

De Novo Transcriptome Analysis of Solanum lycopersicum cv. Super Strain B under Drought Stress

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Sequencing and Data Processing
2.2.1. RNA Extraction
2.2.2. Illumina Paired-End cDNA Library Construction and Sequencing
2.2.3. cDNA Sequencing and De Novo Transcriptome Assembly
2.3. Functional Annotation
2.4. Trait Genes
2.4.1. Comparison of Tomato cv. Super Strain B Transcriptome with the Tomato Genome
2.4.2. Molecular Phylogeny Relationships
2.4.3. Discovery of SSR and Nucleotide-Level Variants
3. Results and Discussion
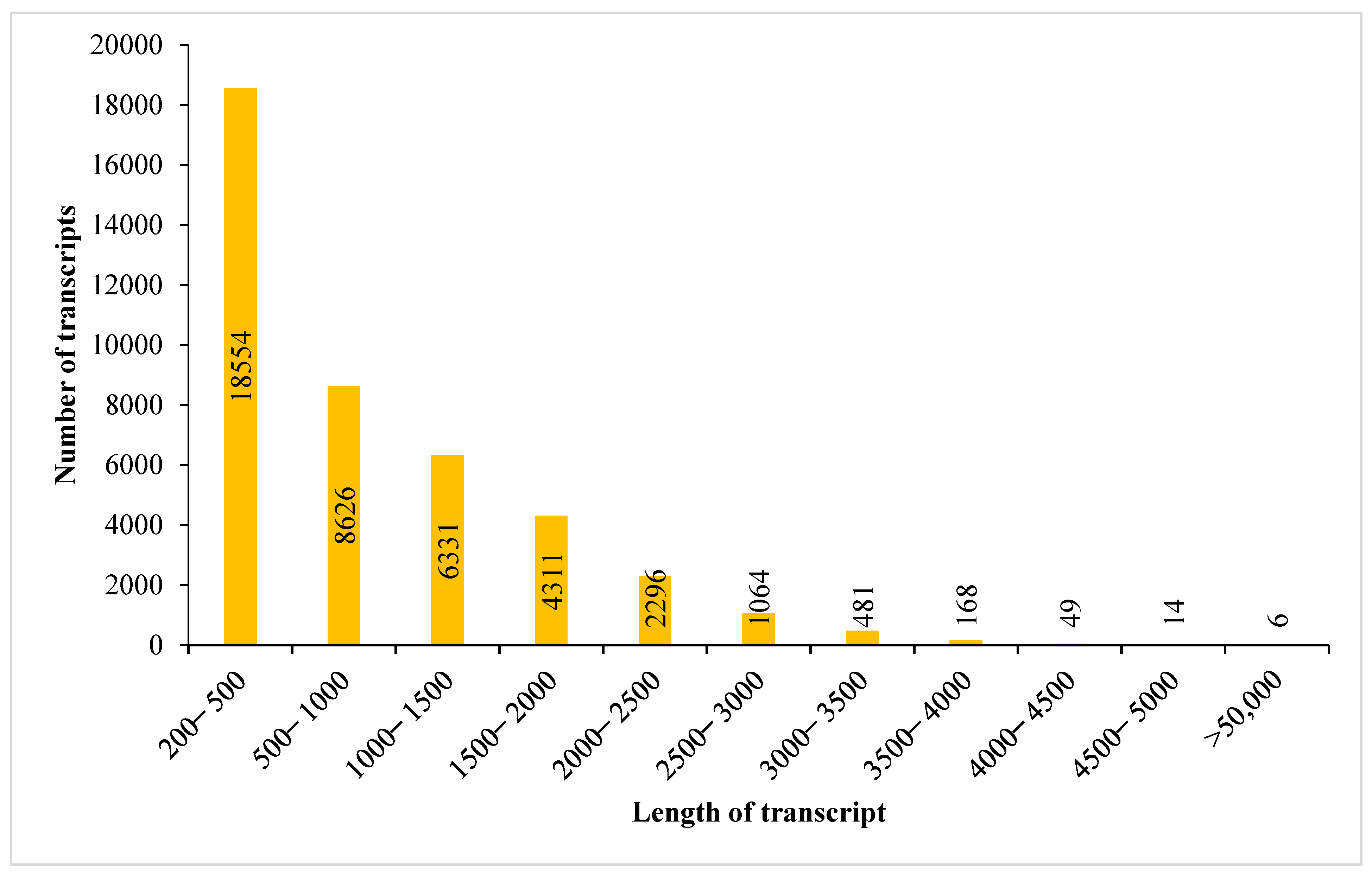
3.1. Transcriptome Sequencing Output and Assembly
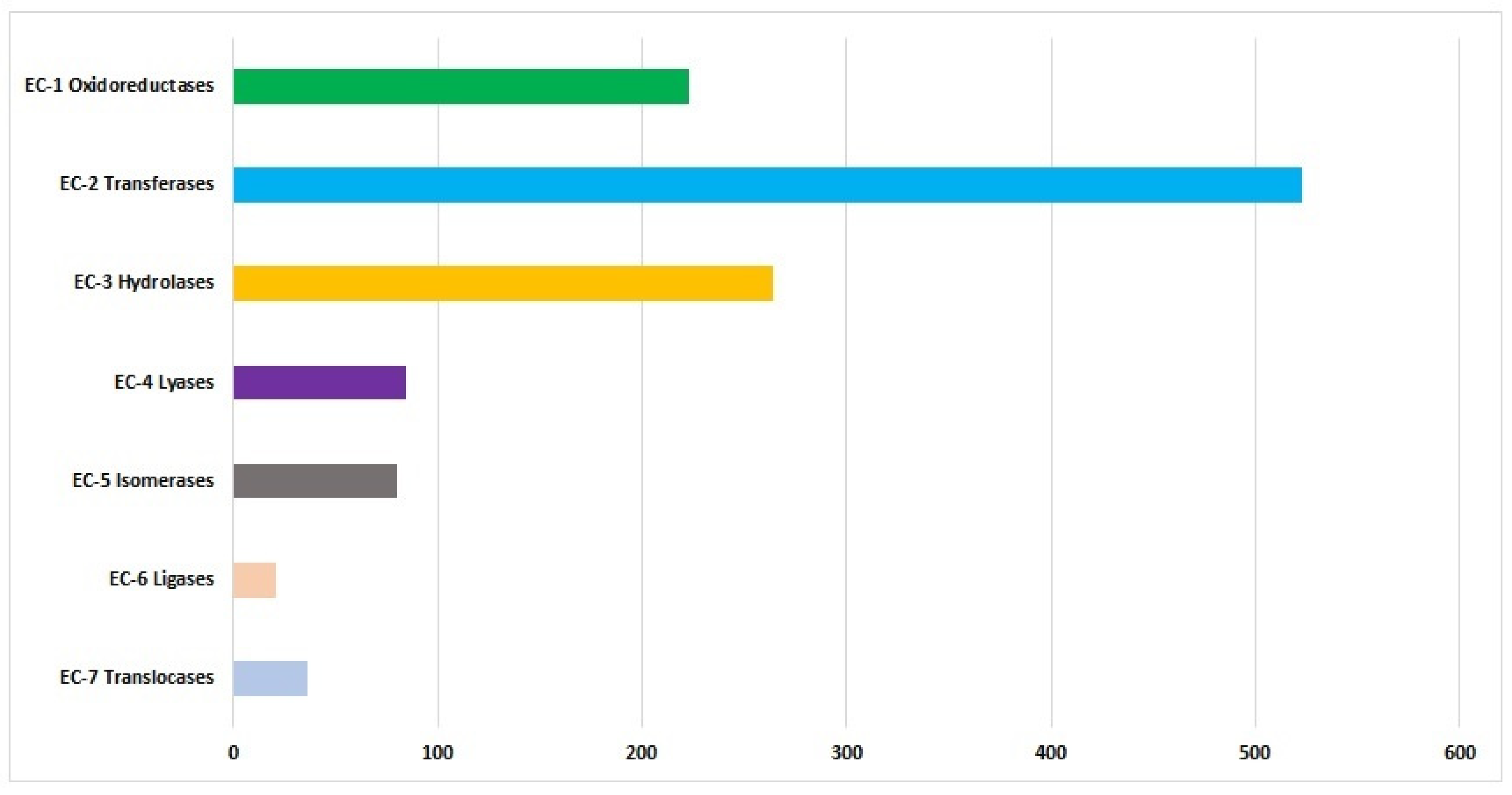
3.1.1. Functional Annotation
3.1.2. Trait Genes
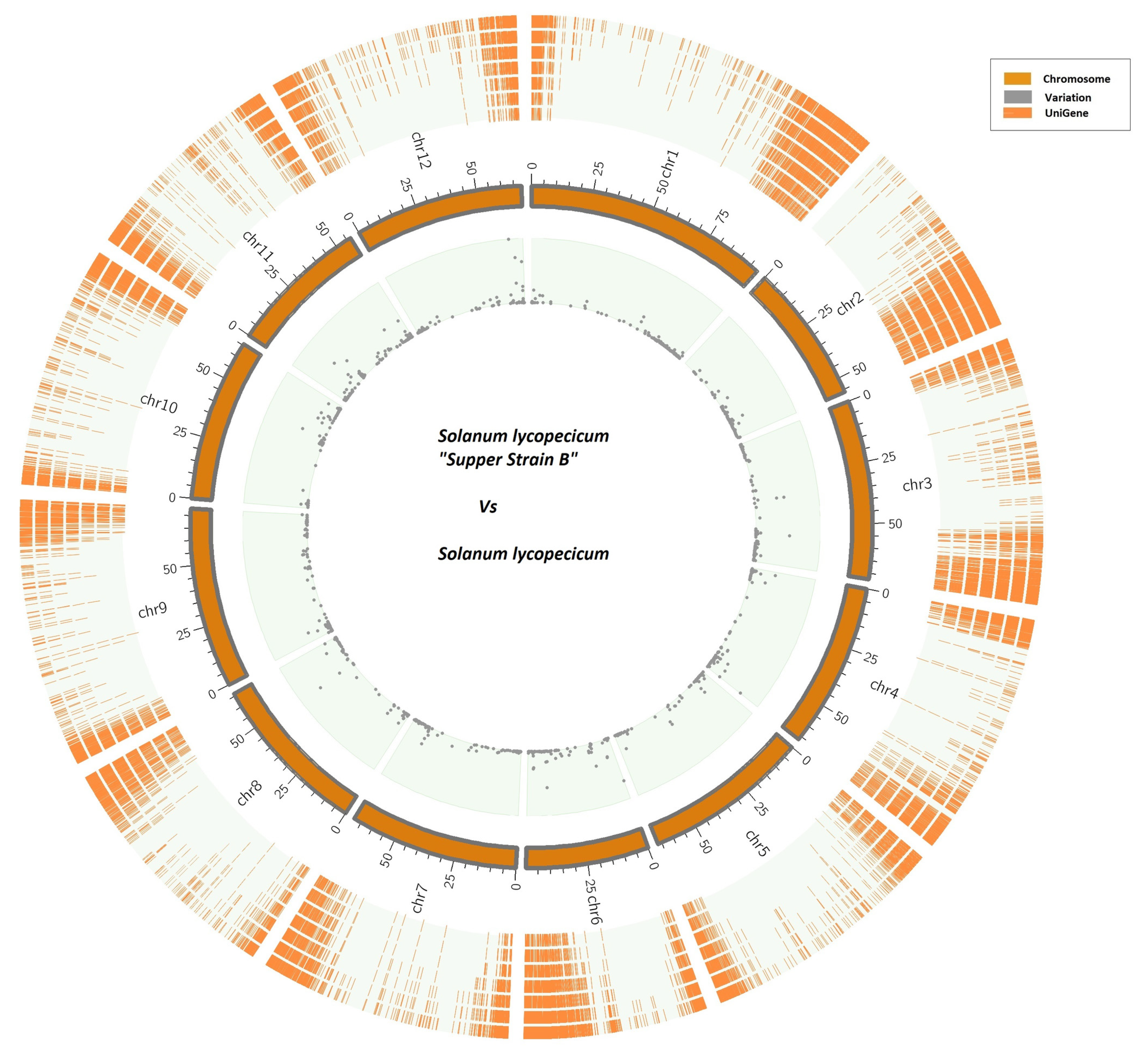
3.1.3. Matching with Tomato Genome
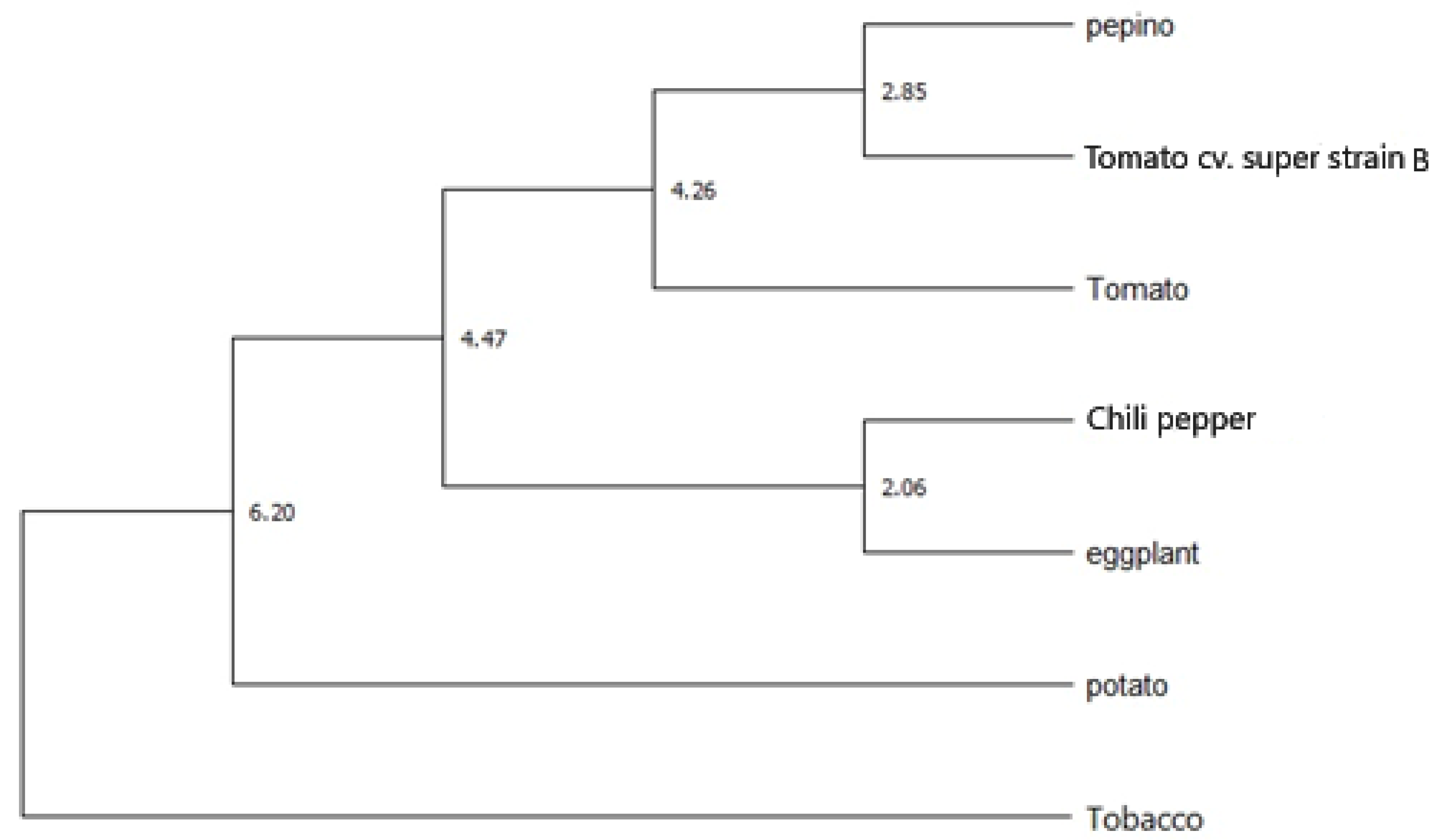
3.1.4. Molecular Phylogeny among Solanaceae Species
3.1.5. SSR and Nucleotide-Level Variants Discovery
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ercolano, M.R.; Sanseverino, W.; Carli, P.; Ferriello, F.; Frusciante, L. Genetic and Genomic Approaches for R-Gene Mediated Disease Resistance in Tomato: Retrospects and Prospects. Plant Cell Rep. 2012, 31, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-H.; Liu, X.-Y.; Wang, Y.; Hua, Q.; Song, X.-M.; Gu, Z.; Pu, D.-Z. Effect of Water Stress on Yield and Nutrition Quality of Tomato Plant Overexpressing StAPX. Biol. Plant. 2014, 58, 99–104. [Google Scholar] [CrossRef]
- Boureau, L.; How-Kit, A.; Teyssier, E.; Drevensek, S.; Rainieri, M.; Joubès, J.; Stammitti, L.; Pribat, A.; Bowler, C.; Hong, Y. A CURLY LEAF Homologue Controls Both Vegetative and Reproductive Development of Tomato Plants. Plant Mol. Biol. 2016, 90, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Olmstead, R.G.; Bohs, L. A Summary of Molecular Systematic Research in Solanaceae: 1982–2006. In Proceedings of the VI International Solanaceae Conference: Genomics Meets Biodiversity 745, Madison, WI, USA, 23–27 July 2006; pp. 255–268. [Google Scholar]
- Olmstead, R.G.; Bohs, L.; Migid, H.A.; Santiago-Valentin, E.; Garcia, V.F.; Collier, S.M. A Molecular Phylogeny of the Solanaceae. Taxon 2008, 57, 1159–1181. [Google Scholar] [CrossRef]
- Clarkson, J.J.; Knapp, S.; Garcia, V.F.; Olmstead, R.G.; Leitch, A.R.; Chase, M.W. Phylogenetic Relationships in Nicotiana (Solanaceae) Inferred from Multiple Plastid DNA Regions. Mol. Phylogenet. Evol. 2004, 33, 75–90. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, Z.Y.; Olmstead, R.G. A Retroposon Insertion to the Waxy Gene: Defining Monophyly of the Tribe Hyoscyameae (Solanaceae) and Revealing the Allopolyploid Origin of Atropa Belladonna. Mol. Biol. Evol 2006, 23, 2263–2267. [Google Scholar] [CrossRef]
- Levin, R.A.; Bernardello, G.; Whiting, C.; Miller, J.S. A New Generic Circumscription in Tribe Lycieae (Solanaceae). Taxon 2011, 60, 681–690. [Google Scholar] [CrossRef]
- Filipowicz, N.; Renner, S.S. Brunfelsia (Solanaceae): A Genus Evenly Divided between South America and Radiations on Cuba and Other Antillean Islands. Mol. Phylogenet. Evol. 2012, 64, 1–11. [Google Scholar] [CrossRef]
- Fregonezi, J.N.; Turchetto, C.; Bonatto, S.L.; Freitas, L.B. Biogeographical History and Diversification of Petunia and Calibrachoa (Solanaceae) in the Neotropical Pampas Grassland. Bot. J. Linn. Soc. 2013, 171, 140–153. [Google Scholar] [CrossRef]
- Fregonezi, J.N.; de Freitas, L.B.; Bonatto, S.L.; Semir, J.; Stehmann, J.R. Infrageneric Classification of Calibrachoa (Solanaceae) Based on Morphological and Molecular Evidence. Taxon 2012, 61, 120–130. [Google Scholar] [CrossRef]
- Poczai, P.; Hyvönen, J.; Symon, D.E. Phylogeny of Kangaroo Apples (Solanum Subg. Archaesolanum, Solanaceae). Mol. Biol. Rep. 2011, 38, 5243–5259. [Google Scholar] [CrossRef] [PubMed]
- Tepe, E.J.; Farruggia, F.T.; Bohs, L. A 10-gene Phylogeny of Solanum Section Herpystichum (Solanaceae) and a Comparison of Phylogenetic Methods. Am. J. Bot. 2011, 98, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.; Bohs, L. An Explosive Innovation: Phylogenetic Relationships of Solanum Section Gonatotrichum (Solanaceae). PhytoKeys 2012, 8, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bohs, L.; Olmstead, R.G. A Reassessment of Normania and Triguera (Solanaceae). Plant Syst. Evol. 2001, 228, 33–48. [Google Scholar] [CrossRef]
- Bohs, L. Phylogeny of the Cyphomandra Clade of the Genus Solanum (Solanaceae) Based on ITS Sequence Data. Taxon 2007, 56, 1012–1026. [Google Scholar] [CrossRef]
- Bloom, A.J.; Zwieniecki, M.A.; Passioura, J.B.; Randall, L.B.; Holbrook, N.M.; St. Clair, D.A. Water Relations under Root Chilling in a Sensitive and Tolerant Tomato Species. Plant. Cell Environ. 2004, 27, 971–979. [Google Scholar] [CrossRef]
- Ahmad, N.; Jianyu, L.; Xu, T.; Noman, M.; Jameel, A.; Na, Y.; Yuanyuan, D.; Nan, W.; Xiaowei, L.; Fawei, W.; et al. Overexpression of a novel Cytochrome P450 Promotes Flavonoid Biosynthesis and Osmotic Stress Tolerance in Transgenic Arabidopsis. Genes 2019, 10, 756. [Google Scholar] [CrossRef]
- Hong, Y.; Ahmad, N.; Zhang, J.; Lv, Y.; Zhang, X.; Ma, X.; Xiuming, L.; Na, Y. Genome-wide Analysis and Transcriptional Reprogramming of MYB Superfamily Revealed Positive Insights into Abiotic Stress Responses and Anthocyanin Accumulation in Carthamus tinctorius L. Mol. Genet. Genom. 2022, 297, 125–145. [Google Scholar] [CrossRef]
- Noman, M.; Jameel, A.; Qiang, W.-D.; Ahmad, N.; Liu, W.-C.; Wang, F.-W.; Li, H.-Y. Overexpression of GmCAMTA12 Enhanced Drought Tolerance in Arabidopsis and Soybean. Int. J. Mol. Sci. 2019, 20, 4849. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential Gene and Transcript Expression Analysis of RNA-Seq Experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Rensink, W.A.; Lee, Y.; Liu, J.; Iobst, S.; Ouyang, S.; Buell, C.R. Comparative analyses of six solanaceous transcriptomes reveal a high degree of sequence conservation and species-specific transcripts. BMC Genom. 2005, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zeng, R.; Li, Y.; Zhao, M.; Chao, J.; Li, Y.; Wang, K.; Zhu, L.; Tian, W.-M.; Liang, C. Gene Expression Analysis and SNP/InDel Discovery to Investigate Yield Heterosis of Two Rubber Tree F1 Hybrids. Sci. Rep. 2016, 6, 24984. [Google Scholar] [CrossRef]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X. Genomic Analyses Provide Insights into the History of Tomato Breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Tabata, S. Tomato Genome Sequence. In Functional Genomics and Biotechnology in Solanaceae and Cucurbitaceae Crops; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–13. [Google Scholar]
- Jung, Y.J.; Nou, I.S.; Cho, Y.G.; Kim, M.K.; Kim, H.-T.; Kang, K.K. Identification of an SNP Variation of Elite Tomato (Solanum lycopersicum L.) Lines Using Genome Resequencing Analysis. Hortic. Environ. Biotechnol. 2016, 57, 173–181. [Google Scholar] [CrossRef]
- Kim, B.; Hwang, I.S.; Lee, H.-J.; Oh, C.-S. Combination of Newly Developed SNP and InDel Markers for Genotyping the Cf-9 Locus Conferring Disease Resistance to Leaf Mold Disease in the Tomato. Mol. Breed. 2017, 37, 59. [Google Scholar] [CrossRef]
- Gupta, P.; Dholaniya, P.S.; Devulapalli, S.; Tawari, N.R.; Sreelakshmi, Y.; Sharma, R. Reanalysis of Genome Sequences of Tomato Accessions and Its Wild Relatives: Development of Tomato Genomic Variation (TGV) Database Integrating SNPs and INDELs Polymorphisms. Bioinformatics 2020, 36, 4984–4990. [Google Scholar] [CrossRef] [PubMed]
- Francesca, S.; Vitale, L.; Arena, C.; Raimondi, G.; Olivieri, F.; Cirillo, V.; Paradiso, A.; de Pinto, M.C.; Maggio, A.; Barone, A. The Efficient Physiological Strategy of a Novel Tomato Genotype to Adapt to Chronic Combined Water and Heat Stress. Plant Biol. 2022, 24, 62–74. [Google Scholar] [CrossRef]
- Meher; Shivakrishna, P.; Ashok Reddy, K.; Manohar Rao, D. Effect of PEG-6000 Imposed Drought Stress on RNA Content, Relative Water Content (RWC), and Chlorophyll Content in Peanut Leaves and Roots. Saudi J. Biol. Sci. 2018, 25, 285–289. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Li, Y.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic Distribution, Putative Functions and Mutational Mechanisms: A Review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Blanca, J.M.; Pascual, L.; Ziarsolo, P.; Nuez, F.; Cañizares, J. Ngs_backbone: A Pipeline for Read Cleaning, Mapping and SNP Calling Using Next Generation Sequence. BMC Genom. 2011, 12, 285. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Database and Its Supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pozo, N.; Menda, N.; Edwards, J.D.; Saha, S.; Tecle, I.Y.; Strickler, S.R.; Bombarely, A.; Fisher-York, T.; Pujar, A.; Foerster, H. The Sol Genomics Network (SGN)—From Genotype to Phenotype to Breeding. Nucleic Acids Res. 2015, 43, D1036–D1041. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Mott, R. EST_GENOME: A Program to Align Spliced DNA Sequences to Unspliced Genomic DNA. Bioinformatics 1997, 13, 477–478. [Google Scholar] [CrossRef]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A Program for Detecting, Evaluating, and Reconstructing Potential Coding Regions in EST Sequences. In Proceedings of the International Conference on Intelligent Systems for Molecular Biology ISMB, Heidelberg, Germany, 6–10 August 1999; Volume 99, pp. 138–148. [Google Scholar]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Särkinen, T.; Bohs, L.; Olmstead, R.G.; Knapp, S. A Phylogenetic Framework for Evolutionary Study of the Nightshades (Solanaceae): A Dated 1000-Tip Tree. BMC Evol. Biol. 2013, 13, 214. [Google Scholar] [CrossRef]
- Peralta, I.E.; Spooner, D.M. Granule-Bound Starch Synthase (GBSSI) Gene Phylogeny of Wild Tomatoes (Solanum L. Section Lycopersicon [Mill.] Wettst. Subsection Lycopersicon). Am. J. Bot. 2001, 88, 1888–1902. [Google Scholar] [CrossRef]
- Martins, T.R.; Barkman, T.J. Reconstruction of Solanaceae Phylogeny Using the Nuclear Gene SAMT. Syst. Bot. 2005, 30, 435–447. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Yano, K.; Suzuki, A.; Kawamura, S.; Sakurai, N.; Suda, K.; Kurabayashi, A.; Suzuki, T.; Tsugane, T.; Watanabe, M.; et al. Large-Scale Analysis of Full-Length CDNAs from the Tomato (Solanum lycopersicum) Cultivar Micro-Tom, a Reference System for the Solanaceae Genomics. BMC Genom. 2010, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Lim, J.; Dale, J.M.; Chen, H.; Shinn, P.; Palm, C.J.; Southwick, A.M.; Wu, H.C.; Kim, C.; Nguyen, M. Empirical Analysis of Transcriptional Activity in the Arabidopsis Genome. Science 2003, 302, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Sakurai, T.; Totoki, Y.; Toyoda, A.; Seki, M.; Ishiwata, A.; Akiyama, K.; Kurotani, A.; Yoshida, T.; Mochida, K. Sequencing and Analysis of Approximately 40 000 Soybean CDNA Clones from a Full-Length-Enriched CDNA Library. DNA Res. 2008, 15, 333–346. [Google Scholar] [CrossRef]
- Alexandrov, N.N.; Troukhan, M.E.; Brover, V.V.; Tatarinova, T.; Flavell, R.B.; Feldmann, K.A. Features of Arabidopsis Genes and Genome Discovered Using Full-Length CDNAs. Plant Mol. Biol. 2006, 60, 69–85. [Google Scholar] [CrossRef]
- Ralph, S.G.; Chun, H.J.E.; Cooper, D.; Kirkpatrick, R.; Kolosova, N.; Gunter, L.; Tuskan, G.A.; Douglas, C.J.; Holt, R.A.; Jones, S.J.M. Analysis of 4,664 High-Quality Sequence-Finished Poplar Full-Length CDNA Clones and Their Utility for the Discovery of Genes Responding to Insect Feeding. BMC Genom. 2008, 9, 57. [Google Scholar] [CrossRef]
- Herraiz, F.J.; Blanca, J.; Ziarsolo, P.; Gramazio, P.; Plazas, M.; Anderson, G.J.; Prohens, J.; Vilanova, S. The First de Novo Transcriptome of Pepino (Solanum muricatum): Assembly, Comprehensive Analysis and Comparison with the Closely Related Species S. Caripense, Potato and Tomato. BMC Genom. 2016, 17, 321. [Google Scholar] [CrossRef]
- Wei, D.-D.; Chen, E.-H.; Ding, T.-B.; Chen, S.-C.; Dou, W.; Wang, J.-J. De Novo Assembly, Gene Annotation, and Marker Discovery in Stored-Product Pest Liposcelis entomophila (Enderlein) Using Transcriptome Sequences. PLoS ONE 2013, 8, e80046. [Google Scholar] [CrossRef]
- Tipton, K.; Boyce, S. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB), Enzyme Supplement 5 (1999). Eur. J. Biochem 1999, 264, 610–650. [Google Scholar]
- Garzón-Martínez, G.A.; Zhu, Z.I.; Landsman, D.; Barrero, L.S.; Mariño-Ramírez, L. The Physalis peruviana Leaf Transcriptome: Assembly, Annotation and Gene Model Prediction. BMC Genom. 2012, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Sierro, N.; Battey, J.N.D.; Ouadi, S.; Bovet, L.; Goepfert, S.; Bakaher, N.; Peitsch, M.C.; Ivanov, N.V. Reference Genomes and Transcriptomes of Nicotiana Sylvestris and Nicotiana Tomentosiformis. Genome Biol. 2013, 14, R60. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Zhao, J.; He, C. Evolutionary Developmental Genetics of Fruit Morphological Variation within the Solanaceae. Front. Plant Sci. 2015, 6, 248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, Z.; Chu, G.; Huang, C.; Tian, S.; Zhao, Z.; Chen, G. Anthocyanin Accumulation and Molecular Analysis of Anthocyanin Biosynthesis-Associated Genes in Eggplant (Solanum melongena L.). J. Agric. Food Chem. 2014, 62, 2906–2912. [Google Scholar] [CrossRef]
- Kohara, A.; Nakajima, C.; Hashimoto, K.; Ikenaga, T.; Tanaka, H.; Shoyama, Y.; Yoshida, S.; Muranaka, T. A Novel Glucosyltransferase Involved in Steroid Saponin Biosynthesis in Solanum Aculeatissimum. Plant Mol. Biol. 2005, 57, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Gramazio, P.; Prohens, J.; Plazas, M.; Andújar, I.; Herraiz, F.J.; Castillo, E.; Knapp, S.; Meyer, R.S.; Vilanova, S. Location of Chlorogenic Acid Biosynthesis Pathway and Polyphenol Oxidase Genes in a New Interspecific Anchored Linkage Map of Eggplant. BMC Plant Biol. 2014, 14, 350. [Google Scholar] [CrossRef]
- Klann, E.; Yelle, S.; Bennett, A.B. Tomato Fruit Acid Invertase Complementary DNA: Nucleotide and Deduced Amino Acid Sequences. Plant Physiol. 1992, 99, 351. [Google Scholar] [CrossRef]
- Zhang, N.; Brewer, M.T.; van der Knaap, E. Fine Mapping of Fw3. 2 Controlling Fruit Weight in Tomato. Theor. Appl. Genet. 2012, 125, 273–284. [Google Scholar] [CrossRef]
- Su, L.Y.; Audran, C.; Bouzayen, M.; Roustan, J.-P.; Chervin, C. The Aux/IAA, Sl-IAA17 Regulates Quality Parameters over Tomato Fruit Development. Plant Signal. Behav. 2015, 10, e1071001. [Google Scholar] [CrossRef]
- Gonzalez, N.; Gévaudant, F.; Hernould, M.; Chevalier, C.; Mouras, A. The Cell Cycle-associated Protein Kinase WEE1 Regulates Cell Size in Relation to Endoreduplication in Developing Tomato Fruit. Plant J. 2007, 51, 642–655. [Google Scholar] [CrossRef]
- Outchkourov, N.S.; Karlova, R.; Hölscher, M.; Schrama, X.; Blilou, I.; Jongedijk, E.; Simon, C.D.; van Dijk, A.D.J.; Bosch, D.; Hall, R.D. Transcription Factor-Mediated Control of Anthocyanin Biosynthesis in Vegetative Tissues. Plant Physiol. 2018, 176, 1862–1878. [Google Scholar] [CrossRef]
- Meng, C.; Zhang, S.; Deng, Y.-S.; Wang, G.-D.; Kong, F.-Y. Overexpression of a Tomato Flavanone 3-Hydroxylase-like Protein Gene Improves Chilling Tolerance in Tobacco. Plant Physiol. Biochem. 2015, 96, 388–400. [Google Scholar] [CrossRef]
- Sacco, A.; Raiola, A.; Calafiore, R.; Barone, A.; Rigano, M.M. New Insights in the Control of Antioxidants Accumulation in Tomato by Transcriptomic Analyses of Genotypes Exhibiting Contrasting Levels of Fruit Metabolites. BMC Genom. 2019, 20, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, G.; Li, Y.; Zhang, J.; Shi, M.; Muhammad, T.; Liang, Y. Transcriptome Profiling of Tomato Uncovers an Involvement of Cytochrome P450s and Peroxidases in Stigma Color Formation. Front. Plant Sci. 2017, 8, 897. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Li, W.; Wang, W.; Bai, G. Cloning and Characterization of a CDNA Coding a Hydroxycinnamoyl-CoA Quinate Hydroxycinnamoyl Transferase Involved in Chlorogenic Acid Biosynthesis in Lonicera Japonica. Planta Med. 2010, 76, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Sonnante, G.; D’Amore, R.; Blanco, E.; Pierri, C.L.; De Palma, M.; Luo, J.; Tucci, M.; Martin, C. Novel Hydroxycinnamoyl-Coenzyme A Quinate Transferase Genes from Artichoke Are Involved in the Synthesis of Chlorogenic Acid. Plant Physiol. 2010, 153, 1224–1238. [Google Scholar] [CrossRef]
- Moy, M.; Dai, N.; Cohen, S.; Hadas, R.; Granot, D.; Petrikov, M.; Yeselson, Y.; Shen, S.; Schaf, A.A. The Presence of a Retrotransposon in the Promoter Region of the TIV Gene Encoding for Soluble Acid Invertase Distinguishes between the Sucrose and Hexose Accumulating Species of Lycopersicon. In Proceedings of the VI International Solanaceae Conference: Genomics Meets Biodiversity 745, Madison, WI, USA, 23–27 July 2006; pp. 429–436. [Google Scholar]
- Wang, Y.; Diehl, A.; Wu, F.; Vrebalov, J.; Giovannoni, J.; Siepel, A.; Tanksley, S.D. Sequencing and Comparative Analysis of a Conserved Syntenic Segment in the Solanaceae. Genetics 2008, 180, 391–408. [Google Scholar] [CrossRef]
- Gao, L.; Qi, J. Whole Genome Molecular Phylogeny of Large DsDNA Viruses Using Composition Vector Method. BMC Evol. Biol. 2007, 7, 41. [Google Scholar] [CrossRef]
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within Genes: Structure, Function, and Evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef]
- Kim, T.-S.; Booth, J.G.; Gauch, H.G.; Sun, Q.; Park, J.; Lee, Y.-H.; Lee, K. Simple Sequence Repeats in Neurospora crassa: Distribution, Polymorphism and Evolutionary Inference. BMC Genom. 2008, 9, 31. [Google Scholar] [CrossRef]
- Riley, D.E.; Krieger, J.N. Embryonic Nervous System Genes Predominate in Searches for Dinucleotide Simple Sequence Repeats Flanked by Conserved Sequences. Gene 2009, 429, 74–79. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, Y.; Yang, R.; Feng, H.; Ouyang, Q.; Tian, Y.; Tan, Z.; Li, M.; Niu, Y.; Jiang, J. Coevolution between Simple Sequence Repeats (SSRs) and Virus Genome Size. BMC Genom. 2012, 13, 435. [Google Scholar] [CrossRef]
- Olivieri, F.; Calafiore, R.; Francesca, S.; Schettini, C.; Chiaiese, P.; Rigano, M.M.; Barone, A. High-Throughput Genotyping of Resilient Tomato Landraces to Detect Candidate Genes Involved in the Response to High Temperatures. Genes 2020, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Robbins, M.D.; Sim, S.-C.; Yang, W.; Van Deynze, A.; van der Knaap, E.; Joobeur, T.; Francis, D.M. Mapping and Linkage Disequilibrium Analysis with a Genome-Wide Collection of SNPs That Detect Polymorphism in Cultivated Tomato. J. Exp. Bot. 2011, 62, 1831–1845. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.W.; Jukes, T.H. Rates of Transition and Transversion in Coding Sequences since the Human-Rodent Divergence. Genomics 1994, 20, 386–396. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Reads | |
|---|---|---|
| Raw | Clean | |
| Total reads | 41,779,729 × 2 | 41,083,929 × 2 |
| Total reads data size (Gb) | 31 Gb | 29.6 Gb |
| G/C (%) | 44 | 44 |
| Transcripts | ||
| After Assembly | After Filtration | |
| Number | 98,069 | 41,900 |
| Total length | 75,145,303 | 37,600,138 |
| Average length | 766.25 | 897.37 |
| Maximum length | 6279 | 6279 |
| Number of Transcripts | % | |
|---|---|---|
| Annotated in Swiss-Prot | 21,490 | 94.74 |
| Annotated in ITAG2.4 | 74 | 0.33 |
| Annotated in UniRef90 | 1118 | 4.93 |
| Total annotated in protein databases | 22,682 | 100 |
| Trait | Genes | Gene Name | Bit Score |
|---|---|---|---|
| Fruit shape | Fw3 | Promoter-regulatory | 333 |
| Sl-IAA17 | Solanaceae transcription repressor | 431 | |
| Wee | Protein kinase | 219 | |
| Anthocyanins pathway | SlAT2 | Anthocyanin acyltransferase-like | 331 |
| ANS | Anthocyanidin synthase | 233 | |
| F3H | Flavanone 3-hydroxylase | 491 | |
| CHS2 | Chalcone synthase | 163 | |
| Chlorogenic acid pathway | C3H | Cytochrome P450 | 1068 |
| HCT | Hydroxycinnamoyl transferase | 904 | |
| Saponines pathway | Egp#1-1 | Glycosyltransferase | 326 |
| Sucrose accumulator | TIV1 | Acid invertase | 1310 |
| Nucleotide Motif | Number | % | Unigenes |
|---|---|---|---|
| Dinucleotide | |||
| AT | 509 | 26.22 | |
| TA | 418 | 21.54 | |
| AG | 230 | 11.85 | |
| Other Dinucleotides | 784 | 40.39 | |
| Total | 1941 | 100 | 1826 |
| Trinucleotide | |||
| GAA | 190 | 6.32 | |
| TTC | 183 | 6.09 | |
| TCT | 134 | 4.46 | |
| ATT | 120 | 3.99 | |
| TTG | 119 | 3.96 | |
| TGA | 97 | 3.23 | |
| CTT | 96 | 3.19 | |
| AAG, AGA | 125 | 4.16 | |
| Other Trinucleotides | 1942 | 64.60 | |
| Total | 3006 | 100 | 2873 |
| Tetra-nucleotide | |||
| AAAT | 12 | 12.0 | |
| TTAA | 9 | 9.0 | |
| AAAC | 7 | 7.0 | |
| ATTT, TTTC | 6 | 6.0 | |
| AAAG, AAGA | 4 | 4.0 | |
| Other Tetra-nucleotides | 62 | 62.0 | |
| Total | 100 | 100 | 107 |
| Species | SNPs | INDELs |
|---|---|---|
| Solanum lycopersicum “super strain B” vs. Solanum lycopersicum | 4541 | 744 |
| SNPs Transitions | Number (%) | SNPs Transversions | Number (%) | TS/TV Ratio | Complex |
|---|---|---|---|---|---|
| A<->G | 1315 (28.9) | A<->C | 489 (10.8) | ||
| C<->T | 1404 (30.1) | A<->T | 586 (12.9) | ||
| G<->C | 319 (7.0) | ||||
| G<->T | 400 (8.8) | ||||
| Total | 2719 (59.9) | 11794 (39.5) | 28 (0.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Zahrani, H.S.; Moussa, T.A.A.; Alsamadany, H.; Hafez, R.M.; Fuller, M.P. De Novo Transcriptome Analysis of Solanum lycopersicum cv. Super Strain B under Drought Stress. Agronomy 2023, 13, 2360. https://doi.org/10.3390/agronomy13092360
Al-Zahrani HS, Moussa TAA, Alsamadany H, Hafez RM, Fuller MP. De Novo Transcriptome Analysis of Solanum lycopersicum cv. Super Strain B under Drought Stress. Agronomy. 2023; 13(9):2360. https://doi.org/10.3390/agronomy13092360
Chicago/Turabian StyleAl-Zahrani, Hassan S., Tarek A. A. Moussa, Hameed Alsamadany, Rehab M. Hafez, and Michael P. Fuller. 2023. "De Novo Transcriptome Analysis of Solanum lycopersicum cv. Super Strain B under Drought Stress" Agronomy 13, no. 9: 2360. https://doi.org/10.3390/agronomy13092360
APA StyleAl-Zahrani, H. S., Moussa, T. A. A., Alsamadany, H., Hafez, R. M., & Fuller, M. P. (2023). De Novo Transcriptome Analysis of Solanum lycopersicum cv. Super Strain B under Drought Stress. Agronomy, 13(9), 2360. https://doi.org/10.3390/agronomy13092360