
The Use of Long-Read Sequencing to Study the Phylogenetic Diversity of the Potato Varieties Plastome of the Ural Selection



Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Sequencing
2.2. Quality Control of Reads
2.3. Plastid Genome Assembly and Quality Assessment
2.4. Alignment and Phylogenetic Identification
2.5. Genome Annotation
3. Results
3.1. Sequencing Results
3.2. Phylogenetic Identification
3.3. Long-Reads Confirm the Presence of Two Isoforms of the Plastid Genome in Potatoes
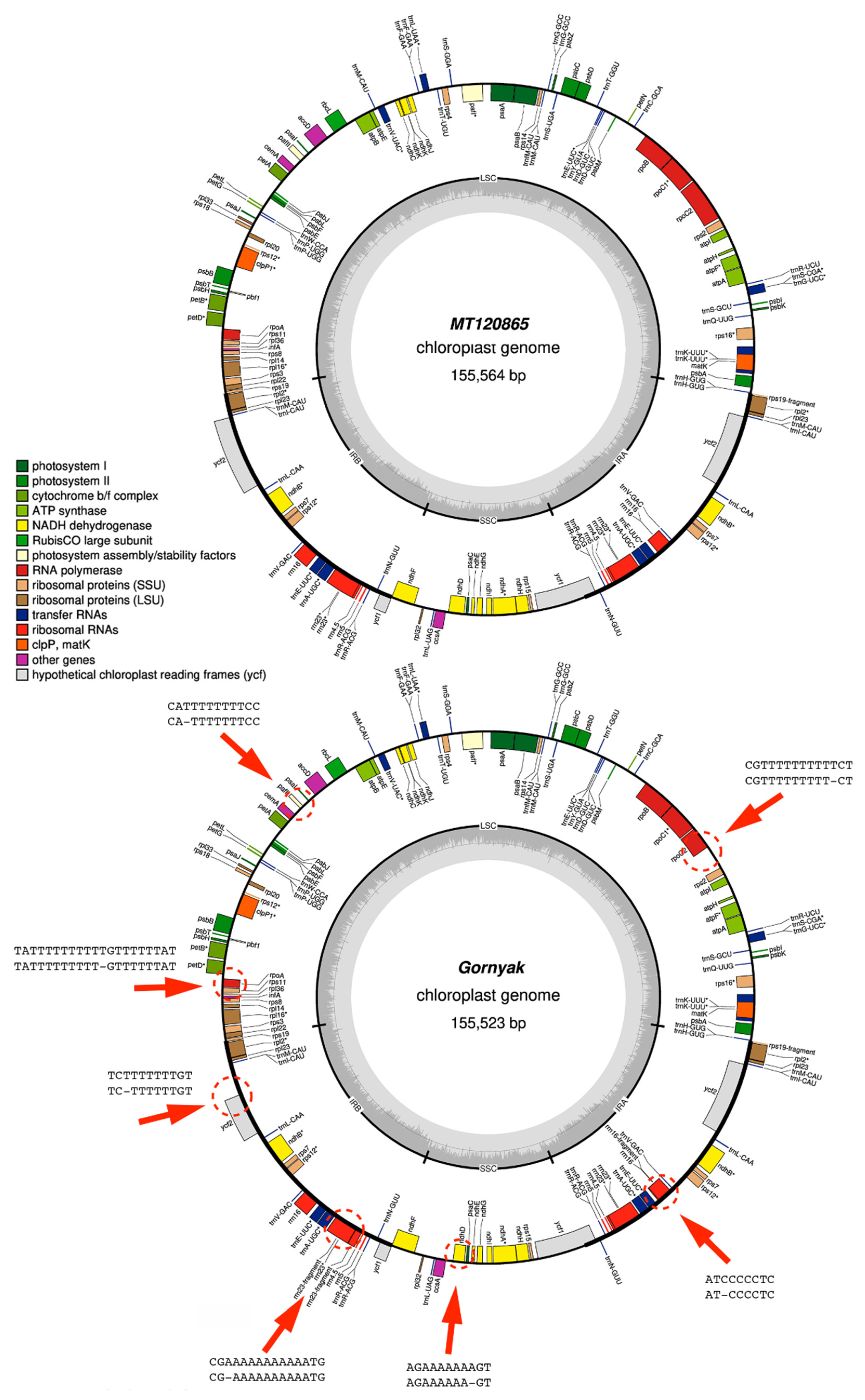
3.4. Nanopore Sequencing Cannot Cope with Homopolymer Repeats
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; de Pamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [PubMed]
- Thode, V.A.; Lohmann, L.G. Comparative Chloroplast Genomics at Low Taxonomic Levels: A Case Study Using Amphilophium (Bignonieae, Bignoniaceae). Front. Plant Sci. 2019, 10, 796. [Google Scholar] [CrossRef]
- Chung, H.-J.; Jung, J.D.; Park, H.-W.; Kim, J.-H.; Cha, H.W.; Min, S.R.; Jeong, W.-J.; Liu, J.R. The complete chloroplast genome sequences of Solanum tuberosum and comparative analysis with Solanaceae species identified the presence of a 241-bp deletion in cultivated potato chloroplast DNA sequence. Plant Cell Rep. 2006, 25, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-S.; Cheon, K.-S.; Hong, S.-Y.; Cho, J.-H.; Im, J.-S.; Mekapogu, M.; Yu, Y.S.; Park, T.H. Complete chloroplast genome sequences of Solanum commersonii and its application to chloroplast genotype in somatic hybrids with Solanum tuberosum. Plant Cell Rep. 2016, 35, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Yang, C.; Liu, J.; Jin, S.; Harijati, N.; Hu, Z.; Diao, Y.; Zhao, L. Comparative analysis of complete chloroplast genome sequences of four major Amorphophallus species. Sci. Rep. 2019, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Achakkagari, S.R.; Kyriakidou, M.; Tai, H.H.; Anglin, N.L.; Ellis, D.; Stromvik, M.V. Complete plastome assemblies from a panel of 13 diverse potato taxa. PLoS ONE 2020, 15, e0240124. [Google Scholar] [CrossRef] [PubMed]
- Spooner, D.M.; Ghislain, M.; Simon, R.; Jansky, S.H.; Gavrilenko, T. Systematics, Diversity, Genetics, and Evolution of Wild and Cultivated Potatoes. Bot. Rev. 2014, 80, 283–383. [Google Scholar] [CrossRef]
- Huang, B.; Ruess, H.; Liang, Q.; Colleoni, C.; Spooner, D.M. Analyses of 202 plastid genomes elucidate the phylogeny of Solanum section Petota. Sci. Rep. 2019, 9, 4454. [Google Scholar] [CrossRef]
- Hosaka, K. Who is the mother of the potato?—Restriction endonuclease analysis of chloroplast DNA of cultivated potatoes. Theor. Appl. Genet. 1986, 72, 606–618. [Google Scholar] [CrossRef]
- Hosaka, K. Successive domestication and evolution of the Andean potatoes as revealed by chloroplast DNA restriction endonuclease analysis. Theor. Appl. Genet. 1995, 90, 356–363. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Available online: https://community.nanoporetech.com/posts/guppy-v6-0-1-patch-release (accessed on 1 December 2021).
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Šikić, M. Time- and memory-efficient genome assembly with Raven. Nat. Comput. Sci. 2021, 1, 332–336. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Cerdeira, L.T.; Hawkey, J.; Méric, G.; Vezina, B.; Wyres, K.L.; Holt, K.E. Trycycler: Consensus long-read assemblies for bacterial genomes. Genome Biol. 2021, 22, 266. [Google Scholar] [CrossRef]
- Available online: https://github.com/nanoporetech/medaka (accessed on 25 December 2021).
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Available online: https://github.com/connor-lab/msa2vcf (accessed on 4 January 2022).
- Available online: https://github.com/RealTimeGenomics/rtg-tools (accessed on 4 January 2022).
- Available online: https://github.com/iliuh/FMAlign (accessed on 15 January 2022).
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucl. Acids Res. 2021, 49, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://chlorobox.mpimp-golm.mpg.de/geseq.html (accessed on 17 January 2022).
- Lihodeevskiy, G.A.; Shanina, E.P. Structural Variations in the Genome of Potato Varieties of the Ural Selection. Agronomy 2021, 11, 1703. [Google Scholar] [CrossRef]
- Gavrilenko, T.A.; Antonova, O.Y.; Kostina, L.I. Study of the genetic diversity of potato varieties using PCR analysis of DNA organelles. Genetika 2007, 43, 1550–1555. [Google Scholar] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Erickson, D.L.; Meng, J. Benchmarking Long-Read Assemblers for Genomic Analyses of Bacterial Pathogens Using Oxford Nanopore Sequencing. Int. J. Mol. Sci. 2020, 21, 9161. [Google Scholar] [CrossRef]
- Tanaka, M.; Mino, S.; Ogura, Y.; Hayashi, T.; Sawabe, T. Availability of Nanopore Sequences in the Genome Taxonomy for Vibrionaceae Systematics: Rumoiensis Clade Species as a Test Case. PeerJ. 2018, 6, e5018. [Google Scholar] [CrossRef]
- Bokma, J.; Vereecke, N.; De Bleecker, K.; Callens, J.; Ribbens, S.; Nauwynck, H.; Haesebrouck, F.; Theuns, S.; Boyen, F.; Pardon, B. Phylogenomic analysis of Mycoplasma bovis from Belgian veal, dairy and beef herds. Vet. Res. 2020, 51, 121. [Google Scholar] [CrossRef]
- Baeza, J.A. Yes, we can use it: A formal test on the accuracy of low-pass nanopore long-read sequencing for mitophylogenomics and barcoding research using the Caribbean spiny lobster Panulirus argus. BMC Genom. 2020, 21, 882. [Google Scholar] [CrossRef]
- Palmer, J.D. Chloroplast DNA exists in two orientations. Nature 1983, 301, 92–93. [Google Scholar] [CrossRef]
- Wang, W.; Lanfear, R. Long-Reads Reveal That the Chloroplast Genome Exists in Two Distinct Versions in Most Plants. Genome Biol. Evol. 2019, 11, 3372–3381. [Google Scholar] [CrossRef] [PubMed]
- Scheunert, A.; Dorfner, M.; Lingl, T.; Oberprieler, C. Can we use it? On the utility of de novo and reference-based assembly of Nanopore data for plant plastome sequencing. PLoS ONE 2020, 15, e0226234. [Google Scholar]
- Delahaye, C.; Nicolas, J. Sequencing DNA with nanopores: Troubles and biases. PLoS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef]
- Wang, W.; Schalamun, M.; Morales-Suarez, A.; Kainer, D.; Schwessinger, B.; Lanfear, R. Assembly of chloroplast genomes with long- and short-read data: A comparison of approaches using Eucalyptus pauciflora as a test case. BMC Genom. 2018, 19, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achakkagari, S.R.; Tai, H.H.; Davidson, C.; Jong, H.; Strömvik, M.V. The complete plastome sequences of nine diploid potato clones. Mitochondrial DNA B Resour. 2021, 6, 811–813. [Google Scholar] [CrossRef]
- Gargano, D.; Scotti, N.; Vezzi, A.; Bilardi, A.; Valle, G.; Grillo, S.; Cozzolino, S.; Cardi, T. Genome-wide analysis of plastome sequence variation and development of plastidial CAPS markers in common potato and related Solanum species. Genet. Resour. Crop Evol. 2012, 59, 419–430. [Google Scholar] [CrossRef]
- Yang, Y.; Dang, Y.; Li, Q.; Lu, J.; Li, X.; Wang, Y. Complete chloroplast genome sequence of poisonous and medicinal plant Datura stramonium: Organizations and implications for genetic engineering. PLoS ONE 2014, 9, e110656. [Google Scholar] [CrossRef]
- Raime, K.; Remm, M. Method for the Identification of Taxon-Specific k-mers from Chloroplast Genome: A Case Study on Tomato Plant (Solanum lycopersicum). Front Plant Sci. 2018, 9, 6. [Google Scholar] [CrossRef]
- Yan, L.; Lai, X.; Li, X.; Wei, C.; Tan, X.; Zhang, Y. Analyses of the complete genome and gene expression of chloroplast of sweet potato [Ipomoea batata]. PLoS ONE 2015, 10, e0124083. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. The chloroplast genome sequence of bittersweet (Solanum dulcamara): Plastid genome structure evolution in Solanaceae. PLoS ONE 2018, 13, e0196069. [Google Scholar] [CrossRef]
- Tamburino, R.; Sannino, L.; Cafasso, D.; Cantarella, C.; Orrù, L.; Cardi, T.; Cozzolino, S.; D’Agostino, N.; Scotti, N. Cultivated Tomato (Solanum lycopersicum L.) Suffered a Severe Cytoplasmic Bottleneck during Domestication: Implications from Chloroplast Genomes. Plants 2020, 9, 1443. [Google Scholar] [CrossRef] [PubMed]
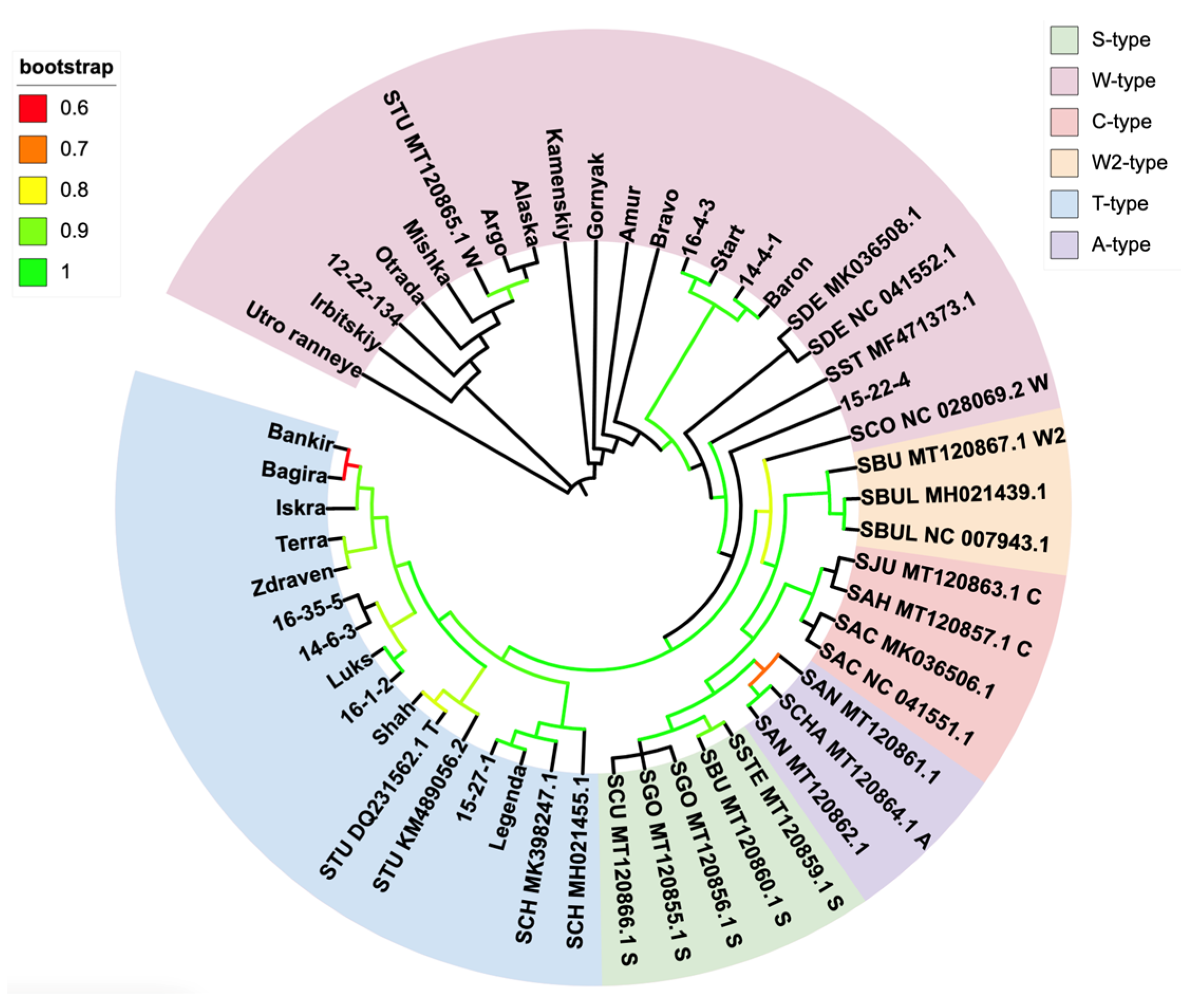

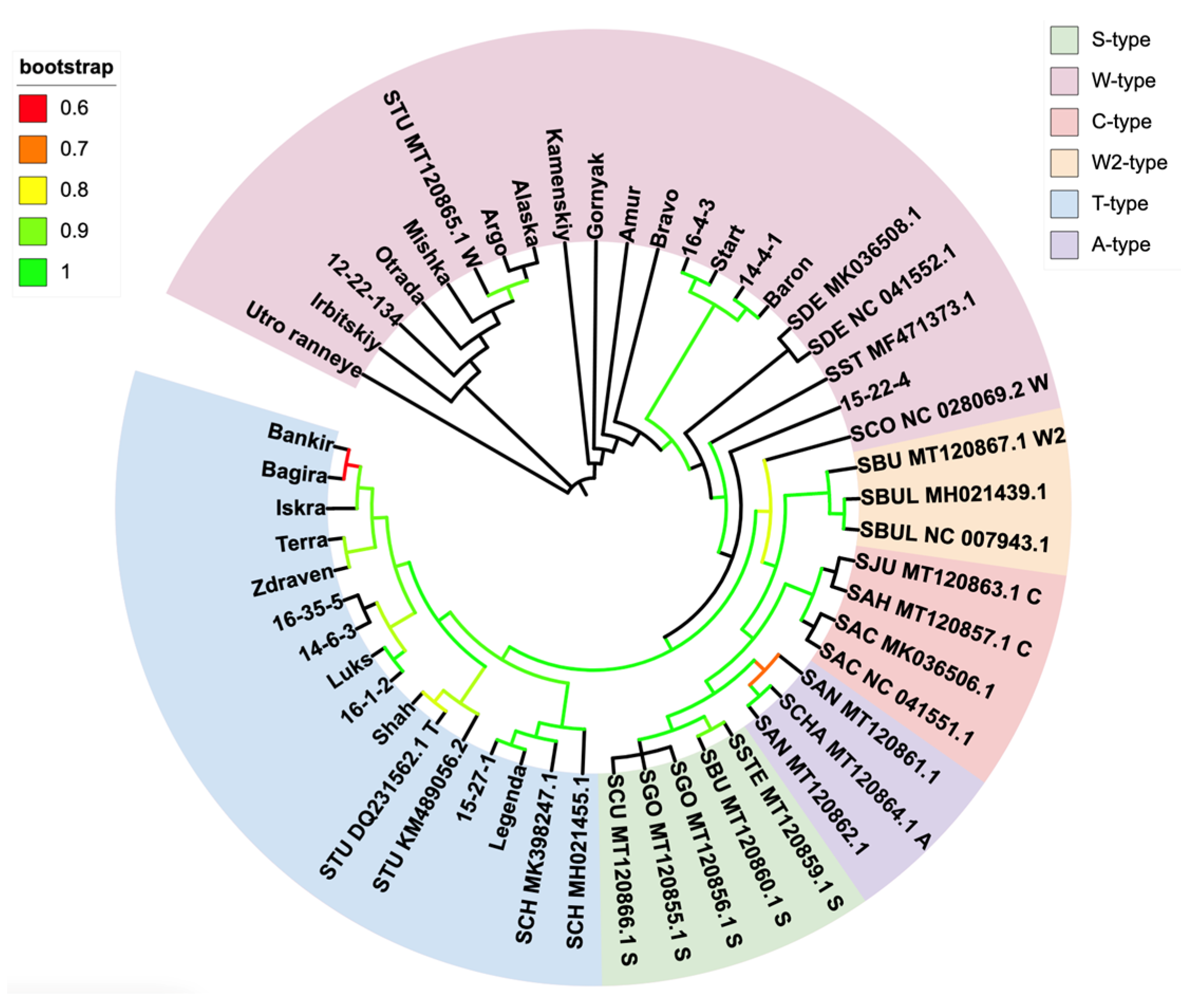{kind=link}
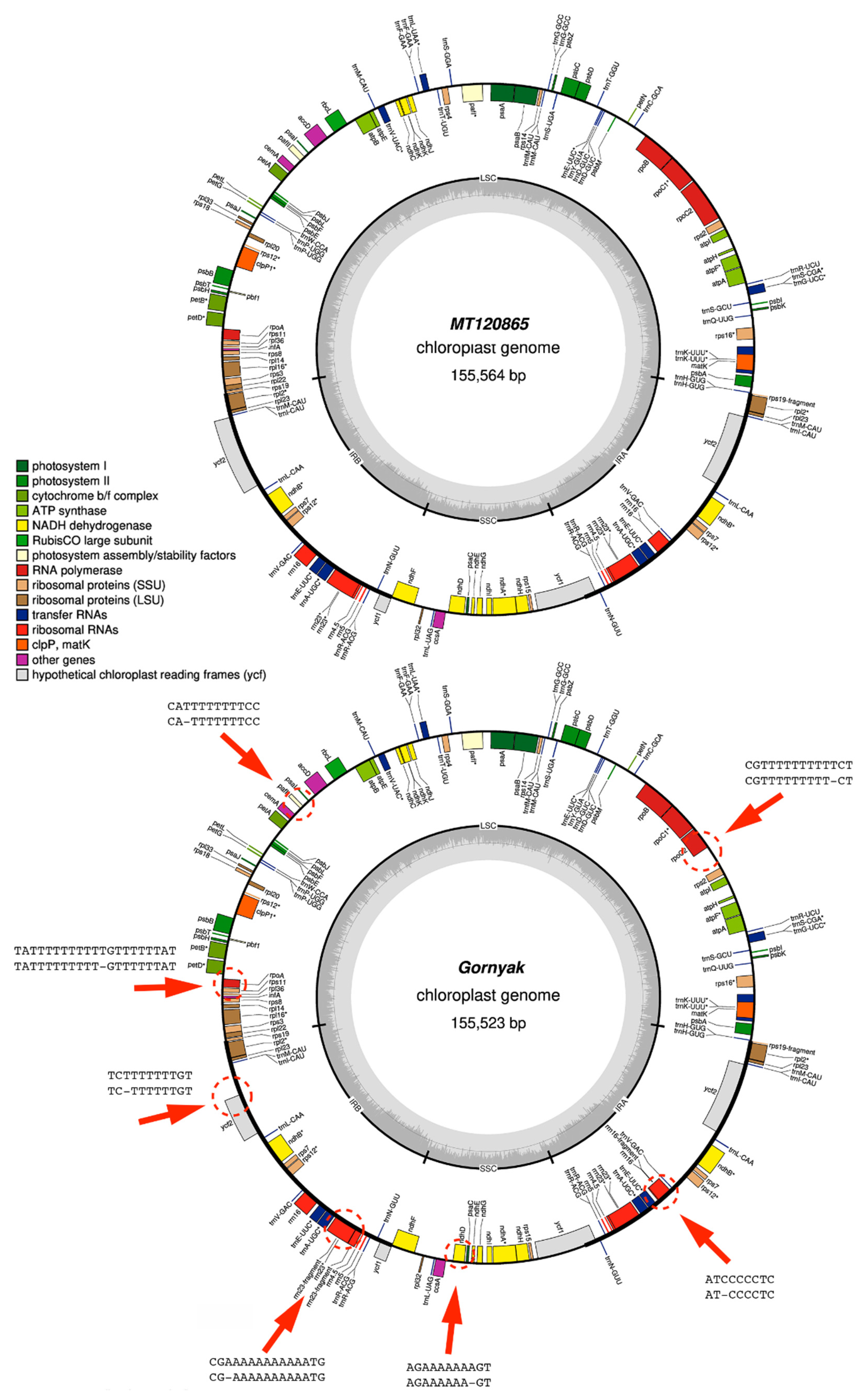{kind=link}
| Variety | Plastome Isoform | Number of Copies |
|---|---|---|
| Alaska | LSC_IR_SSC_IRrc | 18 |
| LSC_IR_SSCrc_IRrc | 21 | |
| Argo | LSC_IR_SSC_IRrc | 14 |
| LSC_IR_SSCrc_IRrc | 19 | |
| Shah | LSC_IR_SSC_IRrc | 19 |
| LSC_IR_SSCrc_IRrc | 14 |
| Reference Sequence | Varieties | SNP/Insertions/Deletions | Total Number of Discovered Variants |
|---|---|---|---|
| S. tuberosum W-type MT120865 | 12-22-134 | 1/2/36 | 110 |
| Alaska | 1/20/10 | ||
| Amur | 1/1/39 | ||
| Argo | 3/6/15 | ||
| Bravo | 0/1/40 | ||
| Gornyak | 1/1/40 | ||
| Irbitskiy | 1/1/38 | ||
| Kamenskiy | 1/0/44 | ||
| Mishka | 1/1/32 | ||
| Otrada | 1/3/44 | ||
| Utro ranneye | 1/1/32 | ||
| S. tuberosum T-type KM489056 | 14-6-3 | 1/1/31 | 104 |
| 16-1-2 | 2/0/37 | ||
| 16-35-5 | 2/0/40 | ||
| Bagira | 3/0/42 | ||
| Bankir | 1/0/38 | ||
| Iskra | 2/1/35 | ||
| Luks | 2/2/36 | ||
| Shah | 3/10/16 | ||
| Terra | 3/0/34 | ||
| Zdraven | 3/0/39 | ||
| S. demissum NC_041552 | 14-4-1 | 9/2/34 | 74 |
| 16-4-3 | 11/1/36 | ||
| Baron | 9/2/33 | ||
| Start | 10/1/36 | ||
| S. stoloniferum MF471373 | 15-22-4 | 8/0/39 | 47 |
| S. chacoense MK398247 | 15-27-1 | 22/2/49 | 86 |
| Legenda | 23/3/43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lihodeevskiy, G.A.; Shanina, E.P. The Use of Long-Read Sequencing to Study the Phylogenetic Diversity of the Potato Varieties Plastome of the Ural Selection. Agronomy 2022, 12, 846. https://doi.org/10.3390/agronomy12040846
Lihodeevskiy GA, Shanina EP. The Use of Long-Read Sequencing to Study the Phylogenetic Diversity of the Potato Varieties Plastome of the Ural Selection. Agronomy. 2022; 12(4):846. https://doi.org/10.3390/agronomy12040846
Chicago/Turabian StyleLihodeevskiy, Georgiy A., and Elena P. Shanina. 2022. "The Use of Long-Read Sequencing to Study the Phylogenetic Diversity of the Potato Varieties Plastome of the Ural Selection" Agronomy 12, no. 4: 846. https://doi.org/10.3390/agronomy12040846
APA StyleLihodeevskiy, G. A., & Shanina, E. P. (2022). The Use of Long-Read Sequencing to Study the Phylogenetic Diversity of the Potato Varieties Plastome of the Ural Selection. Agronomy, 12(4), 846. https://doi.org/10.3390/agronomy12040846