
Transcriptomic and Metabolomic Profiling Provides Insights into Flavonoid Biosynthesis and Flower Coloring in Loropetalum chinense and Loropetalum chinense var. rubrum

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Petal Color Comparison
2.3. Transverse Section Observation
2.4. Total Anthocyanins Analysis
2.5. Flavonoid Extraction and MRM
2.6. RNA Sample Preparation
2.7. PacBio Iso-Seq Library Preparation and SMRT Sequencing
2.8. Illumina RNA-Seq Library Construction and Sequencing
2.9. Gene Functional Annotation, Coding Sequence (CDS) Prediction, Transcription Factor (TF), and Long Non-Coding RNA (lncRNA) Identification
2.10. Quantitation and Differential Expression of Transcripts and Genes Analysis
2.11. The Correlation Analysis between DETs and DCMs
3. Results
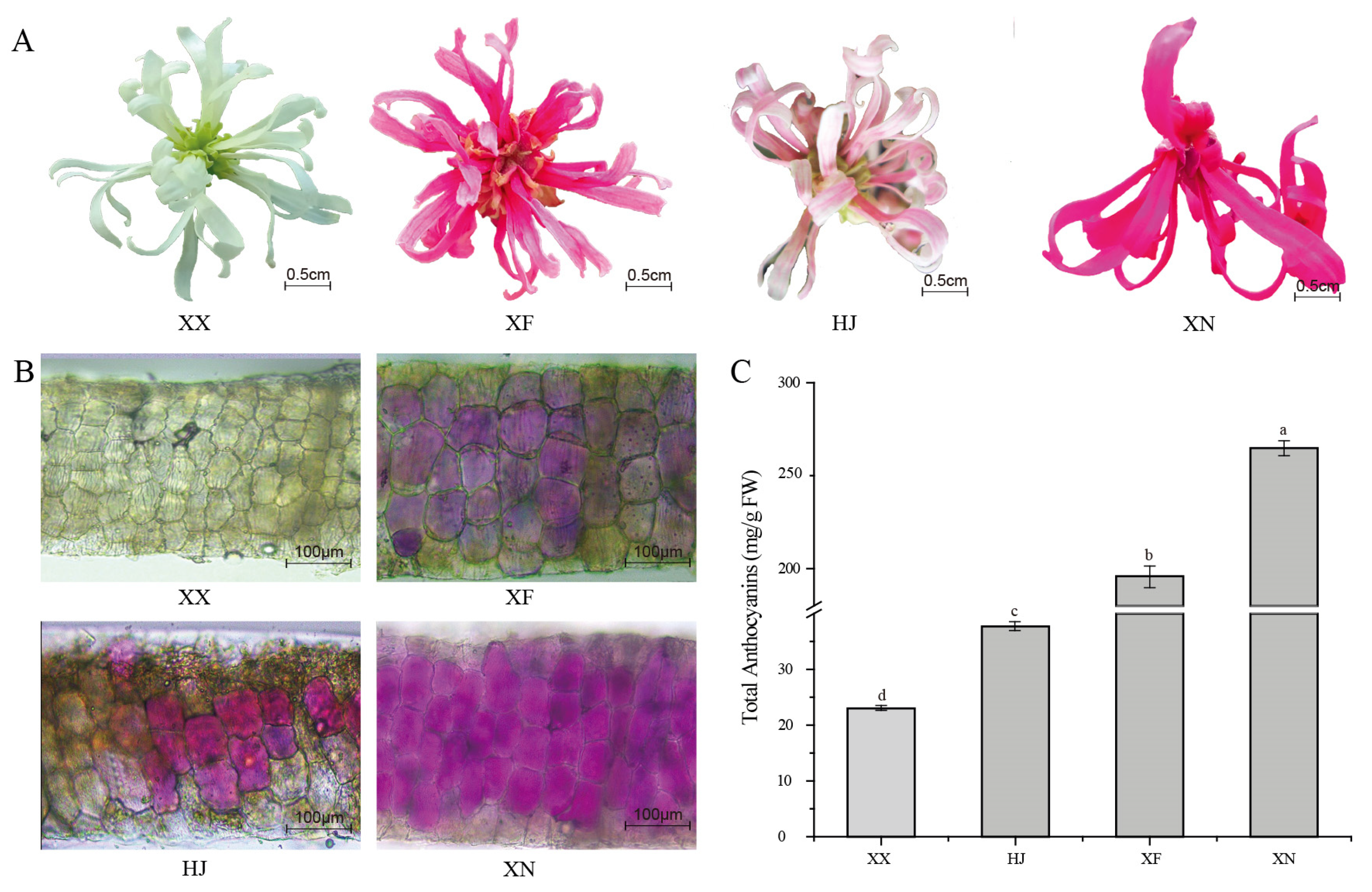
3.1. Petal Color Phenotype and Total Anthocyanins Content among the Four Loropetalum Cultivars
3.2. Identification and Qualification of Flavonoid Metabolite Profiles from the Petals of Loropetalum Cultivars
3.3. Differential Accumulation of Flavonoid Metabolites in Loropetalum Plant’s Petals
3.4. Combined Sequencing Approach to Tissues of L. chinensis var. rubrum
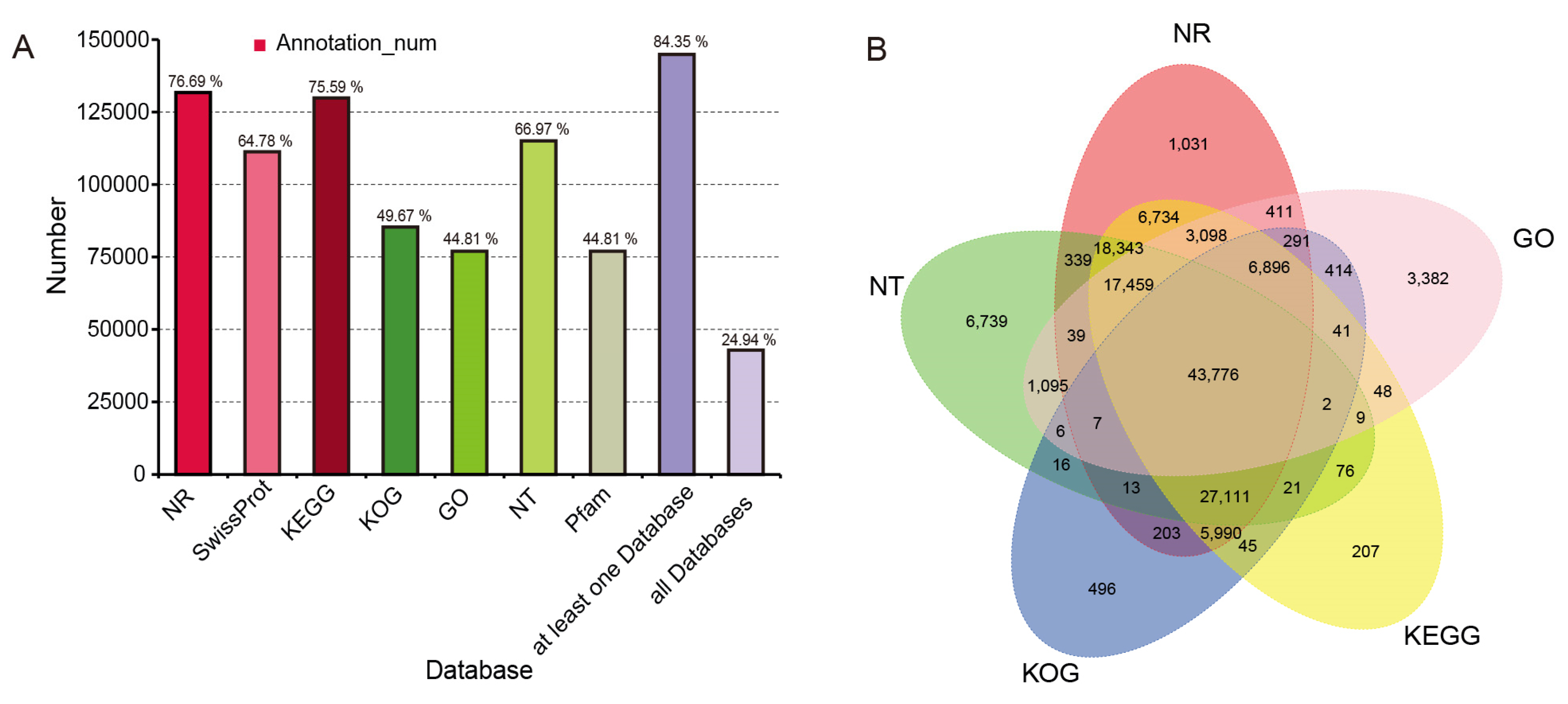
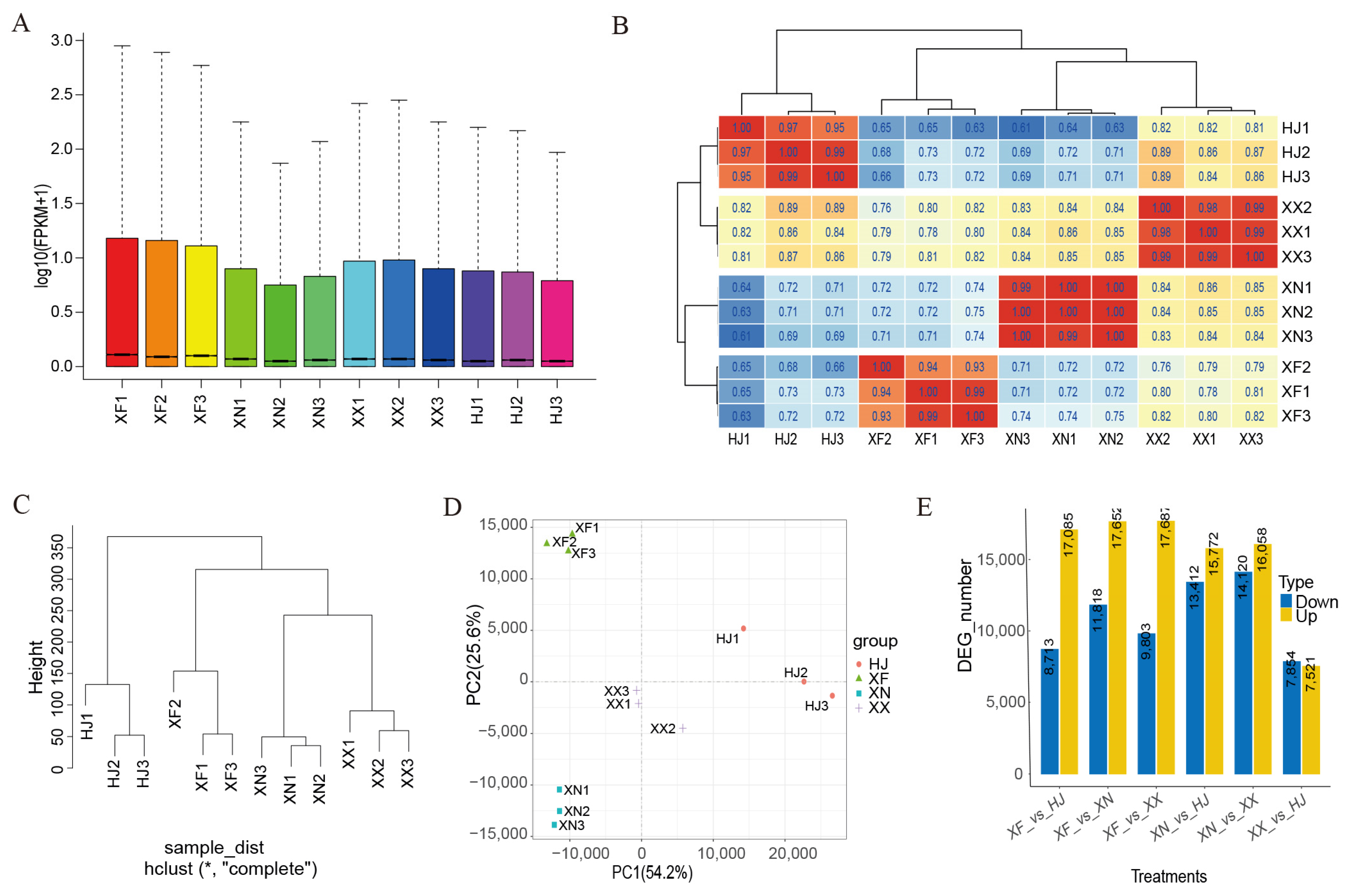
3.5. Global Transcriptomic Characteristics of Flowers during the Full-Bloom Stage
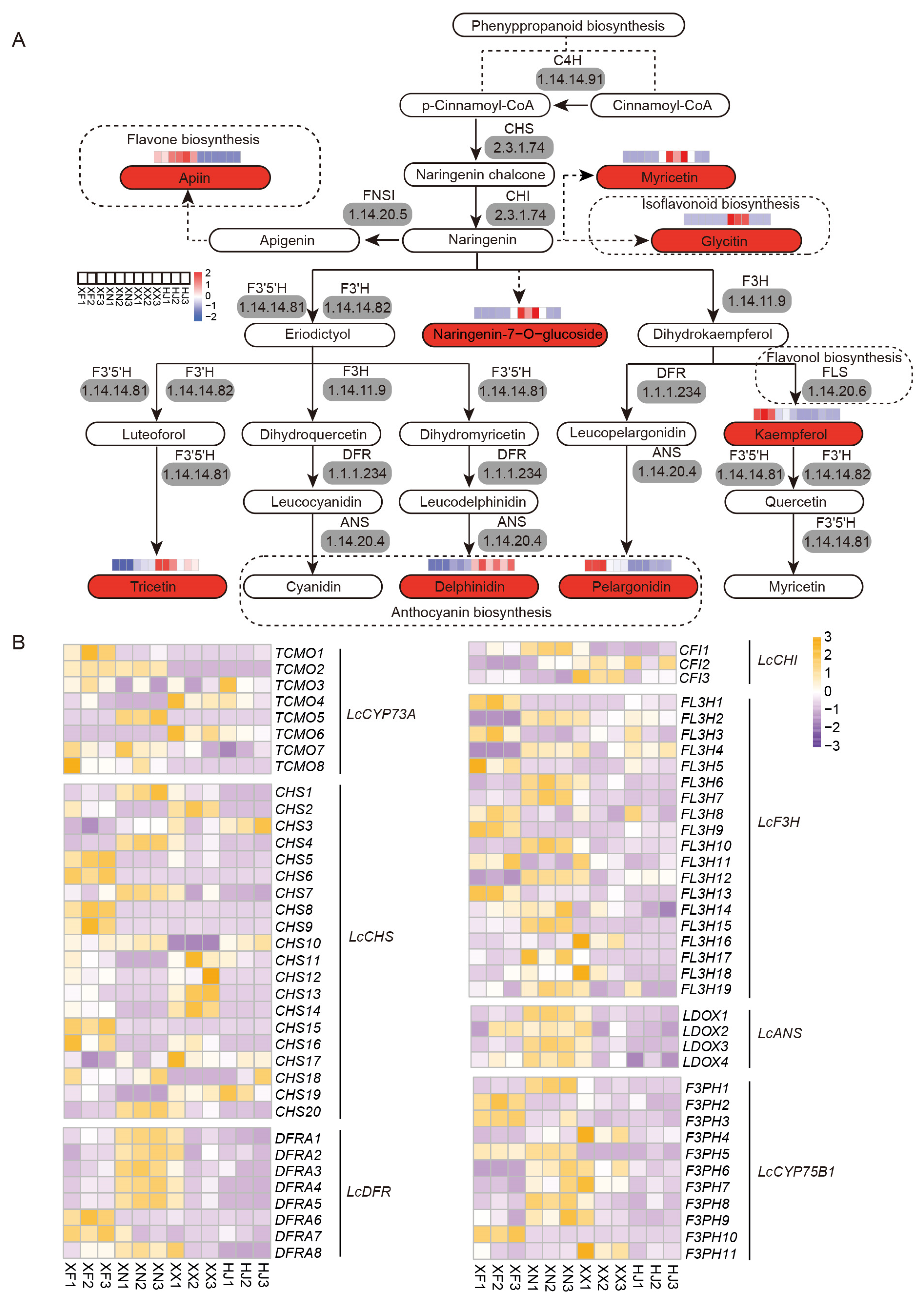
3.6. Expression Analysis Indicates Flavonoid Compounds’ Biosynthesis and Accumulation
3.7. Co-Expression Analysis for the Investigation of Anthocyanin Biosynthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Zhang, D.; Zhang, L.; Wang, X.; Xiong, X.; Gan, D.; Yu, X.; Li, Y. The Whole Genome Analysis of Loropetalum chinense var. Rubrum. Mol. Plant Breed. 2020, 18, 7023–7029. [Google Scholar]
- Shi, F. Comparison and Application of Lorpetalum chinense Var. Rubrum and Photinia Fraseryin Landscape. J. Hengyang Norm. Univ. 2016, 37, 106–111. [Google Scholar]
- Shao, Z.; Hou, W.; Long, X.; Yang, G.; Chen, C.; Zeng, X.; Hou, B.; Yu, G.; Wu, W. The formation and development of the geography symbol product Loropetalum chinense var. rubrum in Liuyang City. Hunan For. Sci. Technol. 2007, 2, 71–73. [Google Scholar]
- Zhang, D.; Cai, W.; Zhang, X.; Li, W.; Zhou, Y.; Chen, Y.; Mi, Q.; Jin, L.; Xu, L.; Yu, X.; et al. Different pruning level effects on flowering period and chlorophyll fluorescence parameters of Loropetalum chinense var. rubrum. PeerJ 2022, 10, 1–17. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, X.; Zhang, X.; Zhang, D.; Li, W.; Xiang, L.; Yang, Y.; Li, Y.; Xu, L. Phenotypic Diversity Analysis of the Progeny Variation of a ‘Mosaic Leaf’ Loropetalum chinense var. rubrum Based on Flower Organ Characteristics. Diversity 2022, 14, 913. [Google Scholar] [CrossRef]
- Chen, Q.; Cai, W.; Zhang, X.; Zhang, D.; Li, W.; Xu, L.; Yu, X.; Li, Y. The Comparative Studies on Phytochemicals of Leaf Coloration of Loropetalum chinense var. rubrum. Acta Hortic. Sin. 2021, 48, 1969–1982. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Zhang, D.; Su, D.; Li, W.; Wang, X.; Chen, Q.; Cai, W.; Xu, L.; Cao, F.; et al. Comprehensive analysis of metabolome and transcriptome reveals the mechanism of color formation in different leave of Loropetalum chinense var. rubrum. BMC Plant Biol. 2023, 23, 133. [Google Scholar] [CrossRef]
- Iwashina, T. Contribution to Flower Colors of Flavonoids Including Anthocyanins: A Review. Nat. Prod. Commun. 2015, 10, 529–544. [Google Scholar] [CrossRef]
- Tanaka, Y.; Brugliera, F. Flower Colour. In Annual Plant Reviews; Wiley: Hoboken, NJ, USA, 2018; Volume 20, pp. 201–239. [Google Scholar]
- Goto, T. Structure, stability and color variation of natural anthocyanins. In Fortschritte der Chemie Organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products; Springer: Berlin/Heidelberg, Germany, 1987; Volume 52, pp. 113–158. [Google Scholar]
- Koes, R.E.; Van Blokland, R.; Quattrocchio, F.; Van Tunen, A.J.; Mol, J.N. Chalcone synthase promoters in petunia are active in pigmented and unpigmented cell types. Plant Cell 1990, 2, 379–392. [Google Scholar] [CrossRef]
- Hůla, M.; Flegr, J. What flowers do we like? The influence of shape and color on the rating of flower beauty. PeerJ 2016, 4, e2106. [Google Scholar] [CrossRef]
- Yin, X.; Wang, T.; Zhang, M.; Zhang, Y.; Irfan, M.; Chen, L.; Zhang, L. Role of core structural genes for flavonoid biosynthesis and transcriptional factors in flower color of plants. Biotechnol. Biotechnol. Equip. 2021, 35, 1214–1229. [Google Scholar] [CrossRef]
- Tanaka, Y.; Brugliera, F.; Chandler, S. Recent progress of flower colour modification by biotechnology. Int. J. Mol. Sci. 2009, 10, 5350–5369. [Google Scholar] [CrossRef]
- Tanaka, Y.; Brugliera, F. Flower colour and cytochromes P450. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120432. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Liang, C.-Y.; Rengasamy, K.P.; Huang, L.-M.; Hsu, C.-C.; Jeng, M.-F.; Chen, W.-H.; Chen, H.-H. Assessment of violet-blue color formation in Phalaenopsis orchids. BMC Plant Biol. 2020, 20, 212. [Google Scholar] [CrossRef]
- Diretto, G.; Jin, X.; Capell, T.; Zhu, C.; Gomez-Gomez, L. Differential accumulation of pelargonidin glycosides in petals at three different developmental stages of the orange-flowered gentian (Gentiana lutea L. var. aurantiaca). PLoS ONE 2019, 14, e0212062. [Google Scholar] [CrossRef]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The flavonoid biosynthesis network in plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef]
- Haslam, E. Practical Polyphenolics: From Structure to Molecular Recognition and Physiological Action; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Winkel-Shirley, B. Molecular genetics and control of anthocyanin expression. Adv. Bot. Res. 2002, 37, 75–94. [Google Scholar]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. Arab. Book 2011, 9, e0152. [Google Scholar] [CrossRef]
- Schröder, G. Stilbene and chalcone synthases: Related enzymes with key functions in plant-specific pathways. Z. Für Nat. C 1990, 45, 1–8. [Google Scholar] [CrossRef]
- Shirley, B.W.; Kubasek, W.L.; Storz, G.; Bruggemann, E.; Koornneef, M.; Ausubel, F.M.; Goodman, H.M. Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. Plant J. 1995, 8, 659–671. [Google Scholar] [CrossRef] [PubMed]
- van Tunen, A.J.; Mur, L.A.; Recourt, K.; Gerats, A.; Mol, J. Regulation and manipulation of flavonoid gene expression in anthers of petunia: The molecular basis of the Po mutation. Plant Cell 1991, 3, 39–48. [Google Scholar] [PubMed]
- Wisman, E.; Hartmann, U.; Sagasser, M.; Baumann, E.; Palme, K.; Hahlbrock, K.; Saedler, H.; Weisshaar, B. Knock-out mutants from an En-1 mutagenized Arabidopsis thaliana population generate phenylpropanoid biosynthesis phenotypes. Proc. Natl. Acad. Sci. 1998, 95, 12432–12437. [Google Scholar] [CrossRef] [PubMed]
- Holton, T.A.; Brugliera, F.; Lester, D.R.; Tanaka, Y.; Hyland, C.D.; Menting, J.G.; Lu, C.-Y.; Farcy, E.; Stevenson, T.W.; Cornish, E.C. Cloning and expression of cytochrome P450 genes controlling flower colour. Nature 1993, 366, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Schoenbohm, C.; Martens, S.; Eder, C.; Forkmann, G.; Weisshaar, B. Identification of the Arabidopsis thaliana flavonoid 3′-hydroxylase gene and functional expression of the encoded P450 enzyme. Bio. Chem. 2000, 381, 749–753. [Google Scholar] [CrossRef]
- Shirley, B.W.; Hanley, S.; Goodman, H.M. Effects of ionizing radiation on a plant genome: Analysis of two Arabidopsis transparent testa mutations. Plant Cell 1992, 4, 333–347. [Google Scholar]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef]
- Zhang, Y.; Chu, G.; Hu, Z.; Gao, Q.; Cui, B.; Tian, S.; Wang, B.; Chen, G. Genetically engineered anthocyanin pathway for high health-promoting pigment production in eggplant. Mol. Breed. 2016, 36, 1–14. [Google Scholar] [CrossRef]
- Tohge, T.; Nishiyama, Y.; Hirai, M.Y.; Yano, M.; Nakajima, J.; Awazuhara, M.; Inoue, E.; Takahashi, H.; Goodenowe, D.B.; Kitayama, M. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 2005, 42, 218–235. [Google Scholar] [CrossRef]
- Veitch, N.C.; Grayer, R.J. Flavonoids and their glycosides, including anthocyanins. Nat. Prod. Rep. 2008, 25, 555–611. [Google Scholar] [CrossRef]
- Wei, X.; Yu, X.; Chen, J.; Fu, H.; Hu, B.; Chen, Y.; Da, L. Cloning and Sequence Analyzing of Chalcone Synthase Gene in Loropetalum chinense var. rubrum. Chin. Agric. Sci. Bull. 2013, 29, 24–28. [Google Scholar]
- Li, C.H.; Duo, D.Y.; Liao, X.S.; Zhang, Y.B. Cloning and Bioinformatic Analysis of Two Chalcone Synthases from Loropetalum chinense var. Rubrum. J. Hunan Univ. Technol. 2020, 34, 92–98. [Google Scholar]
- Rong, D.; Zhang, X.; Pan, T.; Wang, J.; Yang, G.; Zhang, B. Cloning, Expression and Transformation of LcFLS1 Gene from Loropetalum chinense var. rubrum. Acta Bot. Boreali-Occident. Sin. 2019, 28, 1877–1887. [Google Scholar]
- Zhang, B.Y.; Li, C.H.; Liu, X.; Liao, X.S.; Rong, D.Y. Cloning and subcellular localization analysis of LcDFR1 and LcDFR2 in Loropetalum chinense var. rubrum. J. South. Agric. 2020, 51, 2865–2874. [Google Scholar]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant–environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Ma, D.; Constabel, C.P. MYB repressors as regulators of phenylpropanoid metabolism in plants. Trends Plant Sci. 2019, 24, 275–289. [Google Scholar] [CrossRef]
- Ohtani, M.; Demura, T. The quest for transcriptional hubs of lignin biosynthesis: Beyond the NAC-MYB-gene regulatory network model. Curr. Opin. Biotechnol. 2019, 56, 82–87. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Šamec, D.; Tomczyk, M.; Milella, L.; Russo, D.; Habtemariam, S.; Suntar, I.; Rastrelli, L.; Daglia, M.; Xiao, J. Flavonoid biosynthetic pathways in plants: Versatile targets for metabolic engineering. Biotechnol. Adv. 2020, 38, 107316. [Google Scholar] [CrossRef]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Sunil, L.; Shetty, N.P. Biosynthesis and regulation of anthocyanin pathway genes. Appl. Microbiol. Biotechnol. 2022, 106, 1783–1798. [Google Scholar] [CrossRef] [PubMed]
- Kiferle, C.; Fantini, E.; Bassolino, L.; Povero, G.; Spelt, C.; Buti, S.; Giuliano, G.; Quattrocchio, F.; Koes, R.; Perata, P. Tomato R2R3-MYB proteins SlANT1 and SlAN2: Same protein activity, different roles. PLoS ONE 2015, 10, e0136365. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jia, H.; Xing, M.; Jin, R.; Grierson, D.; Gao, Z.; Sun, C.; Chen, K.; Xu, C.; Li, X. Genome-wide analysis of MYB gene family in Chinese bayberry (Morella rubra) and identification of members regulating flavonoid biosynthesis. Front. Plant Sci. 2021, 12, 1244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xu, X.; Huang, R.; Yang, S.; Li, M.; Guo, Y. CRISPR/Cas9-mediated targeted mutation reveals a role for AN4 rather than DPL in regulating venation formation in the corolla tube of Petunia hybrida. Hortic. Res. 2021, 8, 116. [Google Scholar] [CrossRef]
- Lai, Y.-S.; Shimoyamada, Y.; Nakayama, M.; Yamagishi, M. Pigment accumulation and transcription of LhMYB12 and anthocyanin biosynthesis genes during flower development in the Asiatic hybrid lily (Lilium spp.). Plant Sci. 2012, 193, 136–147. [Google Scholar] [CrossRef]
- Nesi, N.; Debeaujon, I.; Jond, C.; Pelletier, G.; Caboche, M.; Lepiniec, L. The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. Plant Cell 2000, 12, 1863–1878. [Google Scholar] [CrossRef]
- Qi, F.; Liu, Y.; Luo, Y.; Cui, Y.; Lu, C.; Li, H.; Huang, H.; Dai, S. Functional analysis of the ScAG and ScAGL11 MADS-box transcription factors for anthocyanin biosynthesis and bicolour pattern formation in Senecio cruentus ray florets. Hortic. Res. 2022, 9, uhac071. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Fang, Z.-Z.; Ye, X.-F.; Pan, S.-L. Identification of candidate genes involved in anthocyanin accumulation in the peel of jaboticaba (Myrciaria cauliflora) fruits by transcriptomic analysis. Gene 2018, 676, 202–213. [Google Scholar] [CrossRef]
- Dong, T.; Han, R.; Yu, J.; Zhu, M.; Li, Z. Anthocyanins Accumulation and Molecular Analysis of Correlated Genes by Metabolome and Transcriptome in Green and Purple Asparaguses (Asparagus officinalis L.). Food Chem. 2018, 271, 18–28. [Google Scholar] [CrossRef]
- Wrolstad, R.E.; Culbertson, J.D.; Cornwell, C.J.; Mattick, L.R. Detection of adulteration in blackberry juice concentrates and wines. J.-Assoc. Off. Anal. Chem. 1982, 65, 1417–1423. [Google Scholar] [CrossRef]
- Leena, S.; Eric, R. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinform. Oxf. 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–73. [Google Scholar]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef]
- Finn, R.D.; Alex, B.; Jody, C.; Penelope, C.; Eberhardt, R.Y.; Eddy, S.R.; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schffer, A.A.; Zhang, J.; Zhang, Z.; Webb, M.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 2006, 4, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Sun, H.H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.F. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Finn, R.; Coggill, P.; Eberhardt, R.; Eddy, S.; Mistry, J.; Mitchell, A.; Potter, S.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Freire-Pritchett, P.; Ray-Jones, H.; Della Rosa, M.; Eijsbouts, C.Q.; Orchard, W.R.; Wingett, S.W.; Wallace, C.; Cairns, J.; Spivakov, M.; Malysheva, V. Detecting chromosomal interactions in Capture Hi-C data with CHiCAGO and companion tools. Nat. Protoc. 2021, 16, 4144–4176. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Qiu, X.H.; Zhang, Y.J.; Yin, G.F.; Shi, C.Y.; Yu, X.Y.; Zhao, N.J.; Liu, W.Q. Photosynthetic Parameters Inversion Algorithm Study Based on Chlorophyll Fluorescence Induction Kinetics Curve. Guang Pu Xue Yu Guang Pu Fen Xi = Guang Pu 2015, 35, 2194–2197. [Google Scholar]
- Spinardi, A.; Cola, G.; Gardana, C.S.; Mignani, I. Variation of Anthocyanin Content and Profile Throughout Fruit Development and Ripening of Highbush Blueberry Cultivars Grown at Two Different Altitudes. Front. Plant Sci. 2019, 10, 1045. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Shi, T.; Chen, M.; Wang, L. Integrating Transcriptomic and GC-MS Metabolomic Analysis to Characterize Color and Aroma Formation during Tepal Development in Lycoris longituba. Plants 2019, 8, 53. [Google Scholar] [CrossRef]
- Chen, S.M.; Li, C.H.; Zhu, X.R.; Deng, Y.M.; Sun, W.; Wang, L.S.; Chen, F.D.; Zhang, Z. The identification of flavonoids and the expression of genes of anthocyanin biosynthesis in the chrysanthemum flowers. Biol. Plant. 2012, 56, 458–464. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, X.; Shu, Q.; Li, H.; Zhong, P.; Zhang, H.; Xu, Y.; Wang, L.; Wang, L. Relationship between the composition of flavonoids and flower colors variation in tropical water lily (Nymphaea) cultivars. PLoS ONE 2012, 7, e34335. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhu, Y.; Cui, G.; Zhang, X.; Hu, R.; Deng, Z.; Lei, L.; Wu, L.; Mei, L. A Comparative Study of Flavonoids and Carotenoids Revealed Metabolite Responses for Various Flower Colorations between Nicotiana tabacum L. and Nicotiana rustica L. Front Plant Sci. 2022, 13, 828042. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, L.; Chen, C.; An, X.; Zhang, Y.; Wang, Y.; Li, Q. Full-length transcriptome analysis of Phytolacca americana and its congener P. icosandra and gene expression normalization in three Phytolaccaceae species. BMC Plant Biol. 2020, 20, 396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhu, C.; Tian, C.; Xu, K.; Huang, L.; Shi, B.; Lai, Z.; Lin, Y.; Guo, Y. Integrated volatile metabolome, multi-flux full-length sequencing, and transcriptome analyses provide insights into the aroma formation of postharvest jasmine (Jasminum sambac) during flowering. Postharvest Biol. Technol. 2022, 183, 111726. [Google Scholar] [CrossRef]
- Chen, J.; Tang, X.; Ren, C.; Wei, B.; Wu, Y.; Wu, Q.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 548. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Z.; He, M.; Gao, H.; Zhu, H.; Yun, Z.; Qu, H.; Jiang, Y. Unveiling the complexity of the litchi transcriptome and pericarp browning by single-molecule long-read sequencing. Postharvest Biol. Technol. 2020, 168, 111252. [Google Scholar] [CrossRef]
- Gong, W.; Qiling, S.; Ji, K.; Gong, S.; Wang, L.; Chen, L.; Zhang, J.; Yuan, D. Full-Length Transcriptome from Camellia oleifera Seed Provides Insight into the Transcript Variants Involved in Oil Biosynthesis. J. Agric. Food Chem. 2020, 68, 14670–14683. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zhang, L.; Zhang, D.; Liu, Y.; Lin, L.; Xiong, X.; Zhang, D.; Sun, M.; Cai, M.; Yu, X.; et al. Transcriptomic and Metabolomic Profiling Provides Insights into Flavonoid Biosynthesis and Flower Coloring in Loropetalum chinense and Loropetalum chinense var. rubrum. Agronomy 2023, 13, 1296. https://doi.org/10.3390/agronomy13051296
Zhang X, Zhang L, Zhang D, Liu Y, Lin L, Xiong X, Zhang D, Sun M, Cai M, Yu X, et al. Transcriptomic and Metabolomic Profiling Provides Insights into Flavonoid Biosynthesis and Flower Coloring in Loropetalum chinense and Loropetalum chinense var. rubrum. Agronomy. 2023; 13(5):1296. https://doi.org/10.3390/agronomy13051296
Chicago/Turabian StyleZhang, Xia, Li Zhang, Damao Zhang, Yang Liu, Ling Lin, Xingyao Xiong, Donglin Zhang, Ming Sun, Ming Cai, Xiaoying Yu, and et al. 2023. "Transcriptomic and Metabolomic Profiling Provides Insights into Flavonoid Biosynthesis and Flower Coloring in Loropetalum chinense and Loropetalum chinense var. rubrum" Agronomy 13, no. 5: 1296. https://doi.org/10.3390/agronomy13051296
APA StyleZhang, X., Zhang, L., Zhang, D., Liu, Y., Lin, L., Xiong, X., Zhang, D., Sun, M., Cai, M., Yu, X., & Li, Y. (2023). Transcriptomic and Metabolomic Profiling Provides Insights into Flavonoid Biosynthesis and Flower Coloring in Loropetalum chinense and Loropetalum chinense var. rubrum. Agronomy, 13(5), 1296. https://doi.org/10.3390/agronomy13051296