Wang, Jie Wei, Fengwang Ma, and Jidi Xu. 2025. "Comparative Transcriptomic Profiling Reveals Divergent Drought-Response Mechanisms Between Resistant and Susceptible Apple Genotype Roots" Agronomy 15, no. 3: 748. https://doi.org/10.3390/agronomy15030748
APA StyleYan, J., Wang, S., Xiao, D., Yang, M., Miao, B., Niu, B., Wang, J., Wei, J., Ma, F., & Xu, J. (2025). Comparative Transcriptomic Profiling Reveals Divergent Drought-Response Mechanisms Between Resistant and Susceptible Apple Genotype Roots. Agronomy, 15(3), 748. https://doi.org/10.3390/agronomy15030748