


Using Isoform Sequencing for De Novo Transcriptome Sequencing and the Identification of Genes Related to Drought Tolerance and Agronomic Traits in Tall Fescue (Festuca arundinacea Schreb.)

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Sampling and RNA Isolation
2.2. cDNA Synthesis and PacBio Iso-Seq
2.3. Iso-Seq Data Processing
2.4. Coding Sequences (CDS) Detection and Functional Annotation
2.5. Simple Sequence Repeat (SSR) and lncRNA Analysis
2.6. Identification of AS Isoforms
2.7. Drought Treatment, TIS108 Treatment, and Illumina Sequencing
2.8. Identification and Validation of Differentially Expressed Isoforms
2.9. Statistical Analysis
3. Results
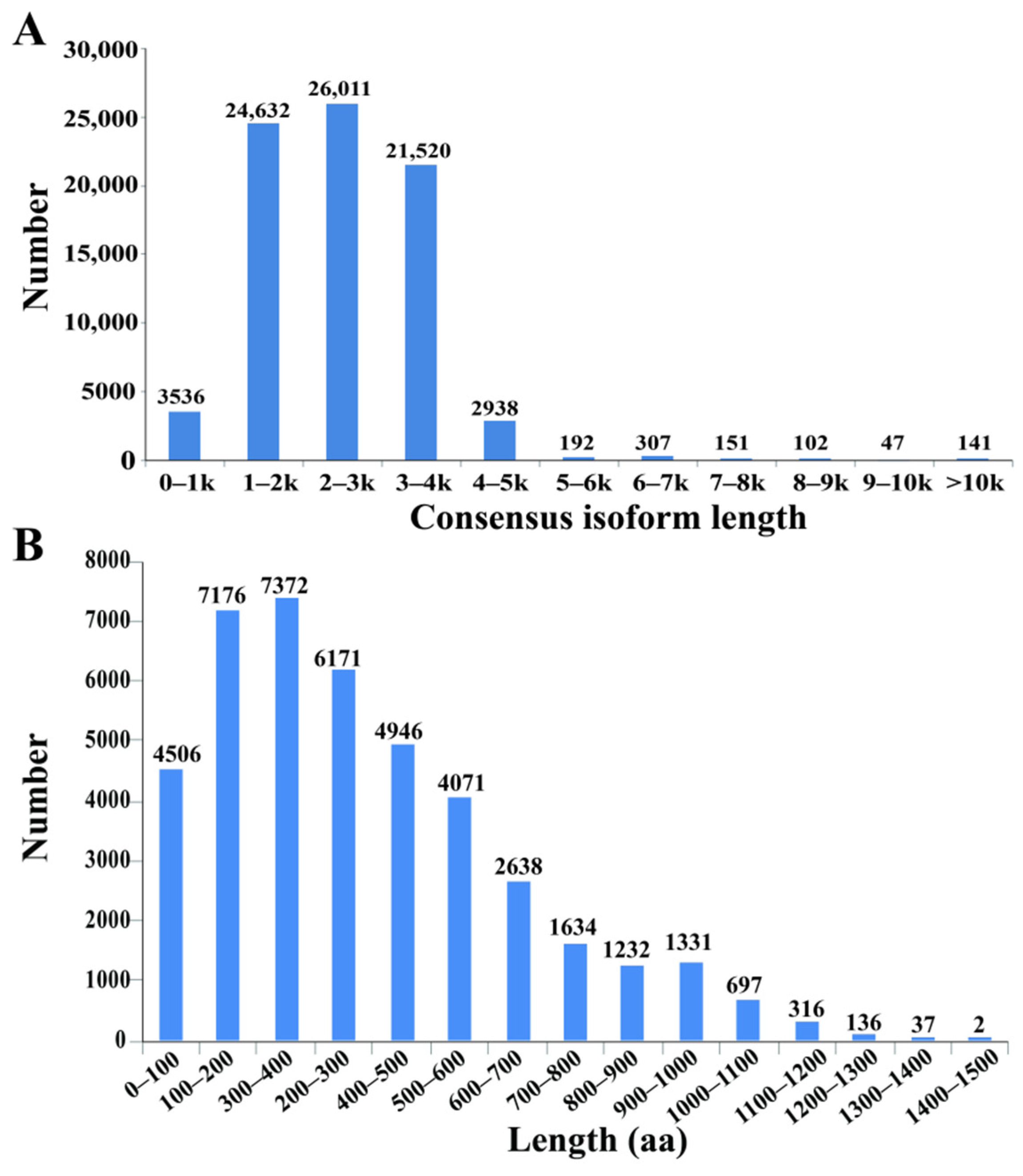
3.1. Transcriptome Sequencing of Tall Fescue Using Iso-Seq
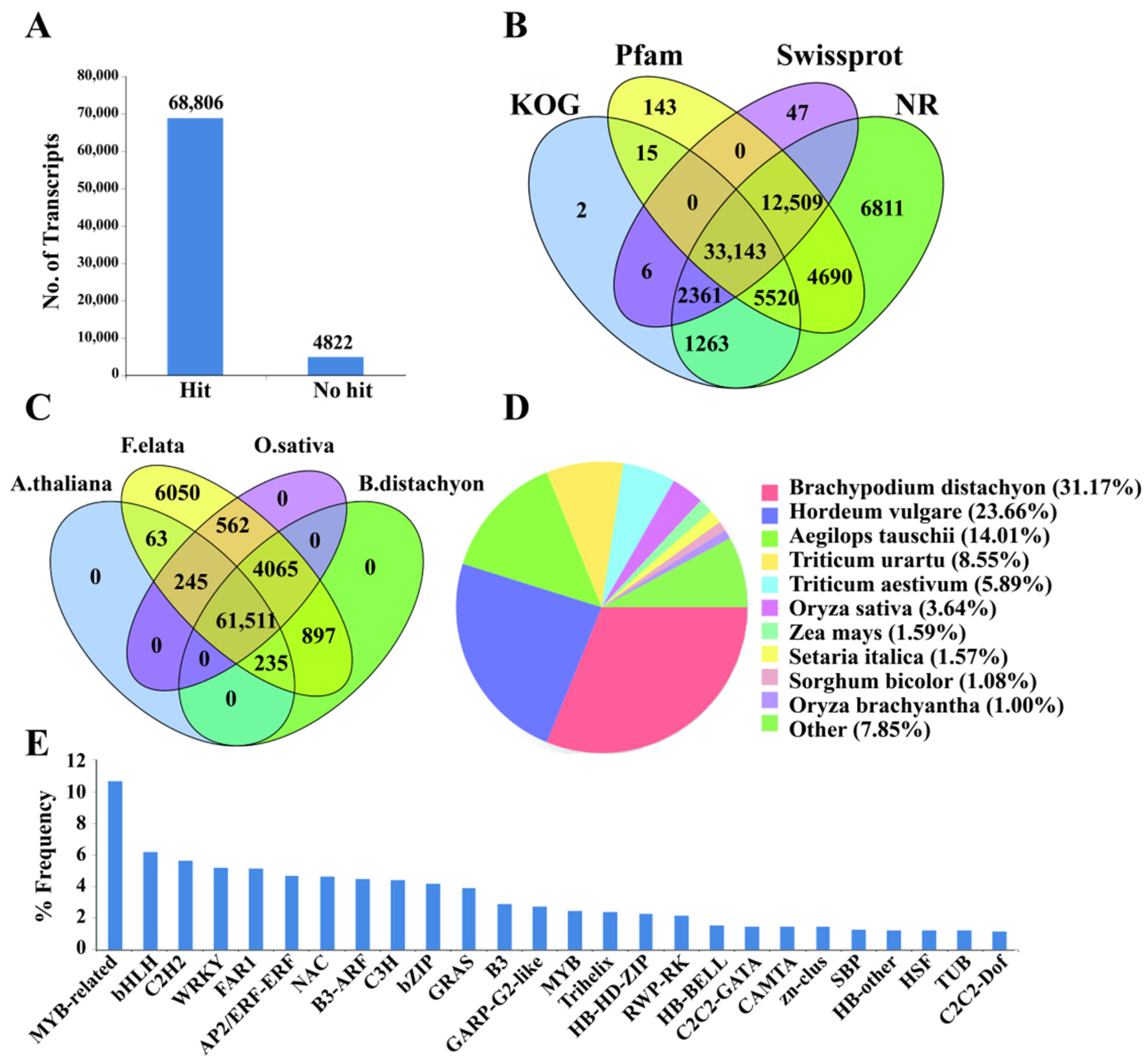
3.2. Functional Annotation and Categorization of the Tall Fescue Transcriptome
3.3. Identification of SSRs, lncRNA, and AS Isoform in the Tall Fescue Transcriptome
3.4. Identification and Analysis of Unigenes Related to Drought Response
3.5. Identification and Functional Analysis of Unigenes Involved in Control of Plant Height and Tillering
4. Discussion
4.1. Iso-Seq Provides a More Comprehensive View of the Tall Fescue Transcriptome
4.2. Unigenes in ABA Pathways under Drought Stress
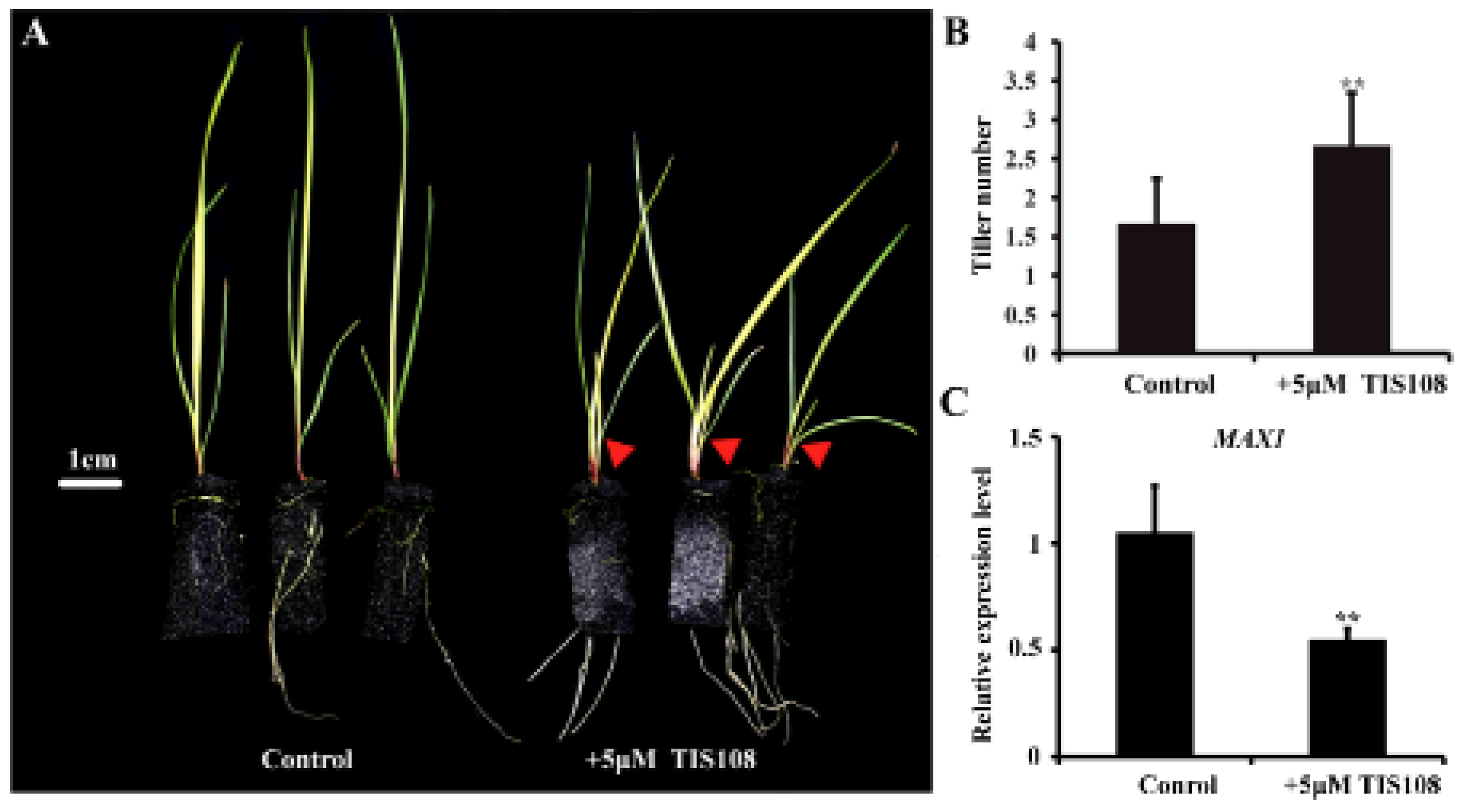
4.3. Unigenes Involved in Regulation of Plant Tillering
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chakrabarti, M.; Nagabhyru, P.; Schardl, C.L.; Dinkins, R.D. Differential gene expression in tall fescue tissues in response to water deficit. Plant Genome 2022, 15, e20199. [Google Scholar] [CrossRef] [PubMed]
- Nagabhyru, P.; Dinkins, R.D.; Schardl, C.L. Transcriptome analysis of Epichloë strains in tall fescue in response to drought stress. Mycologia 2022, 114, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, Y.Y.; Cho, K.S. Effect of Novosphingobium sp. CuT1 inoculation on the rhizoremediation of heavy metal- and diesel-contaminated soil planted with tall fescue. Environ. Sci. Pollut. Res. Int. 2023, 30, 16612–16625. [Google Scholar] [CrossRef]
- Soleimani, M.; Hajabbasi, M.A.; Afyuni, M.; Mirlohi, A.; Borggaard, O.K.; Holm, P.E. Effect of endophytic fungi on cadmium tolerance and bioaccumulation by Festuca arundinacea and Festuca Pratensis. Int. J. Phytoremed. 2010, 12, 535–549. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Z.Z. Phytoremediation of soil co-contaminated with heavy metals and deca-BDE by co-planting of Sedum alfredii with tall fescue associated with Bacillus cereus JP12. Plant Soil 2014, 382, 89–102. [Google Scholar] [CrossRef]
- Dinkins, R.D.; Nagabhyru, P.; Graham, M.A.; Boykin, D.; Schardl, C.L. Transcriptome response of Lolium arundinaceum to its fungal endophyte Epichloë coenophiala. New Phytol. 2017, 213, 324–337. [Google Scholar] [CrossRef]
- Huang, M.; Zhu, H.; Zhang, J.; Tang, D.; Han, X.; Chen, L.; Du, D.; Yao, J.; Chen, K.; Sun, J. Toxic effects of cadmium on tall fescue and different responses of the photosynthetic activities in the photosystem electron donor and acceptor sides. Sci. Rep. 2017, 7, 14387. [Google Scholar] [CrossRef]
- Komatsu, K.; Takezawa, D.; Sakata, Y. Decoding ABA and osmostress signalling in plants from an evolutionary point of view. Plant Cell Environ. 2020, 43, 2894–2911. [Google Scholar] [CrossRef]
- Brookbank, B.P.; Patel, J.; Gazzarrini, S.; Nambara, E. Role of basal ABA in plant growth and development. Genes 2021, 12, 1936. [Google Scholar] [CrossRef]
- Schwachtje, J.; Minchin, P.E.; Jahnke, S.; van Dongen, J.T.; Schittko, U.; Baldwin, I.T. SNF1-related kinases allow plants to tolerate herbivory by allocating carbon to roots. Proc. Natl. Acad. Sci. USA 2006, 103, 12935–12975. [Google Scholar] [CrossRef]
- Baena-González, E.; Rolland, F.; Thevelein, J.M.; Sheen, J. A central integrator of transcription networks in plant stress and energy signalling. Nature 2007, 448, 938–980. [Google Scholar] [CrossRef] [PubMed]
- Purcell, P.C.; Smith, A.M.; Halford, N.G. Antisense expression of a sucrose nonfermenting-1-related protein kinase sequence in potato results in decreased expression of sucrose synthase in tubers and loss of sucrose-inducibility of sucrose synthase transcripts in leaves. Plant J. 1998, 14, 195–202. [Google Scholar] [CrossRef]
- Kanegae, H.; Miyoshi, K.; Hirose, T.; Tsuchimoto, S.; Mori, M.; Nagato, Y.; Takano, M. Expressions of rice sucrose non-fermenting-1 related protein kinase 1 genes are differently regulated during the caryopsis development. Plant Physiol. Biochem. 2005, 43, 669–748. [Google Scholar] [CrossRef] [PubMed]
- McKibbin, R.S.; Muttucumaru, N.; Paul, M.J.; Powers, S.J.; Burrell, M.M.; Coates, S.; Purcell, P.C.; Tiessen, A.; Geigenberger, P.; Halford, N.G. Production of high starch, low glucose potatoes through overexpression of the metabolic regulator, SnRK1. Plant Biotechnol. J. 2006, 4, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Fung, P.; Nishimura, N.; Jensen, D.R.; Fujii, H.; Zhao, Y.; Lumba, S.; Santiago, J.; Rodrigues, A.; Chow, T.F.; et al. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 2009, 324, 1068–1071. [Google Scholar] [CrossRef]
- Rodriguez, P.L.; Benning, G.; Grill, E. ABI2, a second protein phosphatase 2C involved in abscisic acid signal transduction in Arabidopsis. FEBS Lett. 1998, 421, 185–190. [Google Scholar] [CrossRef]
- Gosti, F.; Beaudoin, N.; Serizet, C.; Webb, A.A.; Vartanian, N.; Giraudat, J. ABI1 protein phosphatase 2C is a negative regulator of abscisic acid signaling. Plant Cell 1999, 11, 1897–1910. [Google Scholar] [CrossRef]
- Yoshida, R.; Hobo, T.; Ichimura, K.; Mizoguchi, T.; Takahashi, F.; Aronso, J.; Ecker, J.R.; Shinozaki, K. ABA-activated SnRK2 protein kinase is required for dehydration stress signaling in Arabidopsis. Plant Cell Physiol. 2002, 43, 1473–1483. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yamamoto, S.; Minami, H.; Kagaya, Y.; Hattori, T. Differential activation of the rice sucrose nonfermenting1-related protein kinase2 family by hyperosmotic stress and abscisic acid. Plant Cell 2004, 16, 1163–1177. [Google Scholar] [CrossRef]
- Yoshida, T.; Fujita, Y.; Maruyama, K.; Mogami, J.; Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Four Arabidopsis AREB/ABF transcription factors function predominantly in gene expression downstream of SnRK2 kinases in abscisic acid signalling in response to osmotic stress. Plant Cell Environ. 2015, 38, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Murata, M.; Minami, H.; Yamamoto, S.; Kagaya, Y.; Hobo, T. Abscisic acid-activated SNRK2 protein kinases function in the gene regulation pathway of ABA signal transduction by phosphorylating ABA response element-binding factors. Plant J. 2005, 44, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Seal, A.G. DNA variation in Festuca. Heredity 1983, 50, 225–236. [Google Scholar] [CrossRef]
- Sleper, D.A. Breeding tall fescue. Plant Breed. Rev. 1985, 3, 313–342. [Google Scholar] [CrossRef]
- Mian, M.R.; Zhang, Y.; Wang, Z.Y.; Zhang, J.Y.; Cheng, X.; Chen, L.; Chekhovskiy, K.; Dai, X.B.; Mao, C.H.; Cheung, F.; et al. Analysis of tall fescue ESTs representing different abiotic stresses, tissue types and developmental stages. BMC Plant Biol. 2008, 8, 27. [Google Scholar] [CrossRef]
- Hu, T.; Sun, X.; Zhang, X.; Nevo, E.; Fu, J. An RNA sequencing transcriptome analysis of the high-temperature stressed tall fescue reveals novel insights into plant thermotolerance. BMC Genom. 2014, 15, 1147. [Google Scholar] [CrossRef]
- Wang, Y.; Dai, Y.; Tao, X.; Wang, J.Z.; Cheng, H.Y.; Yang, H.; Ma, X.R. Heat shock factor genes of tall fescue and perennial ryegrass in response to temperature stress by RNA-Seq analysis. Front. Plant Sci. 2015, 6, 1226. [Google Scholar] [CrossRef]
- Lou, Y.; Zhao, P.; Wang, D.; Amombo, E.; Sun, X.; Wang, H.; Zhuge, Y. Germination, physiological responses and gene expression of tall fescue (Festuca arundinacea Schreb.) growing under Pb and Cd. PLoS ONE 2017, 12, e0169495. [Google Scholar] [CrossRef]
- Tao, X.; Wang, M.X.; Dai, Y.; Wang, Y.; Fan, Y.F.; Mao, P.; Ma, X.R. Identification and expression profile of CYPome in perennial ryegrass and tall fescue in response to temperature stress. Front. Plant Sci. 2017, 8, 1519. [Google Scholar] [CrossRef]
- Li, H.; Hu, T.; Amombo, E.; Fu, J. Transcriptome profilings of two tall fescue (Festuca arundinacea) cultivars in response to lead (Pb) stress. BMC Genom. 2017, 18, e145. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Liu, H.; Zhang, J.; Yang, S.; Kong, G.; Chu, J.S.C.; Chen, N.; Wang, D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom. 2015, 16, 1039. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jin, C.; Gong, S.; Wang, X.; Wu, T. RNA-Seq and Iso-Seq Reveal the Important Role of COMT and CCoAOMT Genes in Accumulation of Scopoletin in Noni (Morinda citrifolia). Genes 2022, 13, 1993. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.P.; Lu, Z.Y.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef]
- Jo, I.H.; Lee, J.; Hong, C.E.; Lee, D.J.; Bae, W.; Park, S.G.; Ahn, Y.J.; Kim, Y.C.; Lee, J.W.; Hyun, D.Y.; et al. Isoform sequencing provides a more comprehensive view of the Panax ginseng transcriptome. Genes 2017, 8, 228. [Google Scholar] [CrossRef]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, e17031. [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Blanco, E.; Parra, G.; Guigó, R. Using geneID to identify genes. Curr. Protoc. Bioinform. 2007, 18. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.P.; Lee, S.J.; Park, J.Y.; Plaha, P.; Park, Y.S.; Lee, Y.K.; Choi, J.E.; Kim, K.Y.; Lee, J.H.; Lee, J.; et al. Construction of a BAC library of Korean ginseng and initial analysis of BAC-end sequences. Mol. Genet. Genom. 2004, 271, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Finder, S.S.R. Available online: https://www.gramene.org/pub/gramene/archives/software/scripts/ssr.pl (accessed on 3 July 2017).
- Ito, S.; Umehara, S.; Hanada, A.; Yamaguchi, S.; Asami, T. Effects of strigolactone-biosynthesis inhibitor TIS108 on Arabidopsis. Plant Signal. Behav. 2013, 8, e24193. [Google Scholar] [CrossRef]
- Iuchi, S.; Kobayashi, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K. A stress-inducible gene for 9-cis-epoxycarotenoid dioxygenase involved in abscisic acid biosynthesis under water stress in drought-tolerant cowpea. Plant Physiol. 2000, 123, 553–562. [Google Scholar] [CrossRef]
- Schwartz, S.H.; Qin, X.; Zeevaart, J.A. Elucidation of the indirect pathway of abscisic acid biosynthesis by mutants, genes, and enzymes. Plant Physiol. 2003, 131, 1591–1601. [Google Scholar] [CrossRef]
- Saika, H.; Okamoto, M.; Miyoshi, K.; Kushiro, T.; Shinoda, S.; Jikumaru, Y.; Fujimoto, M.; Arikawa, T.; Takahashi, H.; Ando, M. Ethylene promotes submergence-induced expression of OsABA8ox1, a gene that encodes ABA 8’-hydroxylase in rice. Plant Cell Physiol. 2007, 48, 287–298. [Google Scholar] [CrossRef]
- Zhu, G.; Ye, N.; Zhang, J. Glucose-induced delay of seed germination in rice is mediated by the suppression of ABA catabolism rather than an enhancement of ABA biosynthesis. Plant Cell Physiol. 2009, 50, 644–651. [Google Scholar] [CrossRef]
- Ye, N.; Zhu, G.; Liu, Y.; Li, Y.; Zhang, J. ABA Controls H2O2 accumulation through the induction of OsCATB in rice leaves under water stress. Plant Cell Physiol. 2011, 52, 689–698. [Google Scholar] [CrossRef]
- Sun, H.; Li, W.; Burritt, D.J.; Tian, H.; Zhang, H.; Liang, X.; Miao, Y.; Mostofa, M.G.; Tran, L.S.P. Strigolactones interact with other phytohormones to modulate plant root growth and development. Crop J. 2022, 10, 1517–1527. [Google Scholar] [CrossRef]
- Umehara, M.; Hanada, A.; Yoshida, S.; Akiyama, K.; Arite, T.; Takeda-Kamiya, N.; Magome, H.; Kamiya, Y.; Shirasu, K.; Yoneyama, K.; et al. Inhibition of shoot branching by new terpenoid plant hormones. Nature 2008, 455, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roldan, V.; Fermas, S.; Brewer, P.B.; Puech-Pagès, V.; Dun, E.A.; Pillot, J.P.; Letisse, F.; Matusova, R.; Danoun, S.; Portais, J.C.; et al. Strigolactone inhibition of shoot branching. Nature 2008, 455, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Colasuonno, P.; Lozito, M.L.; Marcotuli, I.; Nigro, D.; Giancaspro, A.; Mangini, G.; De Vita, P.; Mastrangelo, A.M.; Pecchioni, N.; Houston, K.; et al. The carotenoid biosynthetic and catabolic genes in wheat and their association with yellow pigments. BMC Genom. 2017, 18, 122. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, R.; Qian, Q.; Yan, M.; Meng, X.; Fu, Z.; Yan, C.; Jiang, B.; Su, Z.; Li, J.; et al. DWARF27, an iron-containing protein required for the biosynthesis of strigolactones, regulates rice tiller bud outgrowth. Plant Cell 2009, 21, 1512–1525. [Google Scholar] [CrossRef]
- Booker, J.; Auldridge, M.; Wills, S.; McCarty, D.; Klee, H.; Leyser, O. MAX3/CCD7 is a carotenoid cleavage dioxygenase required for the synthesis of a novel plant signaling molecule. Curr. Biol. 2004, 14, 1232–1238. [Google Scholar] [CrossRef]
- Drummond, R.S.; Martínez-Sánchez, N.M.; Janssen, B.J.; Templeton, K.R.; Simons, J.L.; Quinn, B.D.; Karunairetnam, S.; Snowden, K.C. Petunia hybrida CAROTENOID CLEAVAGE DIOXYGENASE7 is involved in the production of negative and positive branching signals in petunia. Plant Physiol. 2009, 151, 1867–1877. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Zhong, L.; Ou, E.; Tian, F.; Yao, M.; Chen, M.; Yan, X.; Li, Y.; Li, X.; He, R.; et al. Using Isoform Sequencing for De Novo Transcriptome Sequencing and the Identification of Genes Related to Drought Tolerance and Agronomic Traits in Tall Fescue (Festuca arundinacea Schreb.). Agronomy 2023, 13, 1484. https://doi.org/10.3390/agronomy13061484
Yang C, Zhong L, Ou E, Tian F, Yao M, Chen M, Yan X, Li Y, Li X, He R, et al. Using Isoform Sequencing for De Novo Transcriptome Sequencing and the Identification of Genes Related to Drought Tolerance and Agronomic Traits in Tall Fescue (Festuca arundinacea Schreb.). Agronomy. 2023; 13(6):1484. https://doi.org/10.3390/agronomy13061484
Chicago/Turabian StyleYang, Chunyan, Li Zhong, Erling Ou, Fang Tian, Mei Yao, Ming Chen, Xu Yan, Yingzheng Li, Xiaofeng Li, Ruyu He, and et al. 2023. "Using Isoform Sequencing for De Novo Transcriptome Sequencing and the Identification of Genes Related to Drought Tolerance and Agronomic Traits in Tall Fescue (Festuca arundinacea Schreb.)" Agronomy 13, no. 6: 1484. https://doi.org/10.3390/agronomy13061484
APA StyleYang, C., Zhong, L., Ou, E., Tian, F., Yao, M., Chen, M., Yan, X., Li, Y., Li, X., He, R., He, J., Tang, Q., & Zhao, D. (2023). Using Isoform Sequencing for De Novo Transcriptome Sequencing and the Identification of Genes Related to Drought Tolerance and Agronomic Traits in Tall Fescue (Festuca arundinacea Schreb.). Agronomy, 13(6), 1484. https://doi.org/10.3390/agronomy13061484