
Full-Length Transcriptome and the Identification of lncRNAs Involved in Salicylic Acid-Induced Flowering in Duckweed (Lemna gibba)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and SA Treatment
2.2. Full-Length Transcriptome Analysis
2.3. Library Preparation and ssRNA-Seq Analysis
2.4. LncRNA Identification
2.5. Prediction of LncRNA Targets
2.6. Identification of LncRNAs Functioning as MiRNA Targets
2.7. Identification of LncRNAs Involved in Flowering
2.8. qRT-PCR Analysis
3. Results
3.1. Full-Length Transcriptome and LncRNA Identification
3.2. ssRNA-Seq and DE LncRNA Identification
3.3. Functional Analysis of LncRNAs in Trans-Acting Regulation
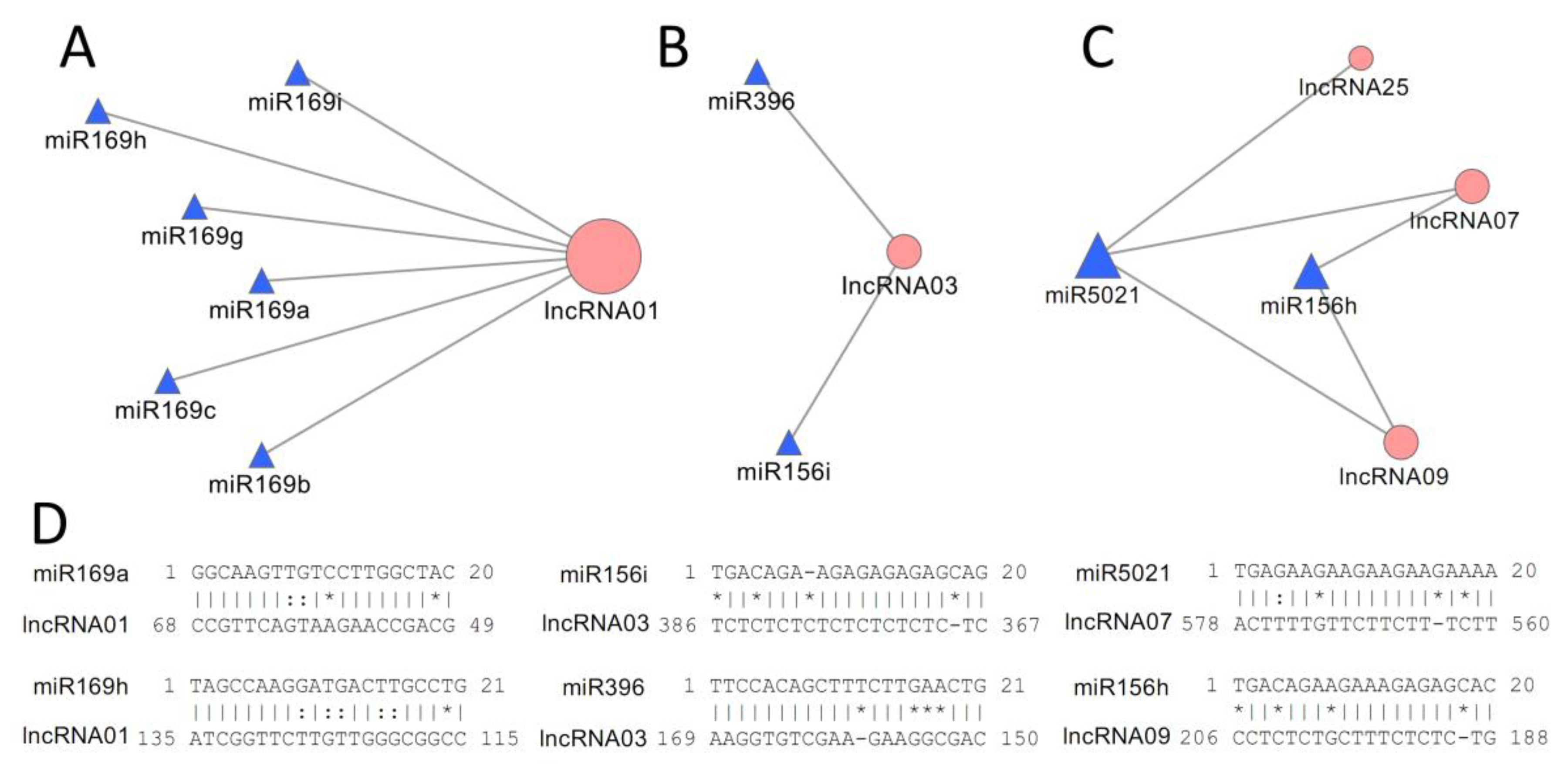
3.4. Functional Analysis of LncRNAs as MiRNA Targets
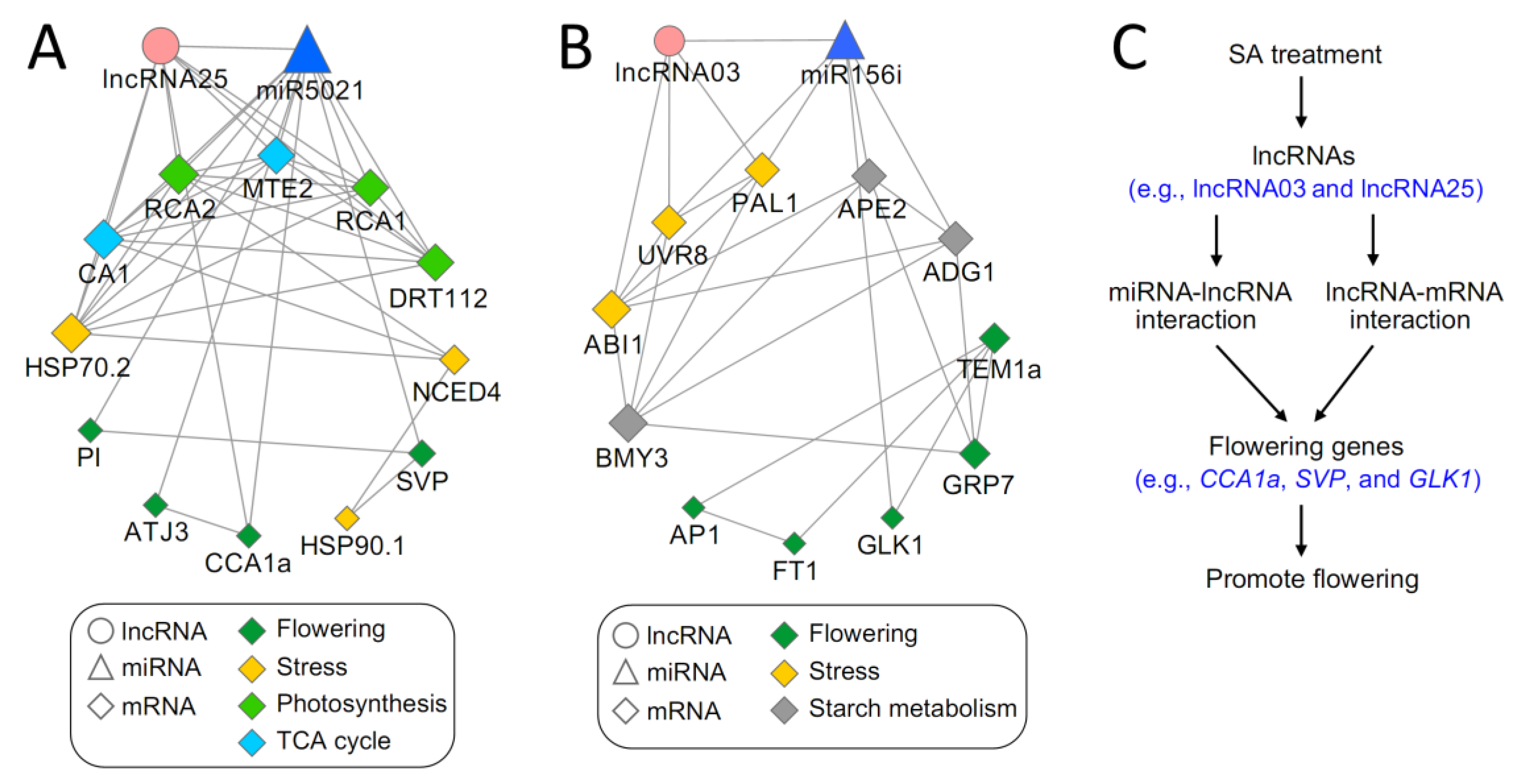
3.5. Identification of LncRNAs Involved in Flowering
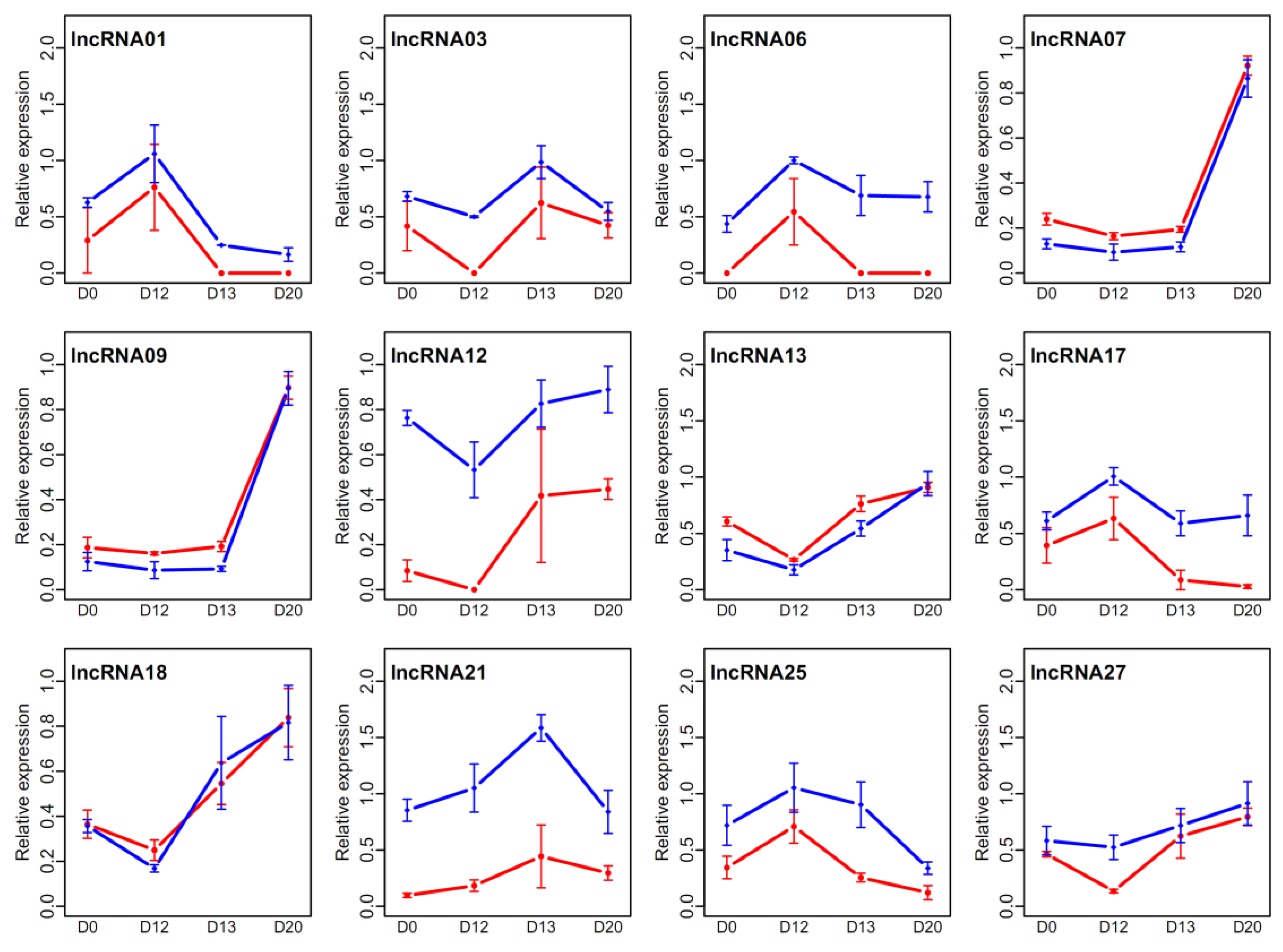
3.6. Expression Verification of LncRNAs
4. Discussion
4.1. LncRNA Is a Critical Player Involved in the Flowering of L. gibba
4.2. Functional Analysis of LncRNAs during SA-Induced Flowering
4.3. Interactive Networks of lncRNA-mRNA-miRNA in SA-Induced Flowering
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Qin, T.; Zhao, H.; Cui, P.; Albesher, N.; Xiong, L. A nucleus-localized long non-coding RNA enhances drought and salt stress tolerance. Plant Physiol. 2017, 175, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, B.; Wirth, S.; Merchan, F.; Laporte, P.; d’Aubenton-Carafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.; Lucas, A.; Deragon, J.M.; et al. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009, 19, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Dong, Q.; Deng, M.; Lin, D.; Xiao, J.; Cheng, P.; Xing, L.; Niu, Y.; Gao, C.; Zhang, W.; et al. The vernalization-induced long non-coding RNA VAS functions with the transcription factor TaRF2b to promote TaVRN1 expression for flowering in hexaploid wheat. Mol. Plant 2021, 14, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.; Jeong, G.; Yang, Y.; Luo, X.; Jeong, D.; Kyung, J.; Hyun, Y.; He, Y.; Lee, I. Vernalization-triggered expression of the antisense transcript COOLAIR is mediated by CBF genes. eLife 2023, 12, e84594. [Google Scholar] [CrossRef]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef]
- Kim, E.D.; Sung, S. Long noncoding RNA: Unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 2012, 17, 16–21. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef]
- Cui, J.; Jiang, N.; Meng, J.; Yang, G.; Liu, W.; Zhou, X.; Ma, N.; Hou, X.; Luan, Y. LncRNA33732-respiratory burst oxidase module associated with WRKY1 in tomato—Phytophthora infestans interactions. Plant J. 2019, 97, 933–946. [Google Scholar] [CrossRef]
- Fukuda, M.; Nishida, S.; Kakei, Y.; Shimada, Y.; Fujiwara, T. Genome-wide analysis of long intergenic noncoding RNAs responding to low-nutrient conditions in Arabidopsis thaliana: Possible involvement of trans-acting siRNA3 in response to low nitrogen. Plant Cell Physiol. 2019, 60, 1961–1973. [Google Scholar] [CrossRef]
- Jin, Y.; Ivanov, M.; Dittrich, A.N.; Nelson, A.D.; Marquardt, S. LncRNA FLAIL affects alternative splicing and represses flowering in Arabidopsis. EMBO J. 2023, 42, e110921. [Google Scholar] [CrossRef]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Hao, Z.; Yan, J.; Li, G. Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize. BMC Genom. 2015, 16, 793. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Wang, Z.M.; Wang, M.; Wang, X.J. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013, 161, 1875–1884. [Google Scholar] [CrossRef]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Severing, E.; Faino, L.; Jamge, S.; Busscher, M.; Kuijer-Zhang, Y.; Bellinazzo, F.; Busscher-Lange, J.; Fernández, V.; Angenent, G.C.; Immink, R.G. Arabidopsis thaliana ambient temperature responsive lncRNAs. BMC Plant Biol. 2018, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, C.; Wang, Z.; Yang, Z.; Chen, D.; Wu, Y. LncRNA expression profile and ceRNA analysis in tomato during flowering. PLoS ONE 2019, 14, e0210650. [Google Scholar] [CrossRef] [PubMed]
- Basu, U.; Hegde, V.S.; Daware, A.; Jha, U.C.; Parida, S.K. Transcriptome landscape of early inflorescence developmental stages identifies key flowering time regulators in chickpea. Plant Mol. Biol. 2022, 108, 565–583. [Google Scholar] [CrossRef]
- Liu, X.; Luo, M.; Li, M.; Wei, J. Transcriptomic analysis reveals lncRNAs associated with flowering of Angelica sinensis during vernalization. Curr. Issues Mol. Biol. 2022, 44, 1867–1888. [Google Scholar] [CrossRef]
- Dai, Y.; Li, G.; Gao, X.; Wang, S.; Li, Z.; Song, C.; Zhang, S.; Li, F.; Fang, Z.; Sun, R. Identification of long noncoding RNAs involved in plumule-vernalization of Chinese cabbage. Front. Plant Sci. 2023, 14, 1147494. [Google Scholar] [CrossRef]
- Xu, W.; Bao, W.; Liu, H.; Chen, C.; Bai, H.; Huang, M.; Zhu, G.; Zhao, H.; Gou, N.; Chen, Y.; et al. Insights into the molecular mechanisms of late flowering in Prunus sibirica by whole-genome and transcriptome analyses. Front. Plant Sci. 2021, 12, 802827. [Google Scholar] [CrossRef]
- An, D.; Li, C.; Zhou, Y.; Wu, Y.; Wang, W. Genomes and transcriptomes of duckweeds. Front. Chem. 2018, 6, 230. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Cheng, J.J. Growing duckweed for biofuel production: A review. Plant Biol. 2015, 17 (Suppl. S1), 16–23. [Google Scholar] [CrossRef]
- Pagliuso, D.; Grandis, A.; Fortirer, J.S.; Camargo, P.; Floh, E.I.S.; Buckeridge, M.S. Duckweeds as promising food feedstocks globally. Agronomy 2022, 12, 796. [Google Scholar] [CrossRef]
- Pieterse, A.H. Is flowering in Lemnaceae stress-induced? A review. Aquat. Bot. 2013, 104, 1–4. [Google Scholar] [CrossRef]
- Cleland, C.F.; Tanaka, O. Effect of daylength on the ability of salicylic acid to induce flowering in the long-day plant Lemna gibba G3 and the short-day plant Lemna paucicostata 6746. Plant Physiol. 1979, 64, 421–424. [Google Scholar] [CrossRef]
- Fu, L.; Huang, M.; Han, B.; Sun, X.; Sree, K.S.; Appenroth, K.J.; Zhang, J. Flower induction, microscope-aided cross-pollination, and seed production in the duckweed Lemna gibba with discovery of a male-sterile clone. Sci. Rep. 2017, 7, 3047. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Tan, D.; Sun, X.; Ding, Z.; Zhang, J. Transcriptional analysis reveals potential genes and regulatory networks involved in salicylic acid-induced flowering in duckweed (Lemna gibba). Plant Physiol. Biochem. 2020, 155, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Wang, M.; Li, X.; Shang, W.; Wang, S.; Han, M.; Ren, X.; Tian, J.; An, N.; Xing, L. Transcriptome analysis reveals dual action of salicylic acid application in the induction of flowering in Malus domestica. Plant Sci. 2022, 324, 111433. [Google Scholar] [CrossRef] [PubMed]
- Martínez, C.; Pons, E.; Prats, G.; León, J. Salicylic acid regulates flowering time and links defence responses and reproductive development. Plant J. 2004, 37, 209–217. [Google Scholar] [CrossRef]
- Yamada, M.; Takeno, K. Stress and salicylic acid induce the expression of PnFT2 in the regulation of the stress-induced flowering of Pharbitis nil. J. Plant Physiol. 2014, 171, 205–212. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Tie, W.; Fu, L.; Yan, Y.; Liu, G.; Yan, W.; Li, Y.; Wu, C.; Zhang, J.; Hu, W. Strand-specific RNA-seq based identification and functional prediction of drought-responsive lncRNAs in cassava. BMC Genom. 2019, 20, 214. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ding, Z.; Tan, D.; Han, B.; Sun, X.; Zhang, J. Genome-wide discovery and functional prediction of salt-responsive lncRNAs in duckweed. BMC Genom. 2020, 21, 212. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Ding, Z.; Fu, L.; Tie, W.; Yan, Y.; Wu, C.; Dai, J.; Zhang, J.; Hu, W. Highly dynamic, coordinated, and stage-specific profiles are revealed by a multi-omics integrative analysis during tuberous root development in cassava. J. Exp. Bot. 2020, 71, 7003–7017. [Google Scholar] [CrossRef]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Bonnet, E.; He, Y.; Billiau, K.; Van de Peer, Y. TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 2010, 26, 1566–1568. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed]
- Bouche, F.; Lobet, G.; Tocquin, P.; Perilleux, C. FLOR-ID: An interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016, 44, D1167–D1171. [Google Scholar] [CrossRef]
- Xu, M.Y.; Zhang, L.; Li, W.W.; Hu, X.L.; Wang, M.B.; Fan, Y.L.; Zhang, C.Y.; Wang, L. Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 89–101. [Google Scholar] [CrossRef]
- Feng, S.; Xu, Y.; Guo, C.; Zheng, J.; Zhou, B.; Zhang, Y.; Ding, Y.; Zhang, L.; Zhu, Z.; Wang, H. Modulation of miR156 to identify traits associated with vegetative phase change in tobacco (Nicotiana tabacum). J. Exp. Bot. 2016, 67, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Li, Z.; Yuan, N.; Hu, Q.; Zhou, M.; Zhao, J.; Li, D.; Luo, H. MiR396 is involved in plant response to vernalization and flower development in Agrostis stolonifera. Hortic. Res. 2020, 7, 173. [Google Scholar] [CrossRef]
- Borges, F.; Pereira, P.A.; Slotkin, R.K.; Martienssen, R.A.; Becker, J.D. MicroRNA activity in the Arabidopsis male germline. J. Exp. Bot. 2011, 62, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Arjmand, M.P.; Lahiji, H.S.; Golfazani, M.M.; Biglouei, M.H. New insights on the regulatory network of drought-responsive key genes in Arabidopsis thaliana. Genetica 2023, 151, 29–45. [Google Scholar] [CrossRef]
- Soorni, A.; Karimi, M.; Al Sharif, B.; Habibi, K. Genome-wide screening and characterization of long noncoding RNAs involved in flowering/bolting of Lactuca sativa. BMC Plant Biol. 2023, 23, 3. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, Y.; Ren, Y.; Wang, S.; Yang, M. Conjoint analysis of genome-wide lncRNA and mRNA expression during the salicylic acid response in Populus× euramericana. Plants 2023, 12, 1377. [Google Scholar] [CrossRef]
- Takeno, K. Stress-induced flowering: The third category of flowering response. J. Exp. Bot. 2016, 67, 4925–4934. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Wu, M.-F.; Yang, L.; Wu, G.; Poethig, R.S.; Wagner, D. The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell 2009, 17, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, C.; Fu, H.; Yang, M.; Wu, H.; Ding, Y.; Li, L.; Lin, S. Genome-wide analysis of SQUAMOSA-promoter-binding protein-like family in flowering Pleioblastus pygmaeus. Int. J. Mol. Sci. 2022, 23, 14035. [Google Scholar] [CrossRef] [PubMed]
- Cho, L.H.; Pasriga, R.; Yoon, J.; Jeon, J.S.; An, G. Roles of sugars in controlling flowering time. J. Plant Biol. 2018, 61, 121–130. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, L.; Tan, D.; Sun, X.; Ding, Z.; Zhang, J. Full-Length Transcriptome and the Identification of lncRNAs Involved in Salicylic Acid-Induced Flowering in Duckweed (Lemna gibba). Agronomy 2023, 13, 2631. https://doi.org/10.3390/agronomy13102631
Fu L, Tan D, Sun X, Ding Z, Zhang J. Full-Length Transcriptome and the Identification of lncRNAs Involved in Salicylic Acid-Induced Flowering in Duckweed (Lemna gibba). Agronomy. 2023; 13(10):2631. https://doi.org/10.3390/agronomy13102631
Chicago/Turabian StyleFu, Lili, Deguan Tan, Xuepiao Sun, Zehong Ding, and Jiaming Zhang. 2023. "Full-Length Transcriptome and the Identification of lncRNAs Involved in Salicylic Acid-Induced Flowering in Duckweed (Lemna gibba)" Agronomy 13, no. 10: 2631. https://doi.org/10.3390/agronomy13102631
APA StyleFu, L., Tan, D., Sun, X., Ding, Z., & Zhang, J. (2023). Full-Length Transcriptome and the Identification of lncRNAs Involved in Salicylic Acid-Induced Flowering in Duckweed (Lemna gibba). Agronomy, 13(10), 2631. https://doi.org/10.3390/agronomy13102631