
Research Progress of Single-Cell Transcriptome Sequencing Technology in Plants

Abstract
1. Introduction
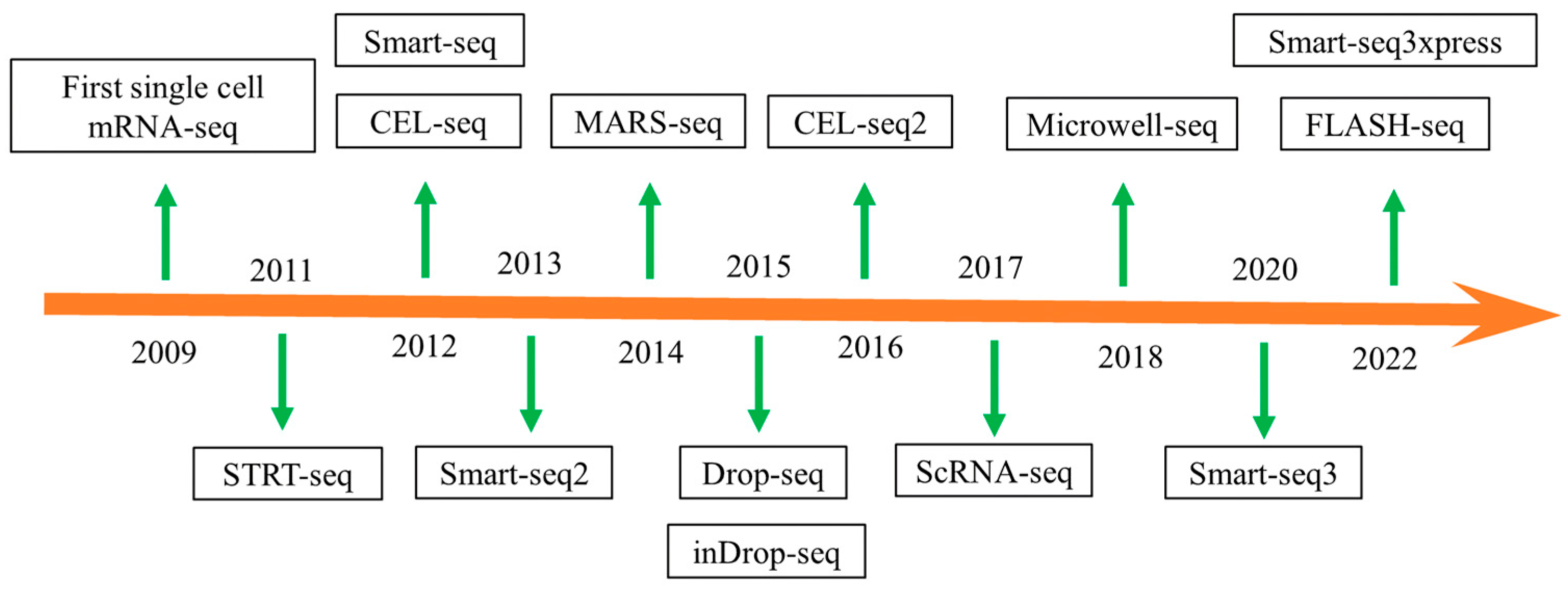
2. Development of Single-Cell Sequencing Technology
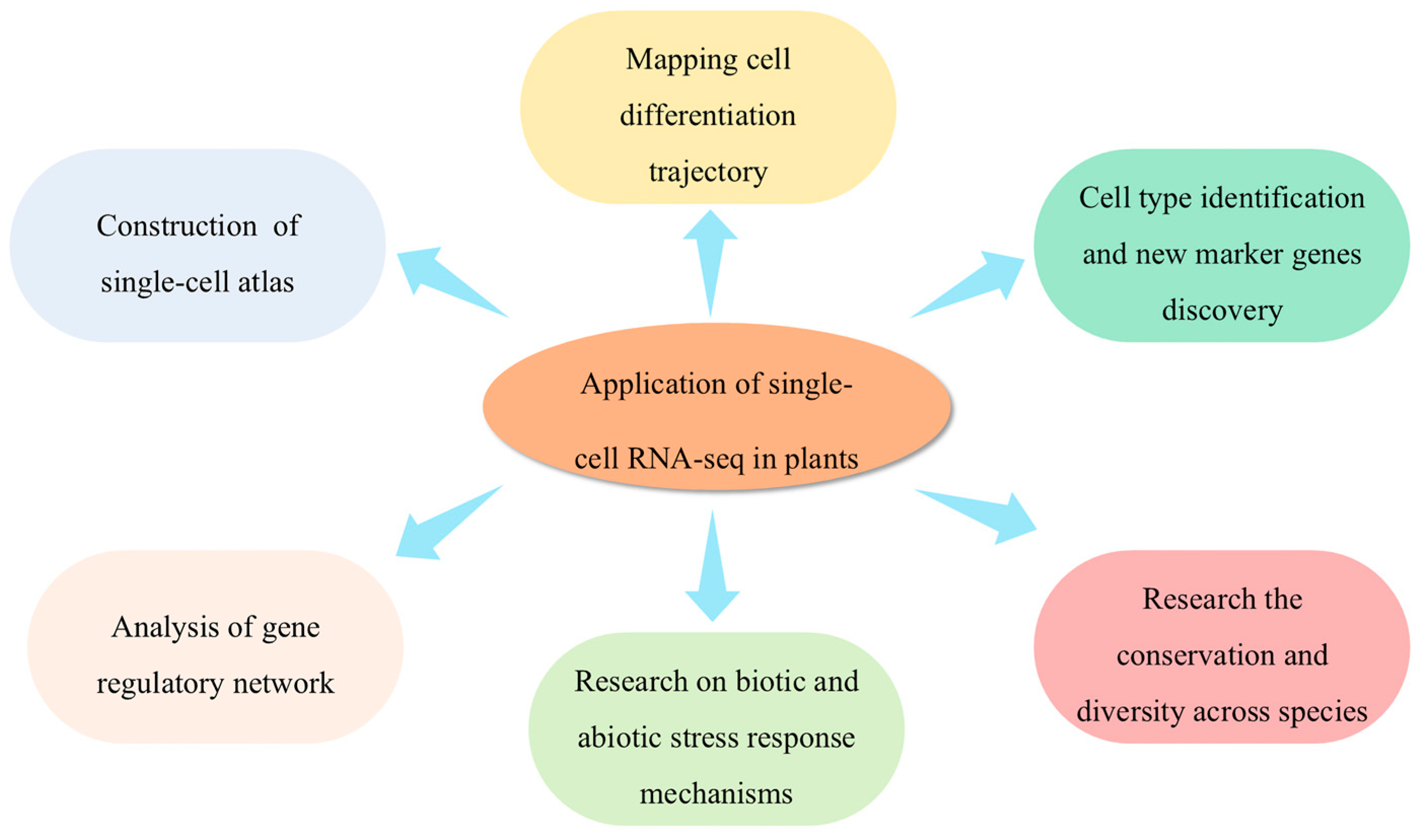
3. Applications of scRNA-seq in Plants
3.1. Construction of a Single-Cell Transcriptome Atlas and Cell Type Identification
3.2. Discovery of New Marker Genes
3.3. Mapping Cell Differentiation Trajectories
3.4. Analysis of Gene Regulatory Networks
3.5. Studying the Conservatism and the Diversity Among Cell Types Across Species
3.6. Research on Biotic and Abiotic Stress Response Mechanisms
3.6.1. Research on Biotic Stress Response Mechanisms
3.6.2. Research on Abiotic Stress Response Mechanisms
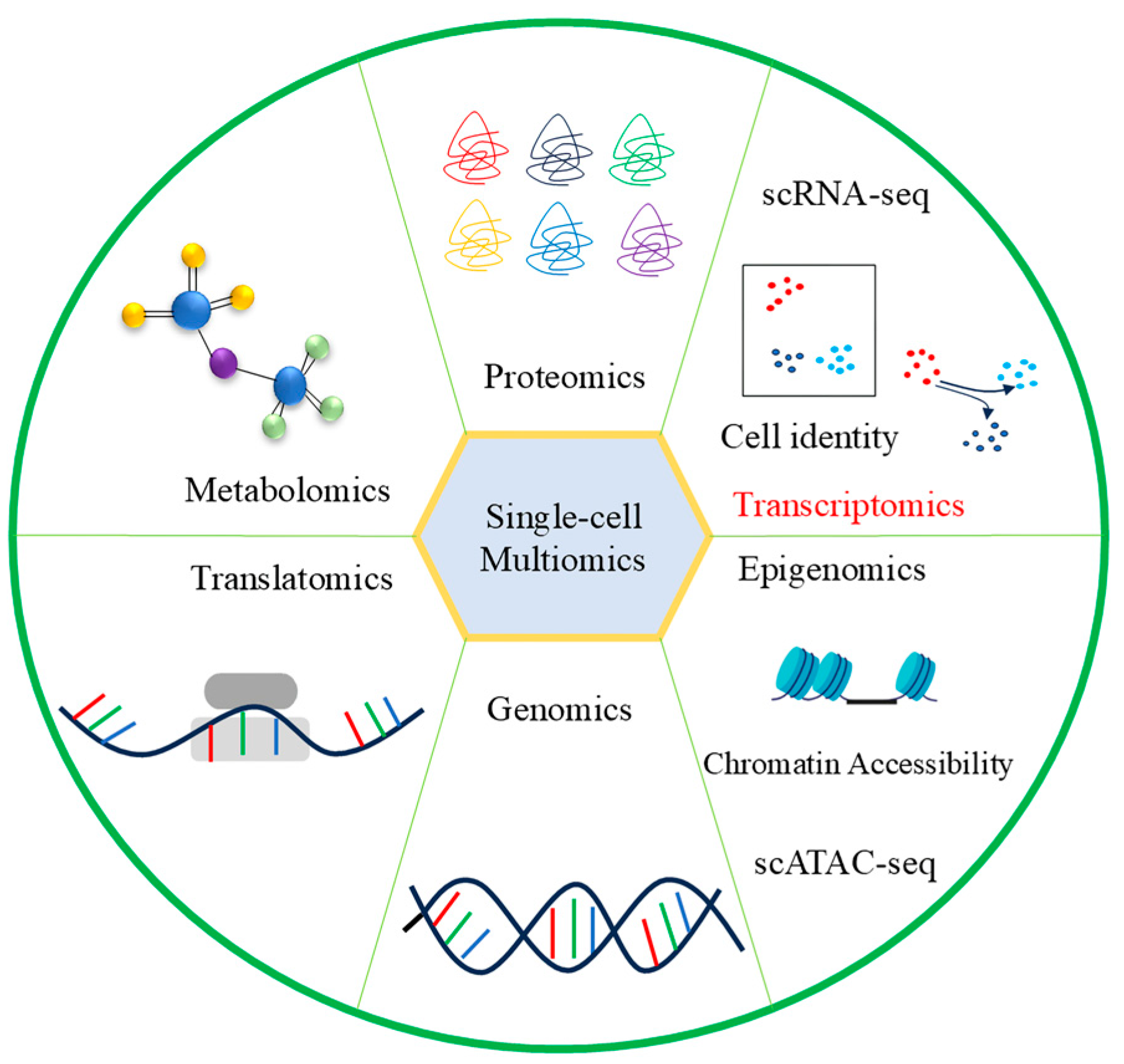
3.7. Combined Analysis of Single-Cell Transcriptome and Other Omics
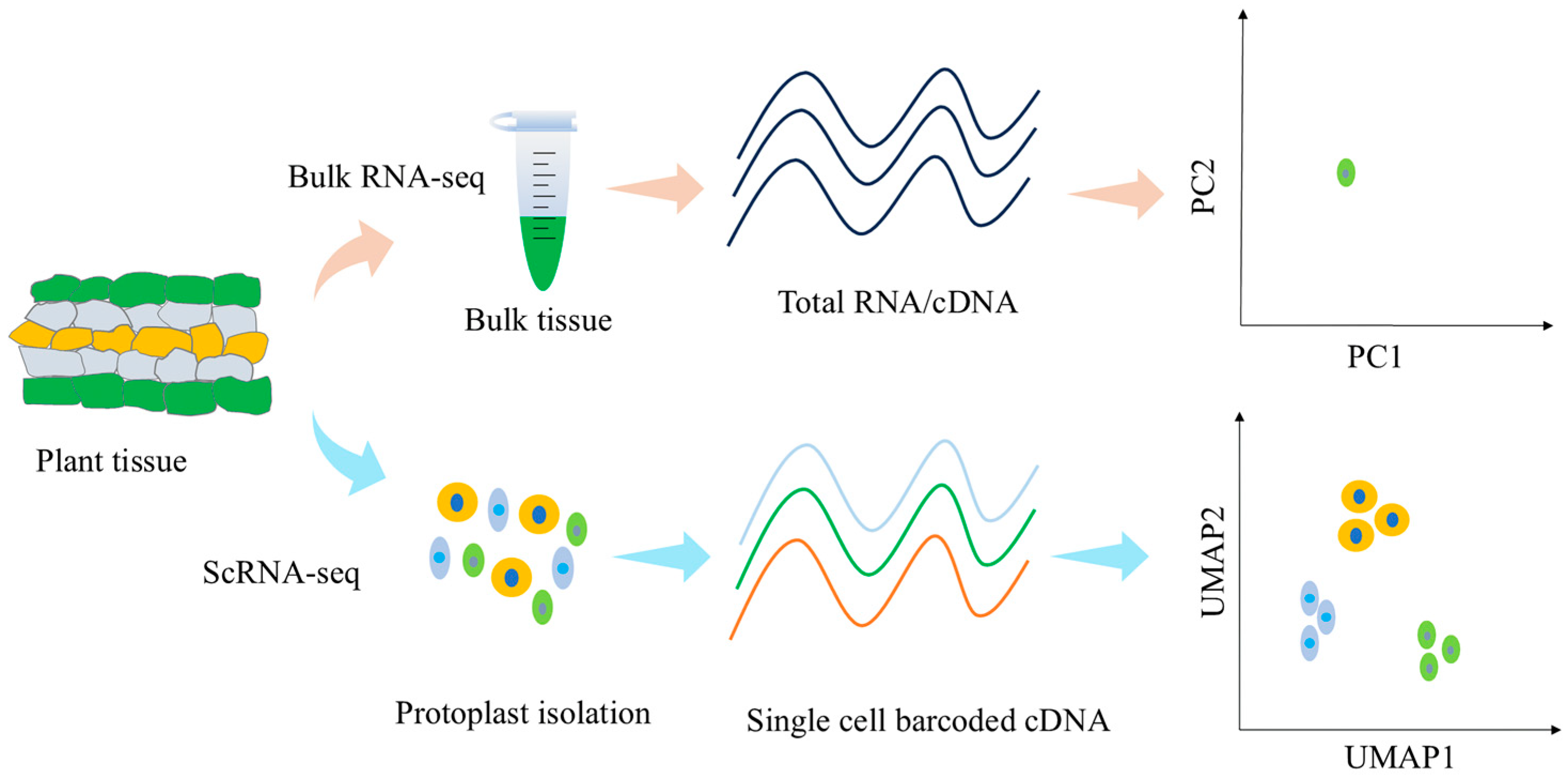
3.7.1. Single-Cell Transcriptional Sequencing and Bulk RNA-seq
3.7.2. Single-Cell Transcriptome and Spatial Transcriptome
3.7.3. Single-Cell Transcriptome and ATAC-seq
4. Challenges and Prospects
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tang, F.; Barbacioru, C.; Nordman, E.; Li, B.; Xu, N.; Bashkirov, V.I.; Lao, K.; Surani, M.A. Rna-Seq Analysis to Capture the Transcriptome Landscape of a Single Cell. Nat. Protoc. 2010, 5, 516–535. [Google Scholar] [CrossRef]
- Gough, A.; Stern, A.M.; Maier, J.; Lezon, T.; Shun, T.Y.; Chennubhotla, C.; Schurdak, M.E.; Haney, S.A.; Taylor, D.L. Biologically Relevant Heterogeneity: Metrics and Practical Insights. SLAS Discov. 2017, 22, 213–237. [Google Scholar] [CrossRef]
- Zhang, S.T.; Zhang, X.Y.; Zhu, C.; Li, Z.Y.; Fu, Z.R.; Zhang, Z.; Lai, Z.X.; Lin, Y.L. Single Cell Transcriptome Sequencing Technology and Its Application in Plants. Acta Hortic. Sin. 2022, 49, 2163–2173. [Google Scholar]
- Cui, K.; Wu, W.W.; Diao, Q.Y. Application and Research Progress on Transcriptomics. Biotechnol. Bull. 2019, 35, 1–9. [Google Scholar]
- Wen, L.; Tang, F.C. Recent Progress in Single-Cell Rna-Seq Analysis. Hereditas 2014, 36, 1069–1076. [Google Scholar]
- Zhu, Z.X.; Chen, X. Single Cell Sequencing Technology and Its Application Progress. Genom. Appl. Biol. 2015, 34, 902–908. [Google Scholar]
- Tanay, A.; Regev, A. Scaling Single-Cell Genomics from Phenomenology to Mechanism. Nature 2017, 541, 331–338. [Google Scholar] [CrossRef]
- Giacomello, S. A New Era for Plant Science: Spatial Single-Cell Transcriptomics. Curr. Opin. Plant Biol. 2021, 60, 102041. [Google Scholar] [CrossRef]
- Aldridge, S.; Teichmann, S.A. Single Cell Transcriptomics Comes of Age. Nat. Commun. 2020, 11, 4307. [Google Scholar] [CrossRef]
- Hedlund, E.; Deng, Q. Single-Cell Rna Sequencing: Technical Advancements and Biological Applications. Mol. Asp. Med. 2018, 59, 36–46. [Google Scholar] [CrossRef]
- Kadam, U.S.; Schulz, B.; Irudayaraj, J.M. Multiplex Single-Cell Quantification of Rare Rna Transcripts from Protoplasts in a Model Plant System. Plant J. 2017, 90, 1187–1195. [Google Scholar] [CrossRef]
- Zheng, H.-X.; Wu, F.-H.; Li, S.-M.; Zhang, X.S.; Sui, N. Single-Cell Profiling Lights Different Cell Trajectories in Plants. aBIOTECH 2021, 2, 64–78. [Google Scholar] [CrossRef]
- Efroni, I.; Ip, P.L.; Nawy, T.; Mello, A.; Birnbaum, K.D. Quantification of Cell Identity from Single-Cell Gene Expression Profiles. Genome Biol. 2015, 16, 9. [Google Scholar] [CrossRef]
- Apelt, F.; Mavrothalassiti, E.; Gupta, S.; Machin, F.; Olas, J.J.; Annunziata, M.G.; Schindelasch, D.; Kragler, F. Shoot and Root Single Cell Sequencing Reveals Tissue- and Daytime-Specific Transcriptome Profiles. Plant Physiol. 2022, 188, 861–878. [Google Scholar] [CrossRef]
- Jean-Baptiste, K.; McFaline-Figueroa, J.L.; Alexandre, C.M.; Dorrity, M.W.; Saunders, L.; Bubb, K.L.; Trapnell, C.; Fields, S.; Queitsch, C.; Cuperus, J.T. Dynamics of Gene Expression in Single Root Cells of Arabidopsis thaliana. Plant Cell 2019, 31, 993–1011. [Google Scholar] [CrossRef]
- Serrano-Ron, L.; Perez-Garcia, P.; Sanchez-Corrionero, A.; Gude, I.; Cabrera, J.; Ip, P.L.; Birnbaum, K.D.; Moreno-Risueno, M.A. Reconstruction of Lateral Root Formation through Single-Cell Rna Sequencing Reveals Order of Tissue Initiation. Mol. Plant 2021, 14, 1362–1378. [Google Scholar] [CrossRef]
- Liu, Q.; Liang, Z.; Feng, D.; Jiang, S.J.; Wang, Y.F.; Du, Z.Y.; Li, R.X.; Hu, G.H.; Zhang, P.X.; Ma, Y.F.; et al. Transcriptional Landscape of Rice Roots at the Single-Cell Resolution. Mol. Plant 2021, 14, 384–394. [Google Scholar] [CrossRef]
- Xu, X.; Crow, M.; Rice, B.R.; Li, F.; Harris, B.; Liu, L.; Demesa-Arevalo, E.; Lu, Z.; Wang, L.; Fox, N.; et al. Single-Cell Rna Sequencing of Developing Maize Ears Facilitates Functional Analysis and Trait Candidate Gene Discovery. Dev. Cell 2021, 56, 557–568.e6. [Google Scholar] [CrossRef]
- Guo, X.L.; Liang, J.L.; Lin, R.M.; Zhang, L.P.; Zhang, Z.C.; Wu, J.; Wang, X.W. Single-Cell Transcriptome Reveals Differentiation between Adaxial and Abaxial Mesophyll Cells in Brassica rapa. Plant Biotechnol. J. 2022, 20, 2233. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, Y.; Peng, A.Q.; Cui, J.L.; Zhao, M.Y.; Pan, Y.T.; Zhang, M.T.; Tian, K.; Schwab, W.; Song, C.K. Single-Cell Transcriptome Atlas Reveals Developmental Trajectories and a Novel Metabolic Pathway of Catechin Esters in Tea Leaves. Plant Biotechnol. J. 2022, 20, 2089–2106. [Google Scholar] [CrossRef]
- Liu, H.; Hu, D.X.; Du, P.X.; Wang, L.P.; Liang, X.Q.; Li, H.F.; Lu, Q.; Li, S.X.; Liu, H.Y.; Chen, X.P. Single-Cell Rna-Seq Describes the Transcriptome Landscape and Identifies Critical Transcription Factors in the Leaf Blade of the Allotetraploid Peanut (Arachis hypogaea L.). Plant Biotechnol. J. 2021, 19, 2261–2276. [Google Scholar] [CrossRef]
- Qin, Y.; Sun, M.L.; Li, W.W.; Xu, M.Q.; Shao, L.; Liu, Y.Q.; Zhao, G.N.; Liu, Z.P.; Xu, Z.P.; You, J.Q. Single-Cell Rna-Seq Reveals Fate Determination Control of an Individual Fibre Cell Initiation in Cotton (Gossypium hirsutum). Plant Biotechnol. J. 2022, 20, 2372–2388. [Google Scholar] [CrossRef]
- Sun, Y.; Han, Y.; Sheng, K.; Yang, P.; Cao, Y.; Li, H.; Zhu, Q.H.; Chen, J.; Zhu, S.; Zhao, T. Single-Cell Transcriptomic Analysis Reveals the Developmental Trajectory and Transcriptional Regulatory Networks of Pigment Glands in Gossypium bickii. Mol. Plant 2023, 16, 694–708. [Google Scholar] [CrossRef]
- Tian, C.H.; Du, Q.W.; Xu, M.X.; Du, F.; Jiao, Y.L. Single-Nucleus Rna-Seq Resolves Spatiotemporal Developmental Trajectories in the Tomato Shoot Apex. BioRxiv 2020. [Google Scholar] [CrossRef]
- Xie, J.B.; Li, M.; Zeng, J.Y.; Li, X.; Zhang, D.Q. Single-Cell Rna Sequencing Profiles of Stem-Differentiating Xylem in Poplar. Plant Biotechnol. J. 2022, 20, 417–419. [Google Scholar] [CrossRef]
- Cuperus, J.T. Single-Cell Genomics in Plants: Current State, Future Directions, and Hurdles to Overcome. Plant Physiol. 2021, 188, 749–755. [Google Scholar] [CrossRef]
- Denyer, T.; Timmermans, M.C. Crafting a Blueprint for Single-Cell Rna Sequencing. Trends Plant Sci. 2022, 27, 92–103. [Google Scholar] [CrossRef]
- Mo, Y.J.; Jiao, Y.L. Advances and Applications of Single-Cell Omics Technologies in Plant Research. Plant J. 2022, 110, 1551–1563. [Google Scholar] [CrossRef]
- Seyfferth, C.; Renema, J.; Wendrich, J.R.; Eekhout, T.; Seurinck, R.; Vandamme, N.; Blob, B.; Saeys, Y.; Helariutta, Y.; Birnbaum, K.D.; et al. Advances and Opportunities in Single-Cell Transcriptomics for Plant Research. Annu. Rev. Plant Biol. 2021, 72, 847–866. [Google Scholar] [CrossRef]
- Xu, X.; Jackson, D. Single-Cell Analysis Opens a Goldmine for Plant Functional Studies. Curr. Opin. Biotechnol. 2023, 79, 102858. [Google Scholar] [CrossRef]
- Das, M.; Burge, C.B.; Park, E.; Colinas, J.; Pelletier, J. Assessment of the Total Number of Human Transcription Units. Genomics 2001, 77, 71–78. [Google Scholar] [CrossRef]
- Feingold, E.; Good, P.; Guyer, M.; Kamholz, S.; Liefer, L.; Wetterstrand, K.; Collins, F.; Gingeras, T.; Kampa, D.; Sekinger, E. The Encode (Encyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-Cell Transcriptomics Reveals Bimodality in Expression and Splicing in Immune Cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef]
- Bengtsson, M.; Stahlberg, A.; Rorsman, P.; Kubista, M. Gene Expression Profiling in Single Cells from the Pancreatic Islets of Langerhans Reveals Lognormal Distribution of Mrna Levels. Genome Res. 2005, 15, 1388–1392. [Google Scholar] [CrossRef]
- Deng, Q.; Ramskold, D.; Reinius, B.; Sandberg, R. Single-Cell Rna-Seq Reveals Dynamic, Random Monoallelic Gene Expression in Mammalian Cells. Science 2014, 343, 193–196. [Google Scholar] [CrossRef]
- Eberwine, J.; Yeh, H.; Miyashiro, K.; Cao, Y.; Nair, S.; Finnell, R.; Zettel, M.; Coleman, P. Analysis of Gene Expression in Single Live Neurons. Proc. Natl. Acad. Sci. USA 1992, 89, 3010–3014. [Google Scholar] [CrossRef]
- Zou, J.-W.; Sun, M.; Yang, H.-Y. Single-Embryo Rt-Pcr Assay to Study Gene Expression Dynamics During Embryogenesis in Arabidopsis thaliana. Plant Mol. Biol. Rep. 2002, 20, 19–26. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of Next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. Mrna-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Islam, S.; Kjallquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lonnerberg, P.; Linnarsson, S. Characterization of the Single-Cell Transcriptional Landscape by Highly Multiplex Rna-Seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef]
- Schmidt, W.M.; Mueller, M.W. Capselect: A Highly Sensitive Method for 5′ Cap-Dependent Enrichment of Full-Length Cdna in Pcr-Mediated Analysis of Mrnas. Nucleic Acids Res. 1999, 27, e31. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. Cel-Seq: Single-Cell Rna-Seq by Multiplexed Linear Amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. Cel-Seq2: Sensitive Highly-Multiplexed Single-Cell Rna-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef]
- Ramsköld, D.; Luo, S.J.; Wang, Y.C.; Li, R.; Deng, Q.L.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C. Full-Length Mrna-Seq from Single-Cell Levels of Rna and Individual Circulating Tumor Cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Goetz, J.J.; Trimarchi, J.M. Transcriptome Sequencing of Single Cells with Smart-Seq. Nat. Biotechnol. 2012, 30, 763–765. [Google Scholar] [CrossRef]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-Seq2 for Sensitive Full-Length Transcriptome Profiling in Single Cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively Parallel Single-Cell Rna-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Fan, H.C.; Fu, G.K.; Fodor, S.P. Expression Profiling: Combinatorial Labeling of Single Cells for Gene Expression Cytometry. Science 2015, 347, 1258367. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Gawad, C.; Koh, W.; Quake, S.R. Single-Cell Genome Sequencing: Current State of the Science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Han, X.P.; Wang, R.Y.; Zhou, Y.C.; Fei, L.J.; Sun, H.Y.; Lai, S.J.; Saadatpour, A.; Zhou, Z.M.; Chen, H.D.; Ye, F. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramsköld, D.; Hendriks, G.J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-Cell Rna Counting at Allele and Isoform Resolution Using Smart-Seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef]
- Sandberg, R. Entering the Era of Single-Cell Transcriptomics in Biology and Medicine. Nat. Methods 2014, 11, 22–24. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Sandberg, R. Scalable Full-Transcript Coverage Single Cell Rna Sequencing with Smart-Seq3xpress. Nat. Biotechnol. 2022, 40, 1452–1457. [Google Scholar] [CrossRef]
- Hahaut, V.; Pavlinic, D.; Carbone, W.; Schuierer, S.; Balmer, P.; Quinodoz, M.; Renner, M.; Roma, G.; Cowan, C.S.; Picelli, S. Fast and Highly Sensitive Full-Length Single-Cell Rna Sequencing Using Flash-Seq. Nat. Biotechnol. 2022, 40, 1447–1451. [Google Scholar] [CrossRef]
- Chang, X.; Zheng, Y.; Xu, K. Single-Cell Rna Sequencing: Technological Progress and Biomedical Application in Cancer Research. Mol. Biotechnol. 2023, 66, 1497–1519. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, S.; Wang, B.; Zhao, J.; Zi, X.; Yan, S.; Dou, T.; Jia, J.; Wang, K.; Ge, C. Advances in Single-Cell Sequencing Technology and Its Application in Poultry Science. Genes 2022, 13, 2211. [Google Scholar] [CrossRef]
- Wu, Y.H.; Zhuang, J.; Song, Y.F.; Gao, X.Y.; Chu, J.; Han, S.W. Advances in Single-Cell Sequencing Technology in Microbiome Research. Genes Dis. 2023, 11, 101129. [Google Scholar] [CrossRef]
- Natarajan, K.N.; Miao, Z.; Jiang, M.; Huang, X.; Zhou, H.; Xie, J.; Wang, C.; Qin, S.; Zhao, Z.; Wu, L. Comparative Analysis of Sequencing Technologies for Single-Cell Transcriptomics. Genome Bio. 2019, 20, 70. [Google Scholar] [CrossRef]
- Wang, Y.; Huan, Q.; Li, K.; Qian, W. Single-Cell Transcriptome Atlas of the Leaf and Root of Rice Seedlings. J. Genet. Genom. 2021, 48, 881–898. [Google Scholar] [CrossRef]
- Bawa, G.; Liu, Z.; Yu, X.; Qin, A.; Sun, X. Single-Cell Rna Sequencing for Plant Research: Insights and Possible Benefits. Int. J. Mol. Sci. 2022, 23, 4497. [Google Scholar] [CrossRef]
- Rich-Griffin, C.; Stechemesser, A.; Finch, J.; Lucas, E.; Ott, S.; Schafer, P. Single-Cell Transcriptomics: A High-Resolution Avenue for Plant Functional Genomics. Trends Plant Sci. 2020, 25, 186–197. [Google Scholar] [CrossRef]
- Birnbaum, K.D.; Otegui, M.S.; Bailey-Serres, J.; Rhee, S.Y. The Plant Cell Atlas: Focusing New Technologies on the Kingdom That Nourishes the Planet. Plant Physiol. 2022, 188, 675–679. [Google Scholar] [CrossRef]
- Zhang, T.Q.; Chen, Y.; Liu, Y.; Lin, W.H.; Wang, J.W. Single-Cell Transcriptome Atlas and Chromatin Accessibility Landscape Reveal Differentiation Trajectories in the Rice Root. Nat. Commun. 2021, 12, 2053. [Google Scholar] [CrossRef]
- Denyer, T.; Ma, X.; Klesen, S.; Scacchi, E.; Nieselt, K.; Timmermans, M.C.P. Spatiotemporal Developmental Trajectories in the Arabidopsis Root Revealed Using High-Throughput Single-Cell Rna Sequencing. Dev. Cell 2019, 48, 840–852.e5. [Google Scholar] [CrossRef]
- Jin, J.; Lu, P.; Xu, Y.; Tao, J.; Li, Z.; Wang, S.; Yu, S.; Wang, C.; Xie, X.; Gao, J. Pcmdb: A Curated and Comprehensive Resource of Plant Cell Markers. Nucleic Acids Res. 2022, 50, 1448–1455. [Google Scholar] [CrossRef]
- Tang, W.; Tang, A.Y. Transcriptional Mechanisms Regulating Gene Expression and Determining Cell Fates in Plant Development. J. For. Res. 2017, 28, 863–880. [Google Scholar] [CrossRef]
- Liu, Z.X.; Zhou, Y.P.; Guo, J.G.; Li, J.A.; Tian, Z.X.; Zhu, Z.N.; Wang, J.J.; Wu, R.; Zhang, B.; Hu, Y.J.; et al. Global Dynamic Molecular Profiling of Stomatal Lineage Cell Development by Single-Cell Rna Sequencing. Mol. Plant 2020, 13, 1178–1193. [Google Scholar] [CrossRef]
- Pierre-Jerome, E.; Drapek, C.; Benfey, P.N. Regulation of Division and Differentiation of Plant Stem Cells. Annu. Rev. Cell Dev. Bi. 2018, 34, 289–310. [Google Scholar] [CrossRef]
- LI, Y.; Sun, C. Research Progress in Single-Cell Rna-Seq of Plant. Biotechnol. Bull. 2021, 37, 60–66. [Google Scholar]
- Li, X.H.; Zhang, X.B.; Gao, S.; Cui, F.Q.; Chen, W.W.; Fan, L.A.; Qi, Y.W. Single-Cell Rna Sequencing Reveals the Landscape of Maize Root Tips and Assists in Identification of Cell Type-Specific Nitrate-Response Genes. Crop J. 2022, 10, 1589–1600. [Google Scholar] [CrossRef]
- Han, X.; Hu, Y.; Zhang, G.; Jiang, Y.; Chen, X.; Yu, D. Jasmonate Negatively Regulates Stomatal Development in Arabidopsis Cotyledons. Plant Physiol. 2018, 176, 2871–2885. [Google Scholar] [CrossRef]
- He, Z.; Lan, Y.; Zhou, X.; Yu, B.; Zhu, T.; Yang, F.; Fu, L.Y.; Chao, H.; Wang, J.; Feng, R.X.; et al. Single-Cell Transcriptome Analysis Dissects Lncrna-Associated Gene Networks in Arabidopsis. Plant Commun. 2024, 5, 100717. [Google Scholar] [CrossRef]
- Wang, R.Y.; Zhang, P.J.; Wang, J.J.; Ma, L.F.; E, W.; Suo, S.B.; Jiang, M.M.; Li, J.Q.; Chen, H.D.; Sun, H.Y. Construction of a Cross-Species Cell Landscape at Single-Cell Level. Nucleic Acids Res. 2023, 51, 501–516. [Google Scholar] [CrossRef]
- Ding, Y.; Gao, W.; Qin, Y.; Li, X.; Zhang, Z.; Lai, W.; Yang, Y.; Guo, K.; Li, P.; Zhou, S.; et al. Single-Cell Rna Landscape of the Special Fiber Initiation Process in Bombax ceiba. Plant Commun. 2023, 4, 100554. [Google Scholar] [CrossRef]
- Atkinson, N.J.; Urwin, P.E. The Interaction of Plant Biotic and Abiotic Stresses: From Genes to the Field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef]
- Tenorio Berrío, R.; Dubois, M. Single-Cell Transcriptomics Reveal Heterogeneity in Plant Responses to the Environment: A Focus on Biotic and Abiotic Interactions. J. Exp. Bot. 2024, 75, erae107. [Google Scholar] [CrossRef]
- Carezzano, M.E.; Paletti Rovey, M.F.; Cappellari, L.d.R.; Gallarato, L.A.; Bogino, P.; Oliva, M.d.l.M.; Giordano, W. Biofilm-Forming Ability of Phytopathogenic Bacteria: A Review of Its Involvement in Plant Stress. Plants 2023, 12, 2207. [Google Scholar] [CrossRef]
- Tang, B.; Feng, L.; Hulin, M.T.; Ding, P.; Ma, W. Cell-Type-Specific Responses to Fungal Infection in Plants Revealed by Single-Cell Transcriptomics. Cell Host Microbe 2023, 31, 1732–1747.e5. [Google Scholar] [CrossRef]
- Ye, J.R.; Zhong, T.; Zhang, D.F.; Ma, C.Y.; Wang, L.N.; Yao, L.S.; Zhang, Q.Q.; Zhu, M.; Xu, M.L. The Auxin-Regulated Protein Zmauxrp1 Coordinates the Balance between Root Growth and Stalk Rot Disease Resistance in Maize. Mol. Plant 2019, 12, 360–373. [Google Scholar] [CrossRef]
- Cao, Y.Y.; Ma, J.; Han, S.B.; Hou, M.W.; Wei, X.; Zhang, X.R.; Zhang, Z.Y.; Sun, S.L.; Ku, L.X.; Tang, J.H. Single-Cell Rna Sequencing Profiles Reveal Cell Type-Specific Transcriptional Regulation Networks Conditioning Fungal Invasion in Maize Roots. Plant Biotechnol. J. 2023, 21, 1839–1859. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, H.; Lyu, H.; Su, L.; Xiong, J.; Cheng, Z.M. Development of a Single-Cell Atlas for Woodland Strawberry (Fragaria vesca) Leaves During Early Botrytis cinerea Infection Using Single Cell Rna-Seq. Hortic. Res. 2022, 9, uhab055. [Google Scholar] [CrossRef]
- Boscaiu, M.; Fita, A. Physiological and Molecular Characterization of Crop Resistance to Abiotic Stresses. Agronomy 2020, 10, 1308. [Google Scholar] [CrossRef]
- Li, P.T.; Liu, Q.K.; Wei, Y.Y.; Xing, C.Z.; Xu, Z.P.; Ding, F.; Liu, Y.L.; Lu, Q.W.; Hu, N.; Wang, T.; et al. Transcriptional Landscape of Cotton Roots in Response to Salt Stress at Single-Cell Resolution. Plant Commun. 2024, 5, 100740. [Google Scholar] [CrossRef]
- Sun, X.; Feng, D.; Liu, M.; Qin, R.; Li, Y.; Lu, Y.; Zhang, X.; Wang, Y.; Shen, S.; Ma, W.; et al. Single-Cell Transcriptome Reveals Dominant Subgenome Expression and Transcriptional Response to Heat Stress in Chinese Cabbage. Genome Biol. 2022, 23, 262. [Google Scholar] [CrossRef]
- Wendrich, J.R.; Yang, B.; Vandamme, N.; Verstaen, K.; Smet, W.; Van de Velde, C.; Minne, M.; Wybouw, B.; Mor, E.; Arents, H.E.; et al. Vascular Transcription Factors Guide Plant Epidermal Responses to Limiting Phosphate Conditions. Science 2020, 370, eaay4970. [Google Scholar] [CrossRef]
- Wang, Y.; Huan, Q.; Chu, X.; Li, K.; Qian, W.F. Single-Cell Transcriptome Analyses Recapitulate the Cellular and Developmental Responses to Abiotic Stresses in Rice. BioRxiv 2020. [Google Scholar] [CrossRef]
- Goldbach, H.E.; Wimmer, M.A. Boron in Plants and Animals: Is There a Role Beyond Cell-Wall Structure? J. Plant Nutr. Soil Sci. 2007, 170, 39–48. [Google Scholar] [CrossRef]
- Chen, X.; Ru, Y.Q.; Takahashi, H.; Nakazono, M.; Shabala, S.; Smith, S.M.; Yu, M. Single-Cell Transcriptomic Analysis of Pea Shoot Development and Cell-Type-Specific Responses to Boron Deficiency. Plant J. 2024, 117, 302–322. [Google Scholar] [CrossRef]
- Chen, C.; Ge, Y.; Lu, L. Opportunities and Challenges in the Application of Single-Cell and Spatial Transcriptomics in Plants. Front. Plant Sci. 2023, 14, 1185377. [Google Scholar] [CrossRef]
- Zhang, J.; Ahmad, M.; Gao, H. Application of Single-Cell Multi-Omics Approaches in Horticulture Research. Mol. Hortic. 2023, 3, 18. [Google Scholar] [CrossRef]
- Peng, T.; Zhu, Q.; Yin, P.; Tan, K. Scrabble: Single-Cell Rna-Seq Imputation Constrained by Bulk Rna-Seq Data. Genome Biol. 2019, 20, 88. [Google Scholar] [CrossRef]
- Yin, R.; Xia, K.; Xu, X. Spatial Transcriptomics Drives a New Era in Plant Research. Plant J. 2023, 116, 1571–1581. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, Z.; Zhu, C. Microarray-Based Analysis of Cadmium-Responsive Micrornas in Rice (Oryza sativa). J. Exp. Bot. 2011, 62, 3563–3573. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef]
- Rozier, F.; Mirabet, V.; Vernoux, T.; Das, P. Analysis of 3d Gene Expression Patterns in Plants Using Whole-Mount Rna in Situ Hybridization. Nat. Protoc. 2014, 9, 2464–2475. [Google Scholar] [CrossRef]
- Blokhina, O.; Valerio, C.; Sokolowska, K.; Zhao, L.; Karkonen, A.; Niittyla, T.; Fagerstedt, K. Laser Capture Microdissection Protocol for Xylem Tissues of Woody Plants. Front. Plant Sci. 2016, 7, 1965. [Google Scholar] [CrossRef]
- Brady, S.M.; Orlando, D.A.; Lee, J.Y.; Wang, J.Y.; Koch, J.; Dinneny, J.R.; Mace, D.; Ohler, U.; Benfey, P.N. A High-Resolution Root Spatiotemporal Map Reveals Dominant Expression Patterns. Science 2007, 318, 801–806. [Google Scholar] [CrossRef]
- Moreno-Romero, J.; Santos-Gonzalez, J.; Hennig, L.; Kohler, C. Applying the Intact Method to Purify Endosperm Nuclei and to Generate Parental-Specific Epigenome Profiles. Nat. Protoc. 2017, 12, 238–254. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Nicolas, P.; Fernandez-Pozo, N.; Ma, Q.; Evanich, D.J.; Shi, Y.; Xu, Y.; Zheng, Y.; Snyder, S.I.; Martin, L.B.B.; et al. High-Resolution Spatiotemporal Transcriptome Mapping of Tomato Fruit Development and Ripening. Nat. Commun. 2018, 9, 364. [Google Scholar] [CrossRef]
- Shahan, R.; Nolan, T.M.; Benfey, P.N. Single-Cell Analysis of Cell Identity in the Arabidopsis Root Apical Meristem: Insights and Opportunities. J. Exp. Bot. 2021, 72, 6679–6686. [Google Scholar] [CrossRef]
- Gurazada, S.G.R.; Cox Jr, K.L.; Czymmek, K.J.; Meyers, B.C. Space: The Final Frontier—Achieving Single-Cell, Spatially Resolved Transcriptomics in Plants. Emerg. Top. Life Sci. 2021, 5, 179–188. [Google Scholar]
- Shi, J.; Pan, Y.; Liu, X.; Cao, W.; Mu, Y.; Zhu, Q. Spatial Omics Sequencing Based on Microfluidic Array Chips. Biosensors 2023, 13, 712. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Xia, K.; Sun, H.X.; Li, J.; Li, J.; Zhao, Y.; Chen, L.; Qin, C.; Chen, R.; Chen, Z.; Liu, G.; et al. The Single-Cell Stereo-Seq Reveals Region-Specific Cell Subtypes and Transcriptome Profiling in Arabidopsis Leaves. Dev. Cell 2022, 57, 1299–1310.e4. [Google Scholar] [CrossRef]
- Li, R.; Wang, Z.; Wang, J.W.; Li, L. Combining Single-Cell Rna Sequencing with Spatial Transcriptome Analysis Reveals Dynamic Molecular Maps of Cambium Differentiation in the Primary and Secondary Growth of Trees. Plant Commun. 2023, 4, 100665. [Google Scholar] [CrossRef]
- Liu, Z.; Kong, X.; Long, Y.; Liu, S.; Zhang, H.; Jia, J.; Cui, W.; Zhang, Z.; Song, X.; Qiu, L.; et al. Integrated Single-Nucleus and Spatial Transcriptomics Captures Transitional States in Soybean Nodule Maturation. Nat. Plants 2023, 9, 515–524. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Li, Z.; Schulz, M.H.; Look, T.; Begemann, M.; Zenke, M.; Costa, I.G. Identification of Transcription Factor Binding Sites Using Atac-Seq. Genome Biol. 2019, 20, 45. [Google Scholar] [CrossRef]
- Bajic, M.; Maher, K.A.; Deal, R.B. Identification of Open Chromatin Regions in Plant Genomes Using Atac-Seq. Methods Mol. Biol. 2018, 1675, 183–201. [Google Scholar]
- Feng, D.; Liang, Z.; Wang, Y.; Yao, J.; Yuan, Z.; Hu, G.; Qu, R.; Xie, S.; Li, D.; Yang, L.; et al. Chromatin Accessibility Illuminates Single-Cell Regulatory Dynamics of Rice Root Tips. BMC Biol. 2022, 20, 274. [Google Scholar] [CrossRef]
- Wang, F.X.; Shang, G.D.; Wu, L.Y.; Mai, Y.X.; Gao, J.; Xu, Z.G.; Wang, J.W. Protocol for Assaying Chromatin Accessibility Using Atac-Seq in Plants. STAR Protoc. 2021, 2, 100289. [Google Scholar] [CrossRef]
- Farmer, A.; Thibivilliers, S.; Ryu, K.H.; Schiefelbein, J.; Libault, M. Single-Nucleus Rna and Atac Sequencing Reveals the Impact of Chromatin Accessibility on Gene Expression in Arabidopsis Roots at the Single-Cell Level. Mol. Plant 2021, 14, 372–383. [Google Scholar] [CrossRef]
- Wang, D.; Hu, X.; Ye, H.; Wang, Y.; Yang, Q.; Liang, X.; Wang, Z.; Zhou, Y.; Wen, M.; Yuan, X.; et al. Cell-Specific Clock-Controlled Gene Expression Program Regulates Rhythmic Fiber Cell Growth in Cotton. Genome Biol. 2023, 24, 49. [Google Scholar] [CrossRef]
- Netla, V.R.; Shinde, H.; Kumar, G.; Dudhate, A.; Hong, J.C.; Kadam, U.S. A Comparative Analysis of Single-Cell Transcriptomic Technologies in Plants and Animals. Curr. Plant Biol. 2023, 35, 100289. [Google Scholar] [CrossRef]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W. Single-Cell Profiling of the Developing Mouse Brain and Spinal Cord with Split-Pool Barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Zhang, X.; Liu, Y.; Cai, L.; Zhang, Q.; Chen, Q.; Lin, L.; Lin, S.; Song, Y. Decoding Expression Dynamics of Protein and Transcriptome at the Single-Cell Level in Paired Picoliter Chambers. Anal. Chem. 2022, 94, 8164–8173. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Han, Y.; Li, R.; Cui, F.; Zhang, H.; Su, X.; Liu, X.; Xu, G.; Wan, S. Spatial Transcriptome Analysis on Peanut Tissues Shed Light on Cell Heterogeneity of the Peg. Plant Biotechnol. J. 2022, 20, 1648. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-Length Rna-Seq from Single Cells Using Smart-Seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, J.; Long, Y.; Zhang, C.; Wang, D.; Zhang, X.; Dong, W.; Zhao, L.; Liu, C.; Zhai, J. Single-Nucleus Transcriptomes Reveal Spatiotemporal Symbiotic Perception and Early Response in Medicago. Nat. Plants 2023, 9, 1734–1748. [Google Scholar] [CrossRef]
- Qu, H.-Q.; Kao, C.; Hakonarson, H. Single-Cell Rna Sequencing Technology Landscape in 2023. Stem Cells 2024, 42, 1–12. [Google Scholar] [CrossRef]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T. Systematic Comparison of Single-Cell and Single-Nucleus Rna-Sequencing Methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef]
- Lake, B.B.; Codeluppi, S.; Yung, Y.C.; Gao, D.; Chun, J.; Kharchenko, P.V.; Linnarsson, S.; Zhang, K. A Comparative Strategy for Single-Nucleus and Single-Cell Transcriptomes Confirms Accuracy in Predicted Cell-Type Expression from Nuclear Rna. Sci. Rep. 2017, 7, 6031. [Google Scholar] [CrossRef]
- Blum, J.A.; Klemm, S.; Shadrach, J.L.; Guttenplan, K.A.; Nakayama, L.; Kathiria, A.; Hoang, P.T.; Gautier, O.; Kaltschmidt, J.A.; Greenleaf, W.J. Single-Cell Transcriptomic Analysis of the Adult Mouse Spinal Cord Reveals Molecular Diversity of Autonomic and Skeletal Motor Neurons. Nat. Neurosci. 2021, 24, 572–583. [Google Scholar] [CrossRef]
- Conde, D.; Triozzi, P.M.; Balmant, K.M.; Doty, A.L.; Miranda, M.; Boullosa, A.; Schmidt, H.W.; Pereira, W.J.; Dervinis, C.; Kirst, M. A Robust Method of Nuclei Isolation for Single-Cell Rna Sequencing of Solid Tissues from the Plant Genus Populus. PLoS ONE 2021, 16, e0251149. [Google Scholar] [CrossRef]
- Sunaga-Franze, D.Y.; Muino, J.M.; Braeuning, C.; Xu, X.; Zong, M.; Smaczniak, C.; Yan, W.; Fischer, C.; Vidal, R.; Kliem, M. Single-Nuclei Rna-Sequencing of Plant Tissues. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M. Cellmarker: A Manually Curated Resource of Cell Markers in Human and Mouse. Nucleic Acids Res. 2019, 47, 721–728. [Google Scholar] [CrossRef]
- Ma, X.; Denyer, T.; Timmermans, M.C. Pscb: A Browser to Explore Plant Single Cell Rna-Sequencing Data Sets. Plant Physiol. 2020, 183, 464–467. [Google Scholar] [CrossRef]
- Chen, H.; Yin, X.; Guo, L.; Yao, J.; Ding, Y.; Xu, X.; Liu, L.; Zhu, Q.-H.; Chu, Q.; Fan, L. Plantscrnadb: A Database for Plant Single-Cell Rna Analysis. Mol. Plant 2021, 14, 855–857. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Q.; Zhu, X.; Wang, G.; Qin, Y.; Ding, F.; Tu, L.; Daniell, H.; Zhang, X.; Jin, S. Plant Single Cell Transcriptome Hub (Pscth): An Integrated Online Tool to Explore the Plant Single-Cell Transcriptome Landscape. Plant Biotechnol. J. 2022, 20, 10–12. [Google Scholar] [CrossRef]
- He, Z.; Luo, Y.; Zhou, X.; Zhu, T.; Lan, Y.; Chen, D. Scplantdb: A Comprehensive Database for Exploring Cell Types and Markers of Plant Cell Atlases. Nucleic Acids Res. 2024, 52, 1629–1638. [Google Scholar] [CrossRef]
- Zhang, Q.; Jin, S.; Zou, X. Scab Detects Multiresolution Cell States with Clinical Significance by Integrating Single-Cell Genomics and Bulk Sequencing Data. Nucleic Acids Res. 2022, 50, 12112–12130. [Google Scholar] [CrossRef]
- Przytycki, P.F.; Pollard, K.S. Cellwalker Integrates Single-Cell and Bulk Data to Resolve Regulatory Elements across Cell Types in Complex Tissues. Genome Biol. 2021, 22, 61. [Google Scholar] [CrossRef]
- Ma, Y.; Sun, Z.; Zeng, P.; Zhang, W.; Lin, Z. Jsnmf Enables Effective and Accurate Integrative Analysis of Single-Cell Multiomics Data. Brief. Bioinform. 2022, 23, bbac105. [Google Scholar] [CrossRef]
- Yan, C.; Zhu, Y.; Chen, M.; Yang, K.; Cui, F.; Zou, Q.; Zhang, Z. Integration Tools for Scrna-Seq Data and Spatial Transcriptomics Sequencing Data. Brief. Funct. Genom. 2024, 23, elae002. [Google Scholar] [CrossRef]
- Sahito, J.H.; Zhang, H.; Gishkori, Z.G.N.; Ma, C.; Wang, Z.; Ding, D.; Zhang, X.; Tang, J. Advancements and Prospects of Genome-Wide Association Studies (Gwas) in Maize. Int. J. Mol. Sci. 2024, 25, 1918. [Google Scholar] [CrossRef]
- Calderon, D.; Bhaskar, A.; Knowles, D.A.; Golan, D.; Raj, T.; Fu, A.Q.; Pritchard, J.K. Inferring Relevant Cell Types for Complex Traits by Using Single-Cell Gene Expression. Am. J. Hum. Genet. 2017, 101, 686–699. [Google Scholar] [CrossRef]
- Zhang, M.J.; Hou, K.; Dey, K.K.; Sakaue, S.; Jagadeesh, K.A.; Weinand, K.; Taychameekiatchai, A.; Rao, P.; Pisco, A.O.; Zou, J. Polygenic Enrichment Distinguishes Disease Associations of Individual Cells in Single-Cell Rna-Seq Data. Nat. Genet. 2022, 54, 1572–1580. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell Rna-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 441123. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Year | Cell Sorting Method | Advantage | Disadvantage | Reference |
|---|---|---|---|---|---|
| Smart-seq | 2012 | Fluidigm C1 |
|
| [44] |
| CEL-seq | 2012 | FACS |
|
| [42] |
| Smart-seq2 | 2013 | FACS |
|
| [46] |
| MARS-seq | 2014 | FACS |
|
| [47] |
| Drop-seq | 2015 | Droplet |
|
| [48] |
| inDrop-seq | 2015 | Droplet |
|
| [49] |
| CEL-seq2 | 2016 | Fluidigm C1 |
|
| [43] |
| scRNA-seq | 2017 | Droplet |
|
| [51] |
| Smart-seq3 | 2020 | FACS |
|
| [55] |
| FLASH-seq | 2022 | FACS |
|
| [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, J.; Zhuang, Z.; Ji, X.; Tang, R.; Li, J.; Chen, J.; Li, Z.; Peng, Y. Research Progress of Single-Cell Transcriptome Sequencing Technology in Plants. Agronomy 2024, 14, 2530. https://doi.org/10.3390/agronomy14112530
Bian J, Zhuang Z, Ji X, Tang R, Li J, Chen J, Li Z, Peng Y. Research Progress of Single-Cell Transcriptome Sequencing Technology in Plants. Agronomy. 2024; 14(11):2530. https://doi.org/10.3390/agronomy14112530
Chicago/Turabian StyleBian, Jianwen, Zelong Zhuang, Xiangzhuo Ji, Rui Tang, Jiawei Li, Jiangtao Chen, Zhiming Li, and Yunling Peng. 2024. "Research Progress of Single-Cell Transcriptome Sequencing Technology in Plants" Agronomy 14, no. 11: 2530. https://doi.org/10.3390/agronomy14112530
APA StyleBian, J., Zhuang, Z., Ji, X., Tang, R., Li, J., Chen, J., Li, Z., & Peng, Y. (2024). Research Progress of Single-Cell Transcriptome Sequencing Technology in Plants. Agronomy, 14(11), 2530. https://doi.org/10.3390/agronomy14112530