

Characterization of a Bacillus velezensis with Antibacterial Activity and Its Inhibitory Effect on Gray Mold Germ



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Growth Conditions and DNA Extraction
2.2. Genome Sequencing and Phylogenetic Analysis
2.3. Comparative Analysis of Gene Clusters for Antimicrobial Compounds
2.4. RNA Extraction and Transcriptome Analysis
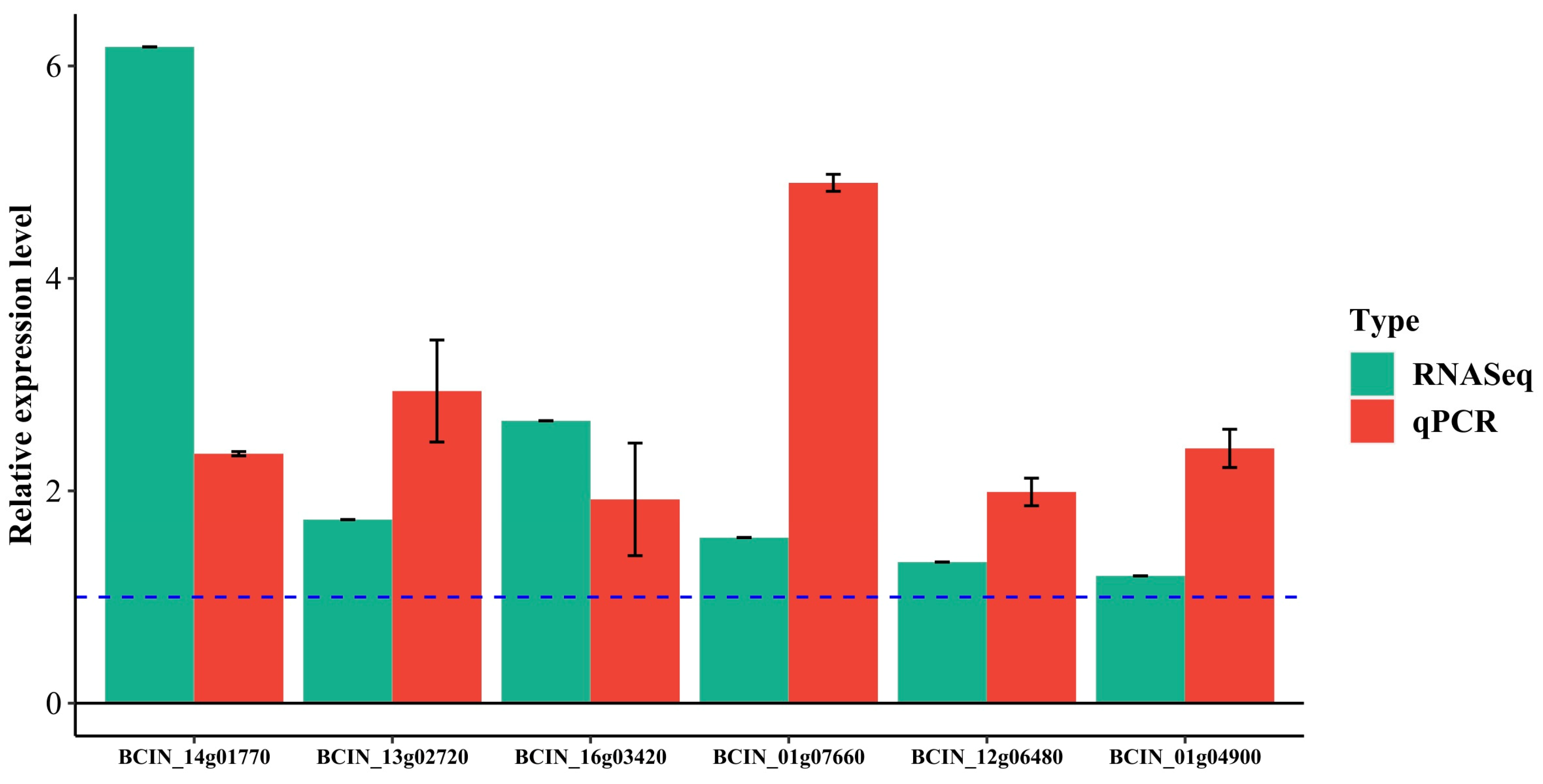
2.5. RT-qPCR Analysis
3. Results
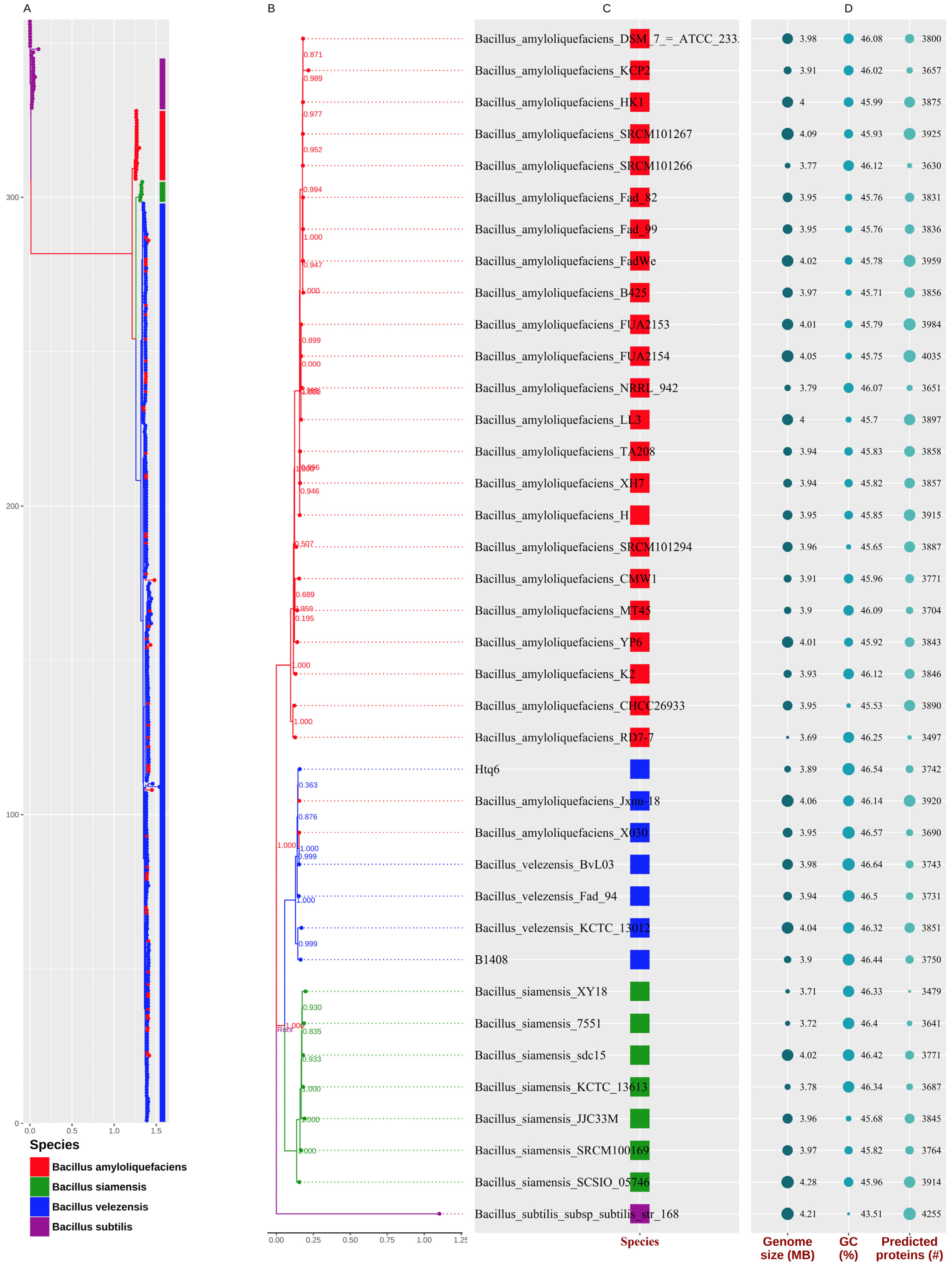
3.1. Species Assignment of the Strain Htq6
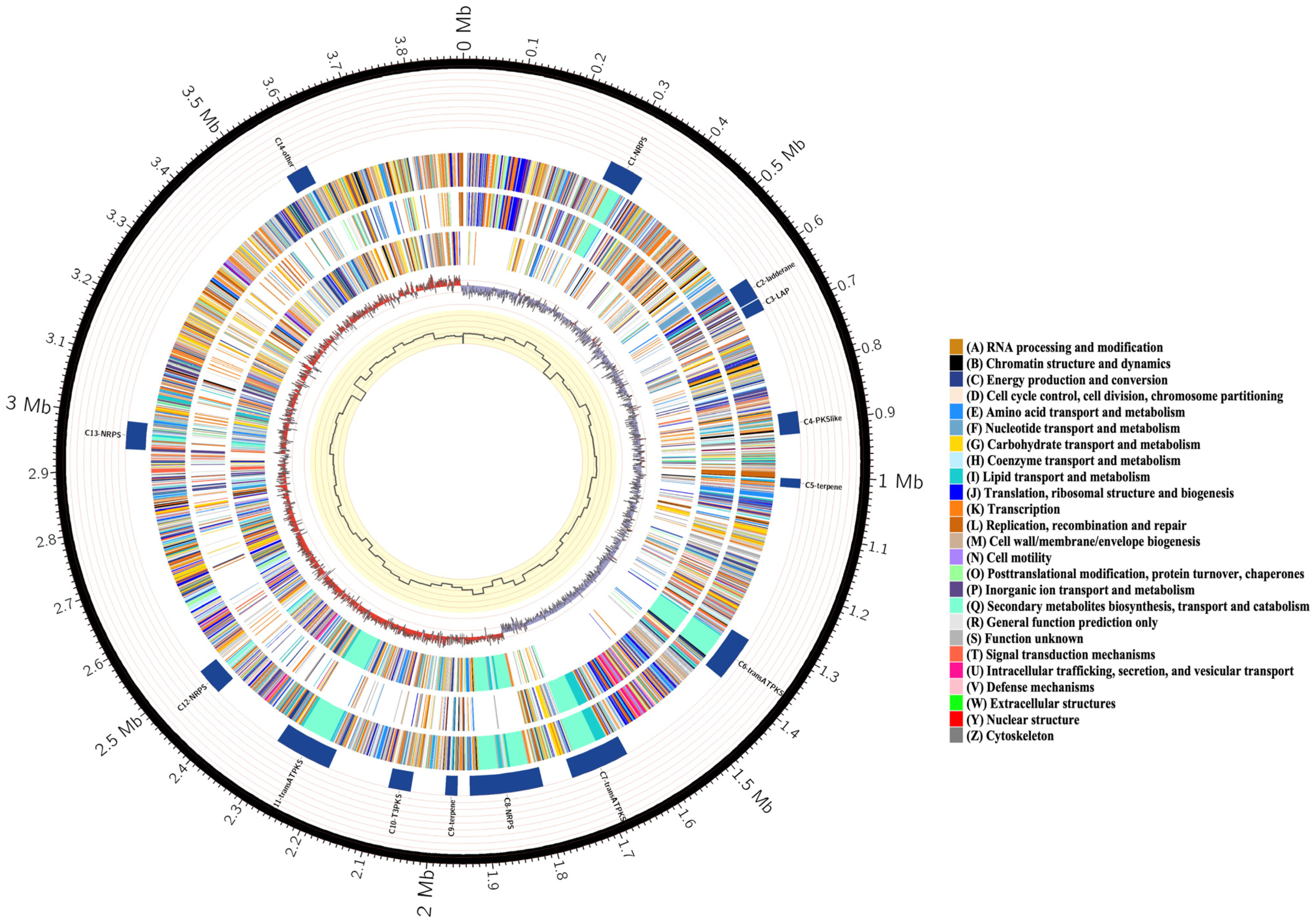
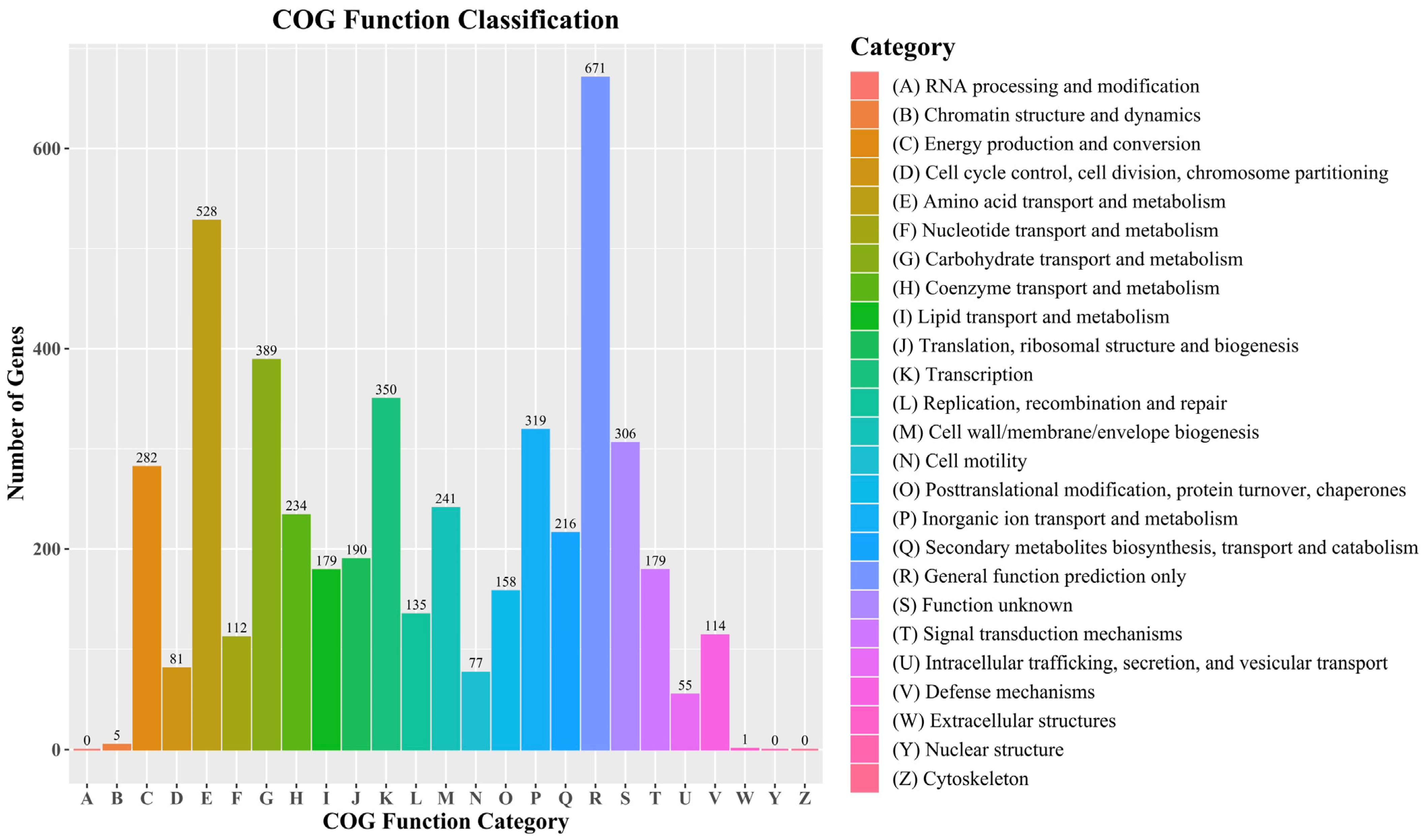
3.2. Features of Bacillus velezensis Htq6 Genome
3.3. Phylogenetic Analysis of Bacillus amyloliquefaciens, Bacillus siamensis, and Bacillus velezensis
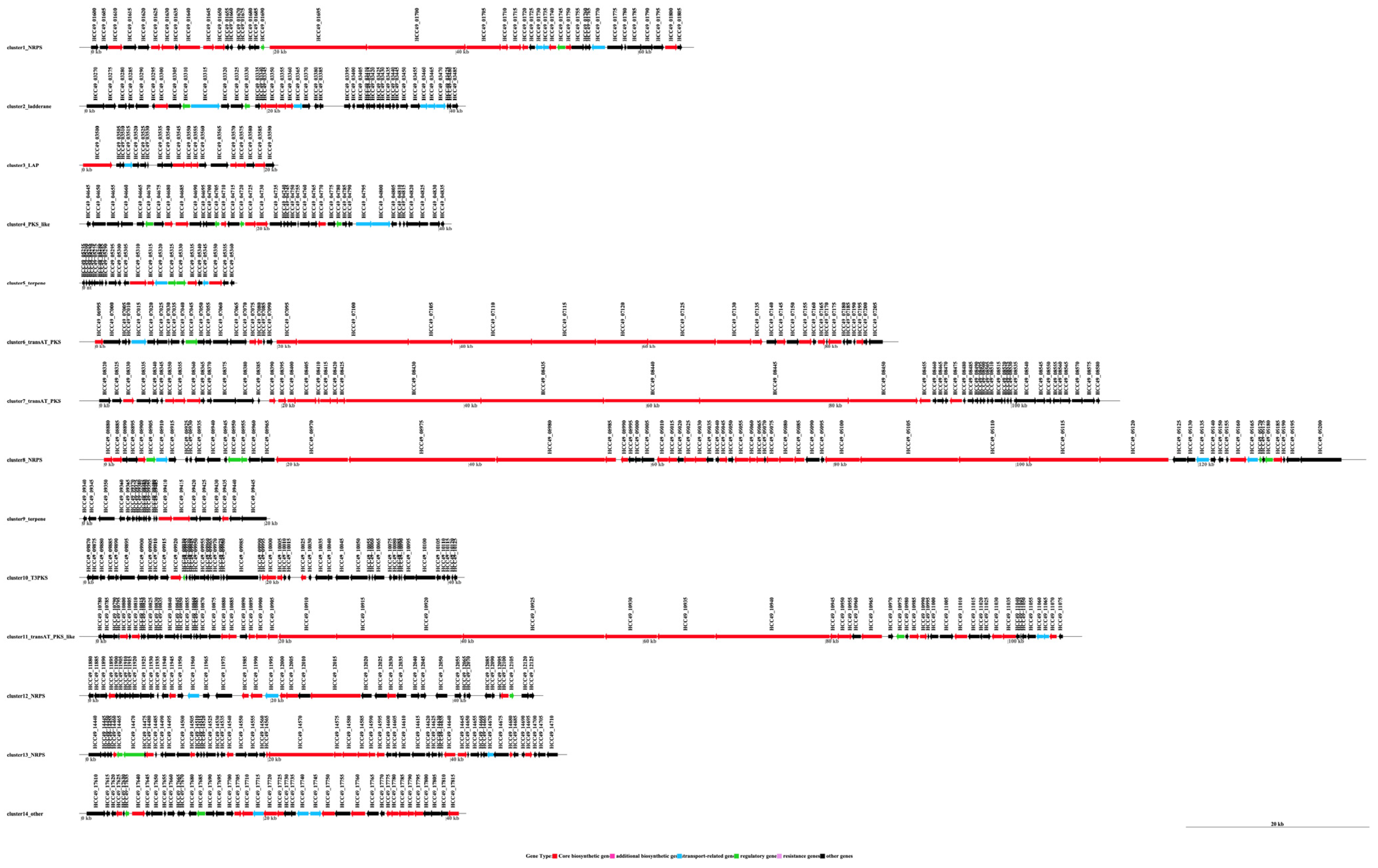
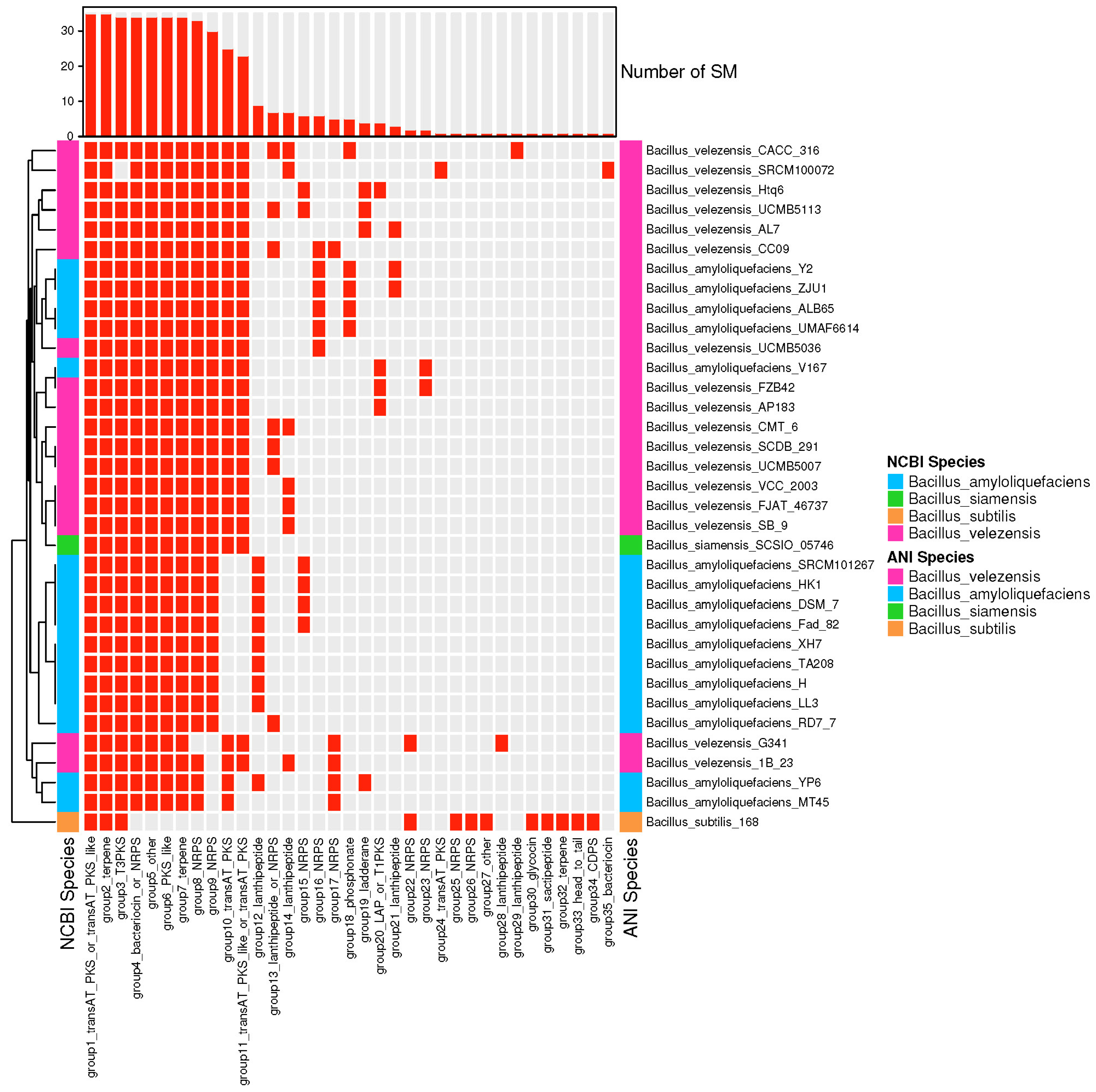
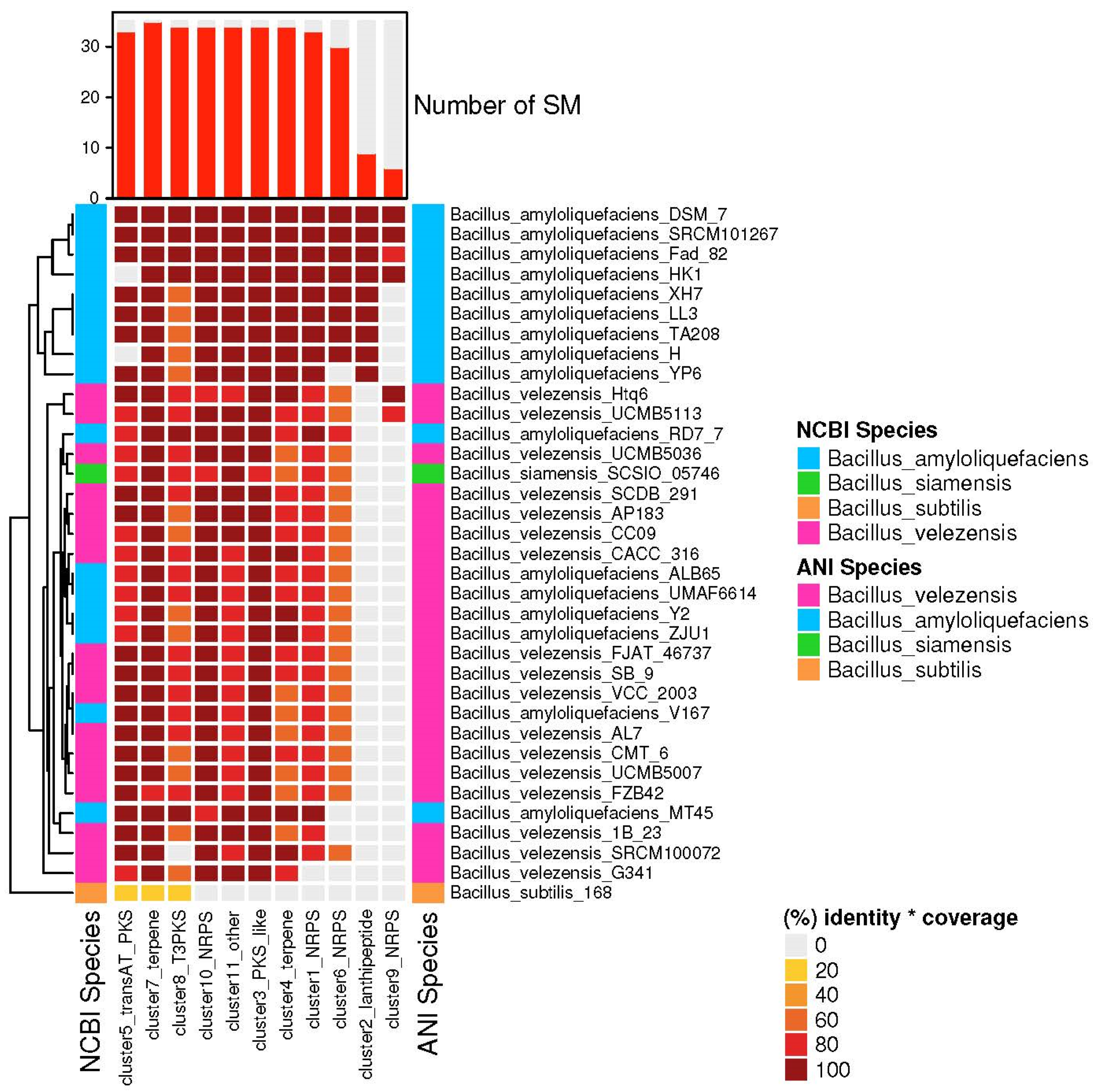
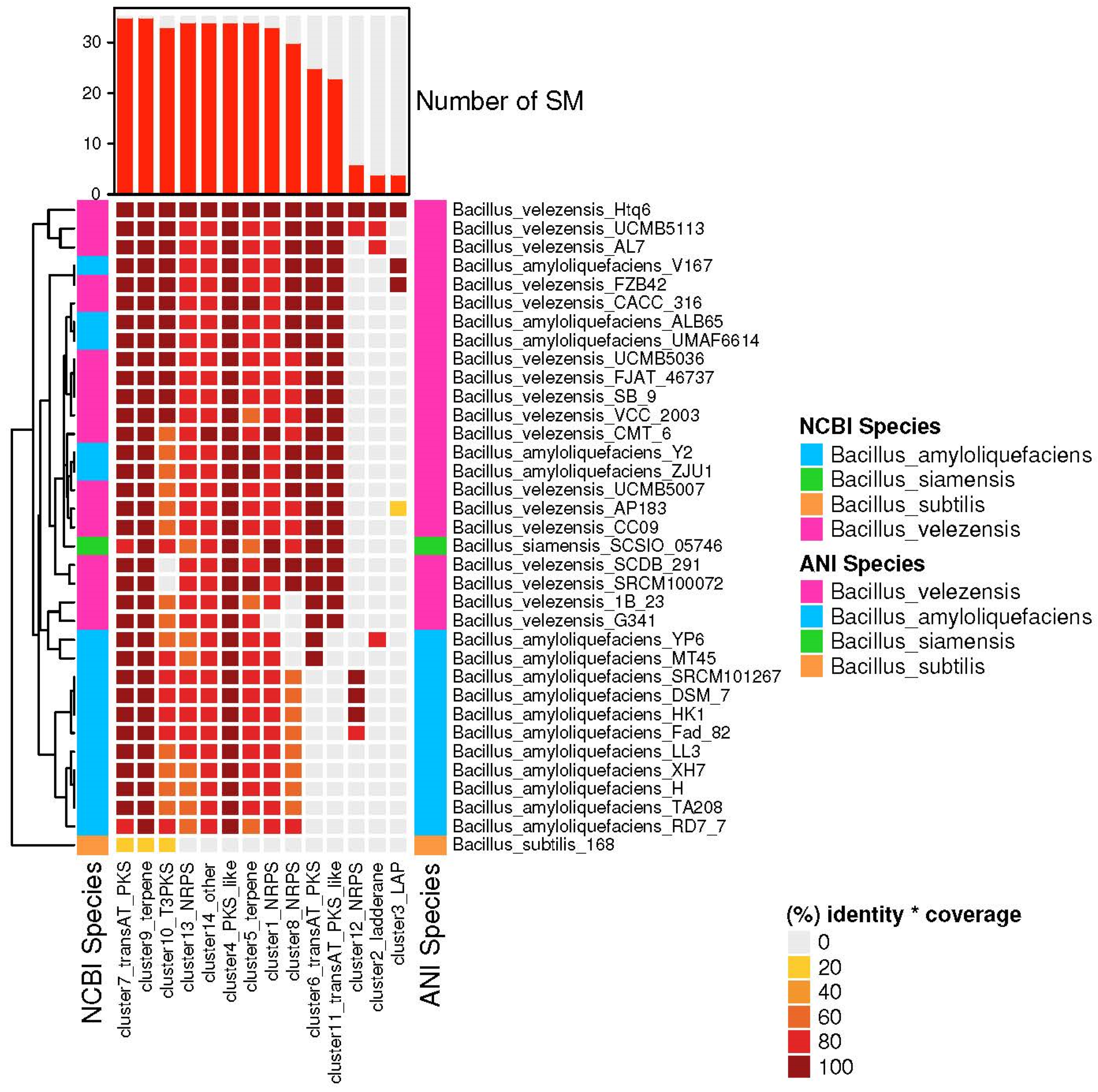
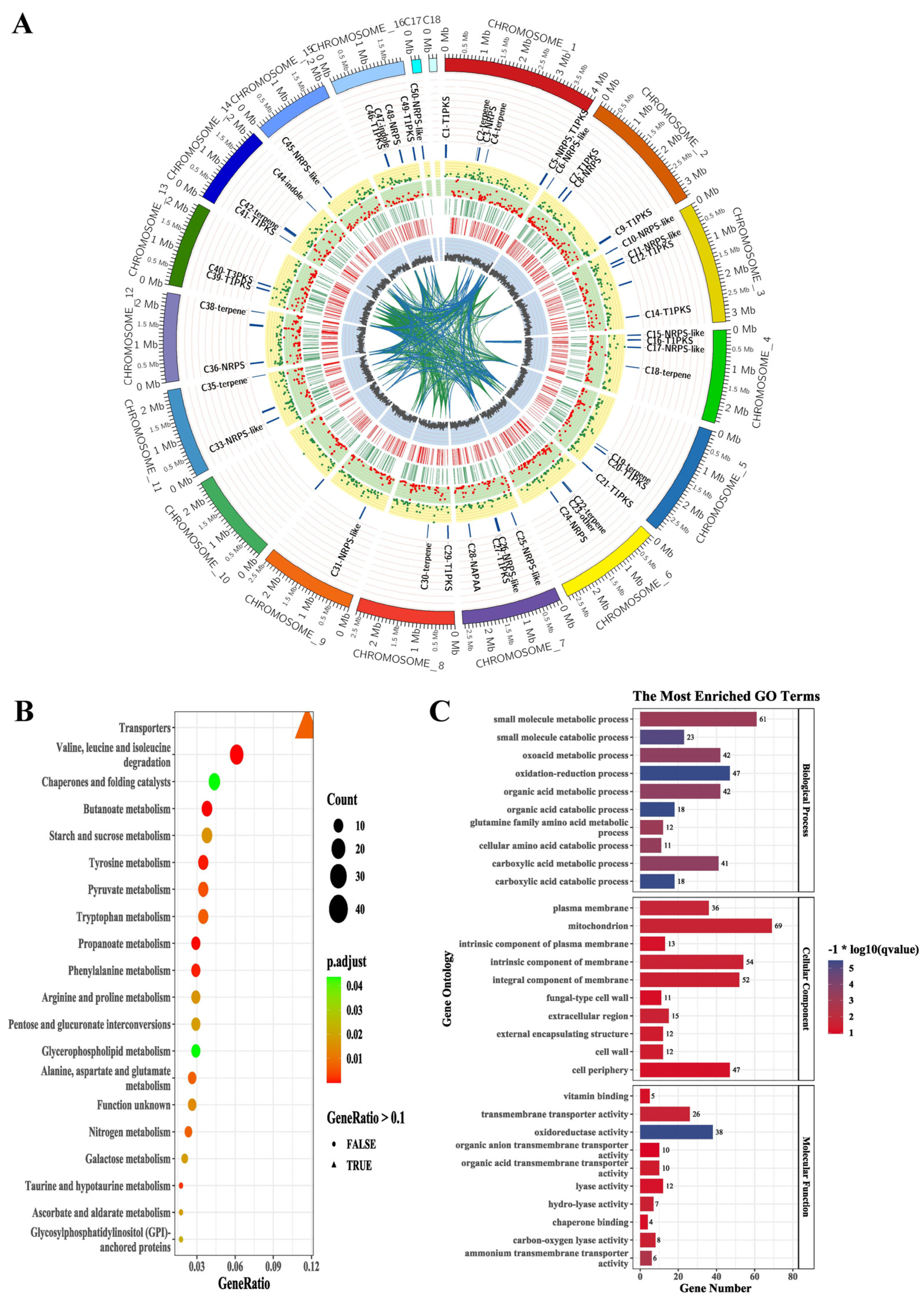
3.4. The Species of Biosynthetic Gene Cluster Were Compared and Analyzed
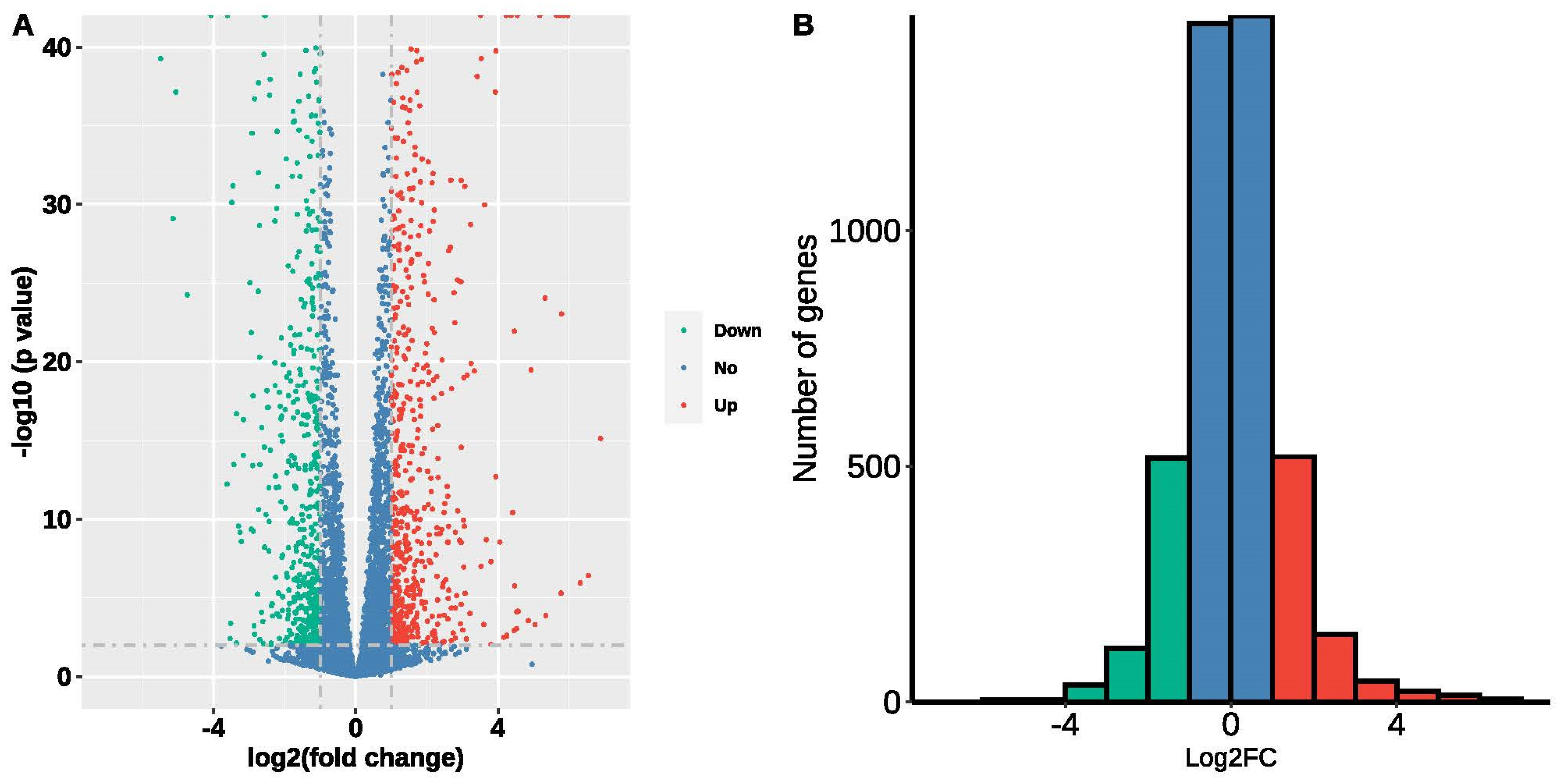
3.5. Data Mining of Transcriptome Profiles
3.6. Effects on Genes Associated with the Carbon Source Utilization and Energy Production of Botrytis cinerea
3.7. Effects on Genes Related to Cell Wall and Plasma Membrane Synthesis of Botrytis cinerea
3.8. Effects on Antioxidant-Related Genes of Botrytis cinerea
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strauch, O.; Strasser, H.; Hauschild, R.; Ehlers, R.U. Proposals for Bacterial and Fungal Biocontrol Agents; Springer Netherlands: Dordrecht, The Netherlands, 2011; pp. 267–288. [Google Scholar]
- Manzarnk, A.S.; Goutam, R.S.; Rajawat, M.V.S.; Sharma, P.K.; Sharma, S.K.; Singh, H.V. Trichoderma: Advent of Versatile Biocontrol Agent, Its Secrets and Insights into Mechanism of Biocontrol Potential. Sustainability 2022, 14, 12786. [Google Scholar] [CrossRef]
- Deravel, J.; Krier, F.; Jacques, P. Biopesticides, a complementary and alternative approach to the use of agrochemicals. A review. Biotechnol. Agron. Soc. Environ. 2014, 18, 220–232. [Google Scholar]
- Gunri, S.K.; Nath, R. Effect of organic manures, biofertilizers and biopesticides on productivity of summer groundnut (Arachis hypogeae L.) in red and laterite zone of west bengal. Legume Res. 2012, 35, 144–148. [Google Scholar]
- Ritika, B.; Utpal, D. An overview of fungal and bacterial biopesticides to control plant pathogens/diseases. Afr. J. Microbiol. Res. 2014, 8, 1749–1762. [Google Scholar] [CrossRef]
- Martyniuk, S. Factors affecting the use of microbial biopesticides in plant protection. Prog. Plant Prot. 2012, 52, 957–962. [Google Scholar]
- Fowler, S.V.; Hayes, L.; Hill; Richard, L.; Paynter, Q. Factors affecting the cost of weed biocontrol programs in New Zealand. Biol. Control 2015, 80, 119–127. [Google Scholar]
- Haripriya, K.; Jeyarani, S.; Mohankumar, S.; Soundararajan, R.P. Field evaluation of biocontrol agents and biopesticides against spotted pod borer, Maruca vitrata (Geyer) on lablab. Indian J. Agric. Res. 2019, 53, 599–603. [Google Scholar] [CrossRef]
- Jorgen, J.; Eilenberg, A.; Vestergaard, S.; Jensen, B. Biocontrol of Pests on Plant Crops in Denmark: Present Status and Future Potential. Biocontrol Sci. Technol. 2010, 10, 703–716. [Google Scholar]
- Peng, Y.; Li, S.J.; Yan, J.; Tang, Y.; Xu, B.L. Research Progress on Phytopathogenic Fungi and Their Role as Biocontrol Agents. Front. Microbiol. 2021, 12, 670135. [Google Scholar] [CrossRef]
- Imran, M.; Abo-Elyousr, K.A.M.; Mousa, M.A.A.; Saad, M.M. A study on the synergetic effect of Bacillus amyloliquefaciens and dipotassium phosphate on Alternaria solani causing early blight disease of tomato. Eur. J. Plant Pathol. 2022, 162, 63–77. [Google Scholar] [CrossRef]
- Xu, X.M.; Jeffries, P.; Pautasso, M.; Jeger, M.J. Combined use of biocontrol agents to manage plant diseases in theory and practice. Phytopathology 2011, 101, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Ayed, B.H.; Jemil, N.; Maalej, H.; Bayoudh, A.; Hmidet, N.; Nasri, M. Enhancement of solubilization and biodegradation of diesel oil by biosurfactant from Bacillus amyloliquefaciens An6. Int. Biodeterior. Biodegrad. 2015, 99, 8–14. [Google Scholar] [CrossRef]
- Lei, X.J.; Ru, Y.J.; Zhang, H.F. Effect of Bacillus amyloliquefaciens-based direct-fed microbials and antibiotic on performance, nutrient digestibility, cecal microflora, and intestinal morphology in broiler chickens. J. Appl. Poult. Res. 2014, 23, 486–493. [Google Scholar] [CrossRef]
- Lu, D.; Zhang, Y.; Niu, S.; Wang, L.; Lin, S.; Wang, C.; Ye, W.; Yan, C. Study of phenol biodegradation using Bacillus amyloliquefaciens strain WJDB-1 immobilized in alginate-chitosan-alginate (ACA) microcapsules by electrochemical method. Biodegradation 2012, 23, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Herrera-Balandrano, D.D.; Wang, Y.X.; Shi, X.C.; Chen, X.; Jin, Y.; Liu, F.Q.; Laborda, P. Biocontrol Ability of the Bacillus amyloliquefaciens Group, B. amyloliquefaciens, B. velezensis, B. nakamurai, and B. siamensis, for the Management of Fungal Postharvest Diseases: A Review. J. Agric. Food Chem. 2022, 70, 6591–6616. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.M.; Mosela, M.; Nicoletto, M.L.A.; Ribeiro, R.A.; Hungria, M.; Youssef, K.; Higashi, A.Y.; Mian, S.; Ferreira, A.S.; Gonçalves, L.S.A.; et al. Genomic Insights into the Antifungal Activity and Plant Growth-Promoting Ability in Bacillus velezensis CMRP 4490. Front. Microbiol. 2020, 11, 618415. [Google Scholar] [CrossRef]
- Tajbakhsh, M.; Karimi, A.; Fallah, F.; Akhavan, M.M. Overview of ribosomal and non-ribosomal antimicrobial peptides produced by Gram positive bacteria. Cell. Mol. Biol. 2017, 63, 20–32. [Google Scholar] [CrossRef]
- Landy, M.; Warren, G.H.; RosenmanM, S.B.; Colio, L.G. Bacillomycin; an antibiotic from Bacillus subtilis active against pathogenic fungi. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1948, 67, 539–541. [Google Scholar] [CrossRef]
- Xiang, Y.P.; Chen, Z.Y.; Luo, C.P.; Zhou, H.F.; Liu, Y.F. The Antifungal Activities of spp. and Its Relationship with Lipopeptide Antibiotics Produced by spp. Sci. Agric. Sin. 2015, 48, 4064–4076. [Google Scholar]
- Dunlap, C.A.; Bowman, M.J.; Schisler, D.A. Genomic analysis and secondary metabolite production in Bacillus amyloliquefaciens AS 43.3: A biocontrol antagonist of Fusarium head blight. Biol. Control 2013, 64, 166–175. [Google Scholar] [CrossRef]
- Zong, G.L.; Zhong, C.Q.; Fu, J.; Zhao, Z.L.; Cao, G.X. Complete Genome Sequence of the High-Natamycin-Producing Strain Streptomyces gilvosporeus F607. Genome Announc. 2018, 6, e01402–e01417. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Huang, T.X.; Xu, Y.K.; Zhou, G.P.; Zou, P.; Zeng, G.F.; Liu, X.J. Genetic and genomic diversity of NheABC locus from Bacillus strains. Arch. Microbiol. 2017, 199, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Aliyu, H.; Gorte, O.; De Maayer, P.; Neumann, A.; Ochsenreither, K. Genomic insights into the lifestyles, functional capacities and oleagenicity of members of the fungal family Trichosporonaceae. Sci. Rep. 2020, 10, 2780. [Google Scholar] [CrossRef]
- Wu, B.; Chen, X.; Yu, M.J.; Ren, J.F.; Hu, J.; Shao, C.W.; Zhou, L.Q.; Sun, X.J.; Yu, T.; Zheng, Y.X.; et al. Chromosome-level genome and population genomic analysis provide insights into the evolution and environmental adaptation of Jinjiang oyster Crassostrea ariakensis. Mol. Ecol. Resour. 2021, 22, 1529–1544. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.C.; Allenby, N.E. Genome mining for the search and discovery of bioactive compounds: The Streptomyces paradigm. FEMS Microbiol. Lett. 2018, 365, fny240. [Google Scholar] [CrossRef]
- Sinha, R.K.; Krishnan, K.P.; Kurian, P.J. Complete genome sequence and comparative genome analysis of Alcanivorax sp. IO_7, a marine alkane-degrading bacterium isolated from hydrothermally-influenced deep seawater of southwest Indian ridge—ScienceDirect. Genomics 2021, 113, 884–891. [Google Scholar] [CrossRef]
- Walker, A.S.; Gautier, A.; Confais, J.; Martinho, D.; Viaud, M.; Le, P.; Cheur, P.; Dupont, J.; Fournier, E. Botrytis pseudocinerea, a new cryptic species causing gray mold in french vineyards in sympatry with Botrytis cinerea. Phytopathology 2011, 101, 1433–1445. [Google Scholar] [CrossRef]
- Tian, H.P.; Li, F.J.; Lu, G.C.; Li, C.H. Green Prevention and Control Technology against Main Diseases and Insect Pests of Facility Tomato. Plant Dis. Pests 2021, 12, 28–29. [Google Scholar]
- Junior, O.J.C.; Youssef, K.; Koyama, R.; Ahmed, S.; Dominguez, A.R.; Mühlbeier, D.T.; Roberto, S.R. Control of Gray Mold on Clamshell-Packaged ‘Benitaka’ Table Grapes Using Sulphur Dioxide Pads and Perforated Liners. Pathogens 2019, 8, 271. [Google Scholar] [CrossRef]
- Wang, M.Q.; Hao, X.J.; Yao, Y.P.; Xu, M. Study on colonization ability in plants of Endophytic Bacillus amyloliquefaciens Ht-q6 isolated from Walnut. Fresenius Environ. Bull. 2018, 27, 1152–1156. [Google Scholar]
- Wang, M.Q.; Hao, X.J.; Yao, Y.P.; Xu, M. Mutation breeding of bacillus amyloliquefaciens HT-Q6. Fresenius Environ. Bull. 2018, 27, 4337–4342. [Google Scholar]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Oh, H.S.; Park, S.C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64 Pt 2, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Y.; Cui, H.H.; Liu, J.W.; Wu, Y.Q.; Cheng, Y.; Xu, H.X.; Huang, X.X.; Li, S.T.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Kim, S.H.; Park, I.H.; Chung, S.Y.; Chandra, M.S.; Choi, Y.L. Isolation, purification, and characterization of novel fengycin S from Bacillus amyloliquefaciens LSC04 degrading-crude oil. Biotechnol. Bioprocess Eng. 2010, 15, 246–253. [Google Scholar]
- Qi, G.F.; Zhu, F.Y.; Du, P.; Yang, X.F.; Qiu, D.W.; Yu, Z.N.; Chen, J.Y.; Zhao, X.Y. Lipopeptide induces apoptosis in fungal cells by a mitochondria-dependent pathway. Peptides 2010, 31, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.P.; Hu, B.X.; Wei, W.; Xiong, Y.; Zhu, W.J.; Peng, F.; Yu, Y.; Zheng, Y.L.; Chen, P. De Novo Analysis of Wolfiporia cocos Transcriptome to Reveal the Differentially Expressed Carbohydrate-Active Enzymes (CAZymes) Genes During the Early Stage of Sclerotial Growth. Front. Microbiol. 2016, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Geiser, E.; Reindl, M.; Blank, L.M.; Feldbrügge, M.; Wierckx, N.; Schipper, K. Activating Intrinsic Carbohydrate-Active Enzymes of the Smut Fungus Ustilago maydis for the Degradation of Plant Cell Wall Components. Appl. Environ. Microbiol. 2016, 82, 5174–5185. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Bhattacharjee, A. Molecular profiling of microbial community structure and their CAZymes via metagenomics, from Tsomgo lake in the Eastern Himalayas. Arch. Microbiol. 2021, 203, 3135–3146. [Google Scholar] [CrossRef]
- Holder-Franklin, M.A.; Thorpe, A.; Cormier, C.J. Comparison of numerical taxonomy and DNA–DNA hybridization in diurnal studies of river bacteria. Can. J. Microbiol. 2011, 27, 1165–1184. [Google Scholar] [CrossRef]
- Borriss, R.; Chen, X.H.; Rueckert, C.; Blom, J.; Becker, A.; Baumgarth, B.; Fan, B.; Pukall, R.; Schumann, P.; Spröer, C.; et al. Relationship of Bacillus amyloliquefaciens clades associated with strains DSM 7T and FZB42T: A proposal for Bacillus amyloliquefaciens subsp. amyloliquefaciens subsp. nov. and Bacillus amyloliquefaciens subsp. plantarum subsp. nov. based on complete genome sequence comparisons. Int. J. Syst. Evol. Microbiol. 2011, 61 Pt 8, 1786–1801. [Google Scholar]
- Zweerink, M.M.; Edison, A. Difficidin and oxydifficidin: Novel broad spectrum antibacterial antibiotics produced by Bacillus subtilis. III. Mode of action of difficidin. J. Antibiot. 1988, 40, 1692–1697. [Google Scholar] [CrossRef]
- Romero Tabarezm, M.; Jansenr, R.; Syllam, M.; Lunsdorf, H.; Haubler, S.; Santosa, D.A.; Timmis, K.N.; Molinari, G. 7-O-Malonyl Macrolactin A, a New Macrolactin Antibiotic from Bacillus subtilis Active against Methicillin-Resistant Staphylococcus aureus, Vancomycin-Resistant Enterococci, and a Small-Colony Variant of Burkholderia cepacia. Antimicrob. Agents Chemother. 2006, 50, 1701. [Google Scholar] [CrossRef]
- Chen, X.H.; Koumoutsi, A.; Scholz, R.; Eisenreich, A.; Schneider, K.; Heinemeyer, I.; Morgenstern, B.; Voss, B.; Hess, W.R.; Reva, O.; et al. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat. Biotechnol. 2007, 25, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.S.; Zheng, C.J.; Lee, S.; Kwak, J.H.; Kim, W.G. Macrolactin N, a New Peptide Deformylase Inhibitor Produced by Bacillus subtilis. Bioorganic Med. Chem. Lett. 2006, 16, 4889–4892. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.A. The Inhibiting Effect of Streptococcus Lactis on Lactobacillus Bulgaricus. J. Bacteriol. 1928, 16, 321–325. [Google Scholar] [CrossRef]
- Wiedemann, I.; Breukink, E.; Van Kraaij, C.; Kuipers, O.P.; Bierbaum, G.; de Kruijff, B.; Sahl, H.G. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 2001, 276, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Sussman, I.; Erecińska, M.; Wilson, D.F. Regulation of cellular energy metabolism. Am. J. Physiol. 1978, 591, 209–223. [Google Scholar]
- Fonseca, G.G.; Carvalho NM, B.D.; Gombert, A.K. Growth of the yeast Kluyveromyces marxianus CBS 6556 on different sugar combinations as sole carbon and energy source. Appl. Microbiol. Biotechnol. 2013, 97, 5055–5067. [Google Scholar] [CrossRef] [PubMed]
- Ming, Y.; Wei, Q.L.; Jin, K.; Xia, Y.X. MaSnf1, a sucrose non-fermenting protein kinase gene, is involved in carbon source utilization, stress tolerance, and virulence in Metarhizium acridum. Appl. Microbiol. Biotechnol. 2014, 98, 10153–10164. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.Z.; Lv, W.W.; Zhang, Q.; Zhou, C.Y. Coding the α-subunit of SNF1 kinase, Snf1 is required for the conidiogenesis and pathogenicity of the Alternaria alternata tangerine pathotype. Fungal Biol. 2020, 124, 562–570. [Google Scholar] [CrossRef]
- Jain, S.; Vaishnav, A.; Kumari, S.; Varma, A.; Tuteja, N.; Choudhary, D.K. Chitinolytic Bacillus-Mediated Induction of Jasmonic Acid and Defense-Related Proteins in Soybean (Glycine max L. Merrill) Plant Against Rhizoctonia solani and Fusarium oxysporum. J. Plant Growth Regul. 2016, 36, 200–214. [Google Scholar] [CrossRef]
- Suchodolski, J.; Derkacz, D.; Muraszko, J.; Panek, J.J.; Jezierska, A.; Łukaszewicz, M.; Krasowska, A. Fluconazole and Lipopeptide Surfactin Interplay During Candida albicans Plasma Membrane and Cell Wall Remodeling Increases Fungal Immune System Exposure. Pharmaceutics 2020, 12, 314. [Google Scholar] [CrossRef]
- Hubballi, M.; Sornakili, A.; Nakkeeran, S.; Anand, T.; Raguchander, T. Virulence of Alternaria Alternata Infecting Noni Associated with Production of Cell Wall Degrading Enzymes. J. Plant Prot. Res. 2011, 51, 87–92. [Google Scholar] [CrossRef]
- Meyer, V.; Damveld, R.A.; Arentshorst, M.; Stahl, U.; van den Hondel, C.A.; Ram, A.F. Survival in the presence of antifungals: Genome-wide expression profiling of Aspergillus niger in response to sublethal concentrations of caspofungin and fenpropimorph. J. Biol. Chem. 2007, 282, 32935–32948. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Wang, R.; Liang, X.; Gai, Y.; Jiao, C.; Wang, M. Characterization of a Bacillus velezensis with Antibacterial Activity and Its Inhibitory Effect on Gray Mold Germ. Agronomy 2023, 13, 1553. https://doi.org/10.3390/agronomy13061553
Li L, Wang R, Liang X, Gai Y, Jiao C, Wang M. Characterization of a Bacillus velezensis with Antibacterial Activity and Its Inhibitory Effect on Gray Mold Germ. Agronomy. 2023; 13(6):1553. https://doi.org/10.3390/agronomy13061553
Chicago/Turabian StyleLi, Lei, Rongjie Wang, Xingxing Liang, Yunpeng Gai, Chen Jiao, and Meiqin Wang. 2023. "Characterization of a Bacillus velezensis with Antibacterial Activity and Its Inhibitory Effect on Gray Mold Germ" Agronomy 13, no. 6: 1553. https://doi.org/10.3390/agronomy13061553
APA StyleLi, L., Wang, R., Liang, X., Gai, Y., Jiao, C., & Wang, M. (2023). Characterization of a Bacillus velezensis with Antibacterial Activity and Its Inhibitory Effect on Gray Mold Germ. Agronomy, 13(6), 1553. https://doi.org/10.3390/agronomy13061553