
Deciphering Codon Usage Patterns in Genome of Cucumis sativus in Comparison with Nine Species of Cucurbitaceae



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequences Acquisition
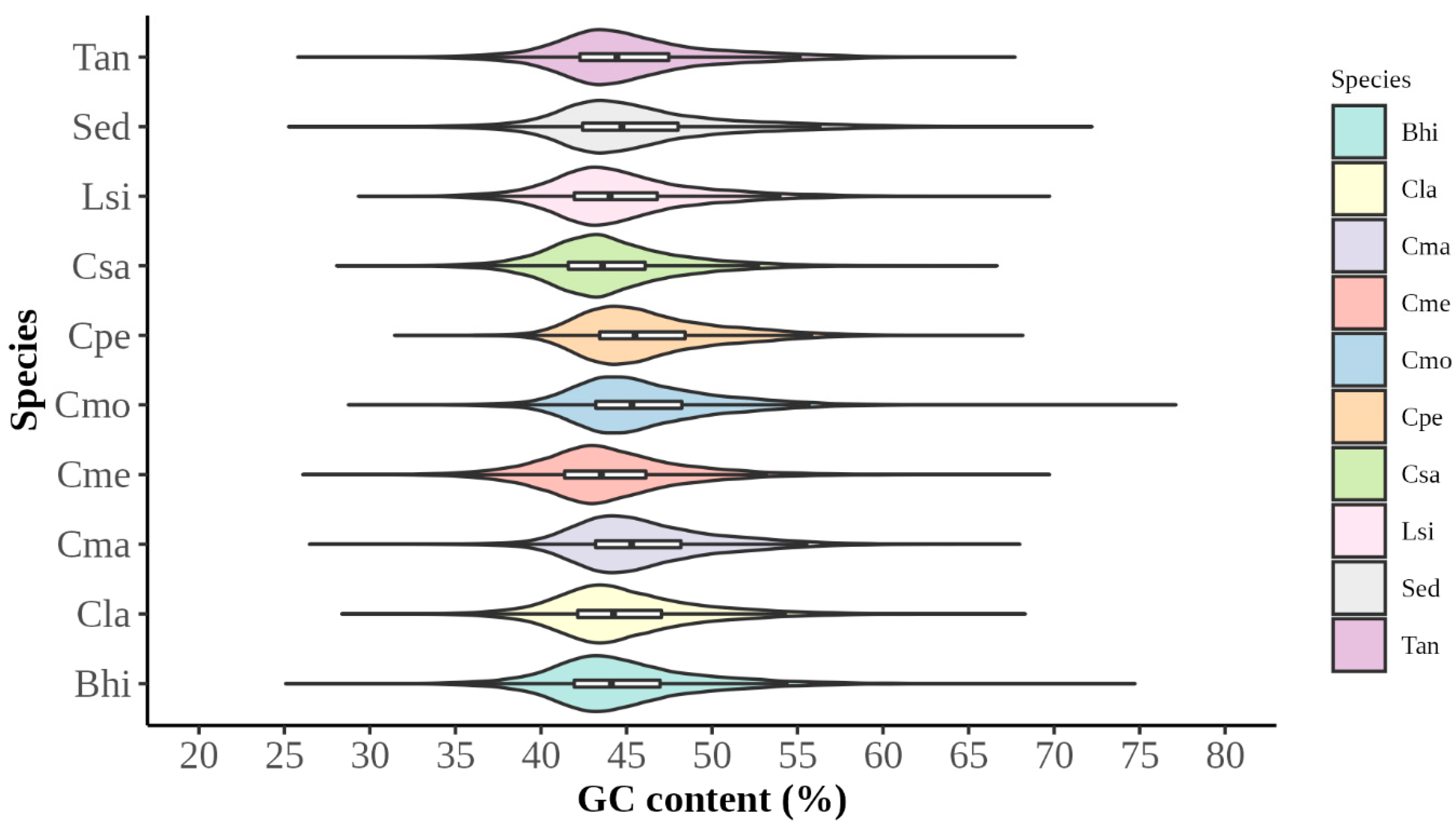
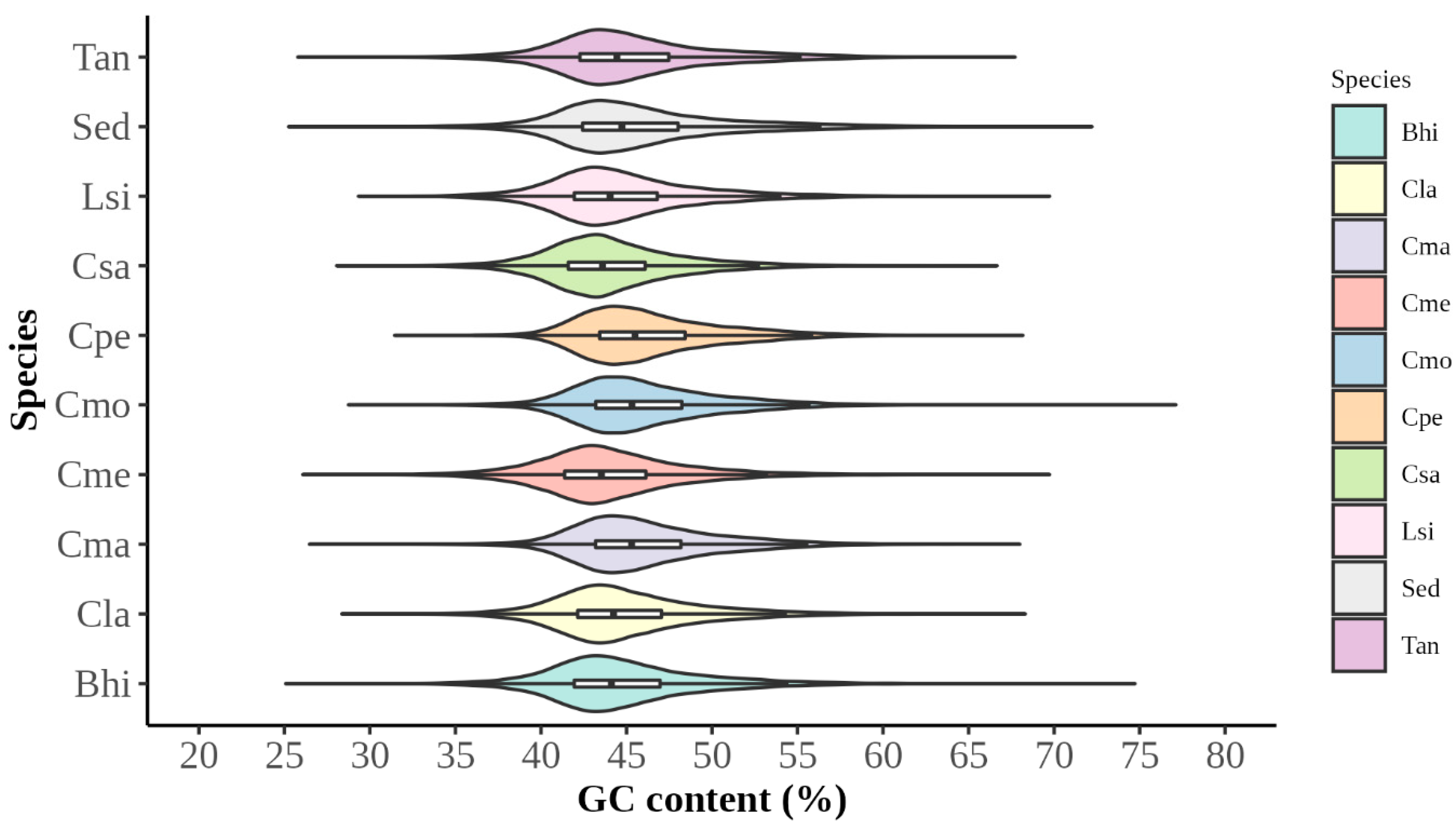
2.2. Nucleotide Composition Analysis
2.3. Indicators of Codon Usage
2.4. Correspondence Analysis and Correlation Analysis
2.5. The Analysis of the Source of CUB
2.6. Identification of Optimal Codons
2.7. RSCU-Based Cluster Analysis
2.8. Statistical Analysis and Graph Drawing
3. Results
3.1. Analysis of Codon Usage Patterns
3.1.1. Analysis of Codon Usage Indicators
3.1.2. RSCU Analysis
3.2. Analysis of Factors for CUB
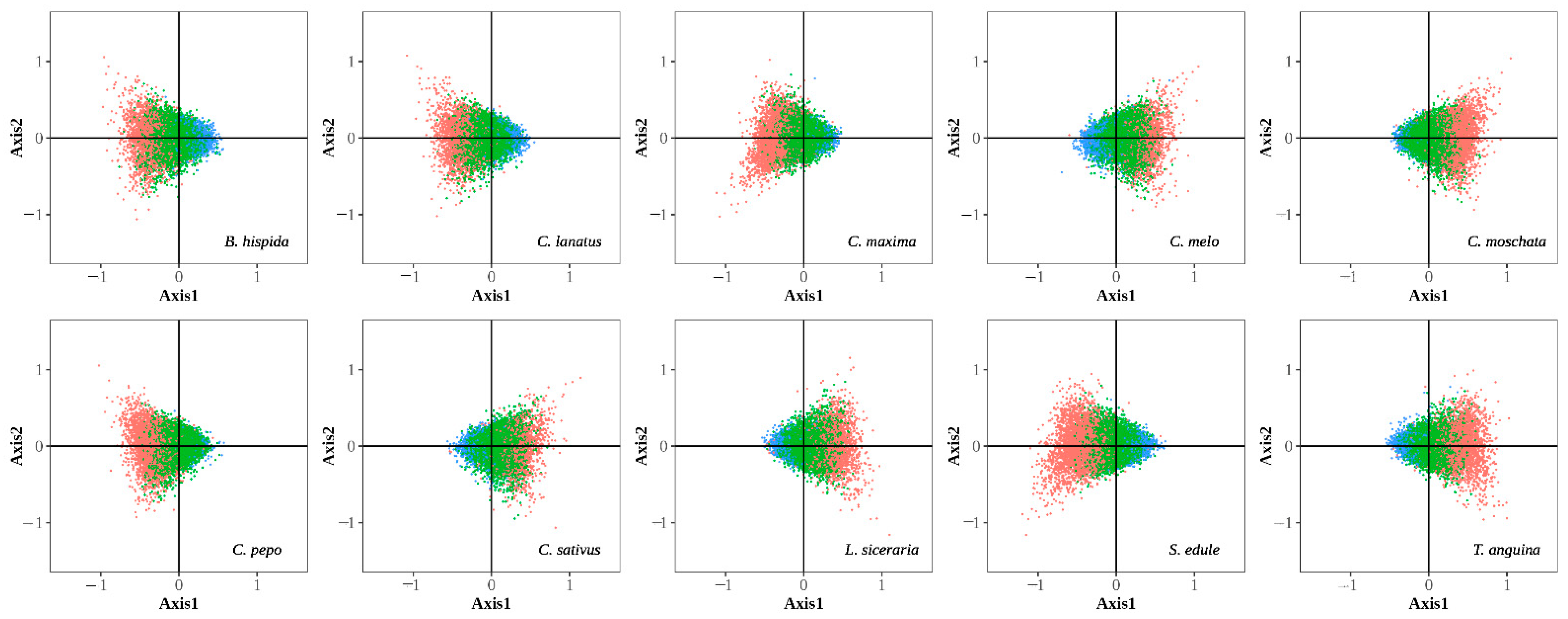
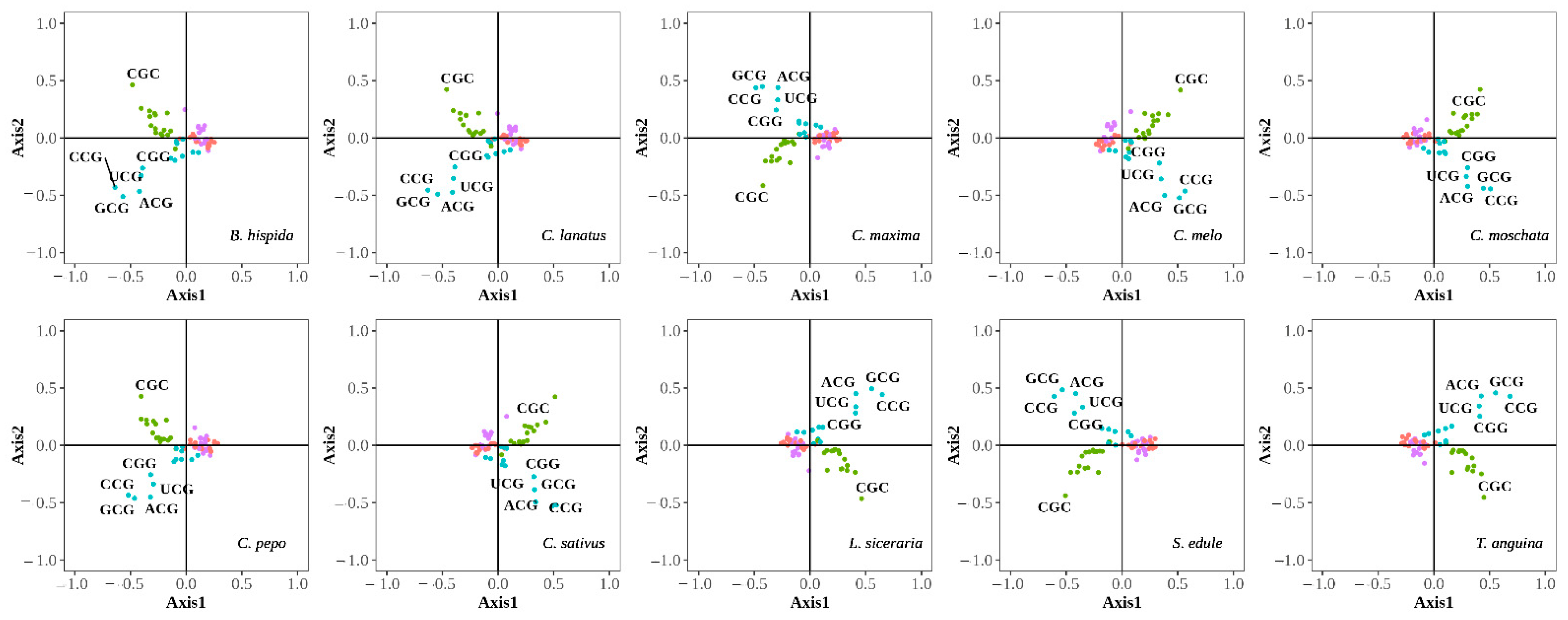
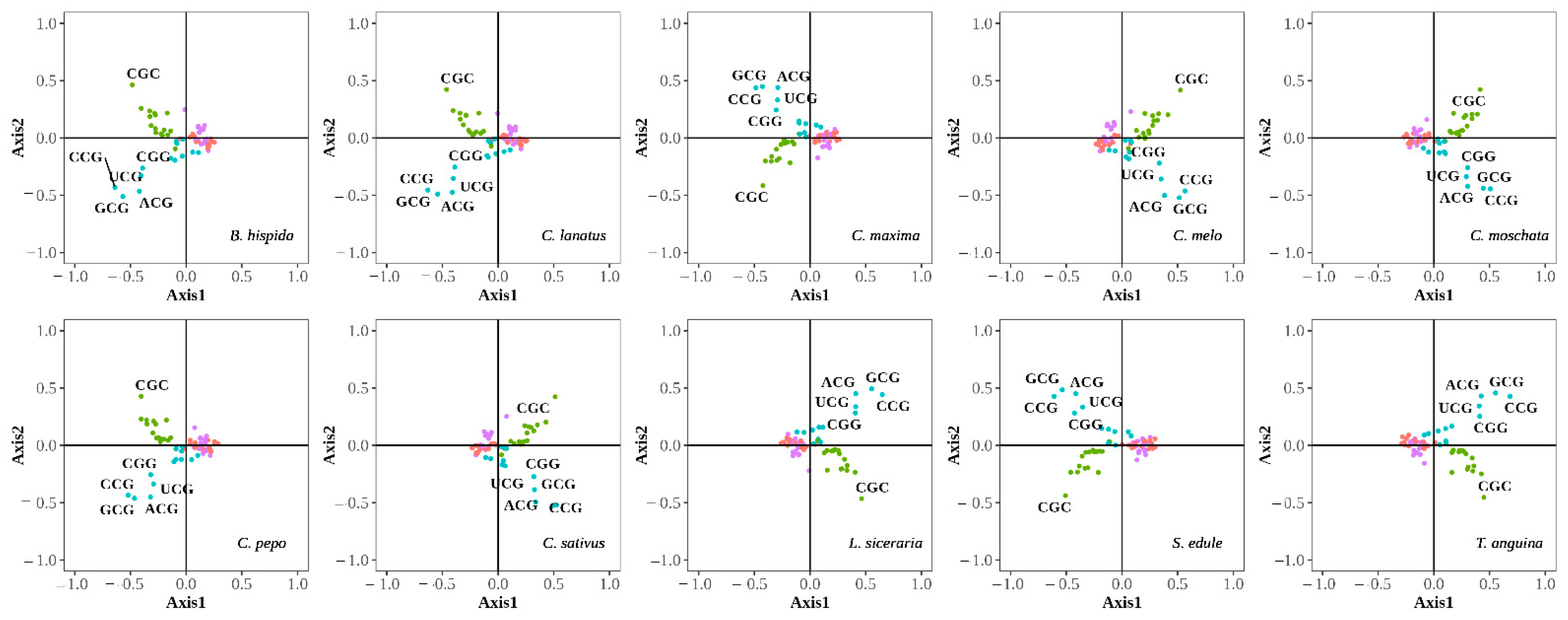
3.2.1. Correspondence Analysis
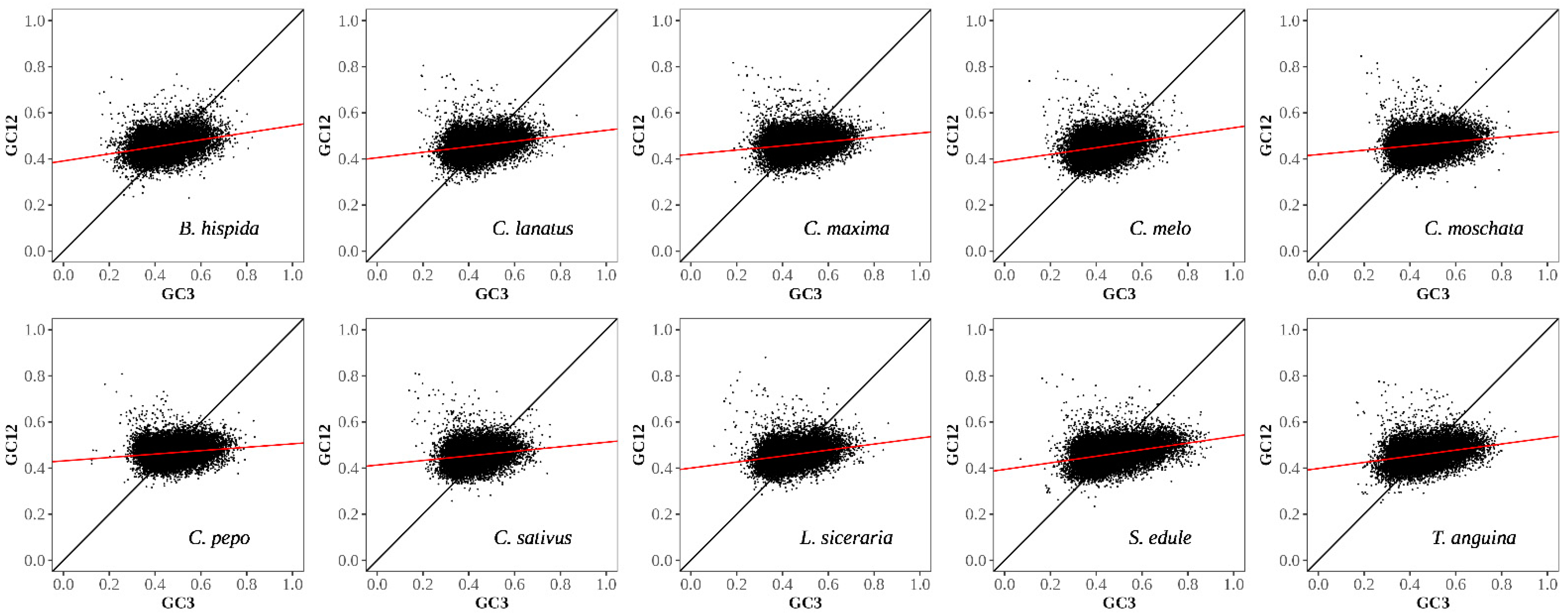
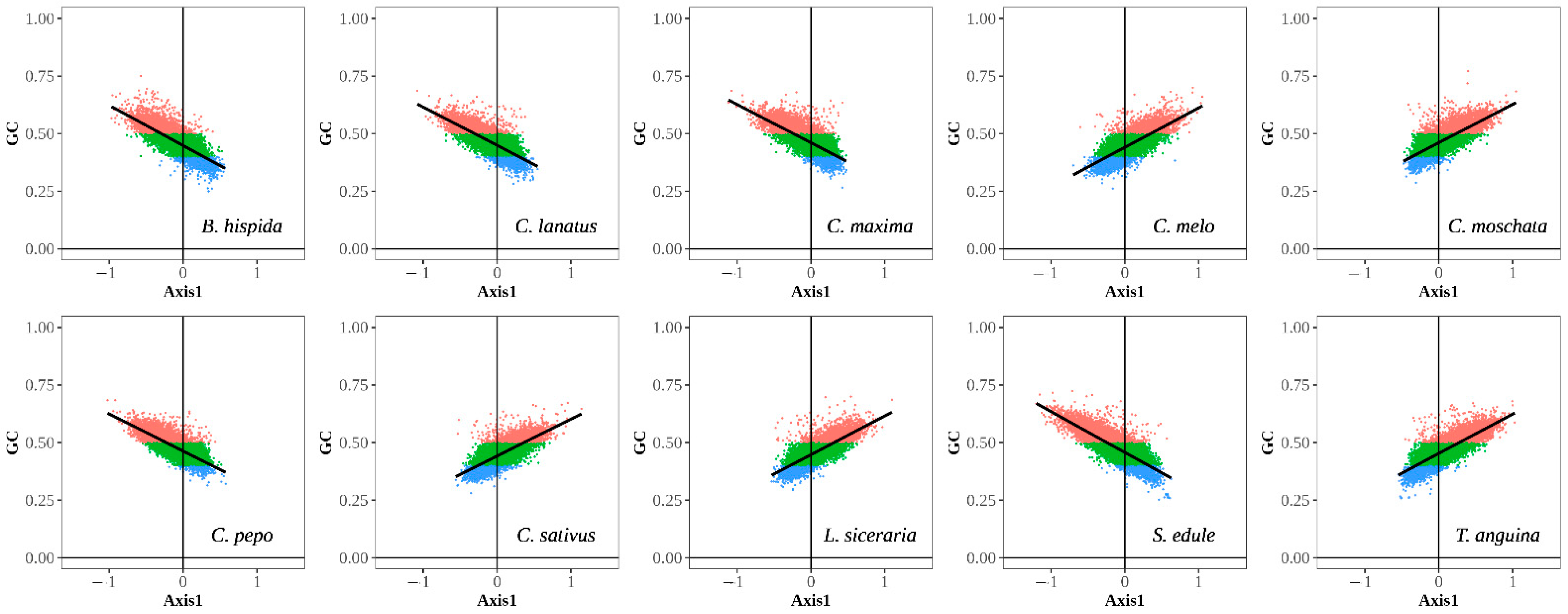
3.2.2. Neutral Plot Analysis
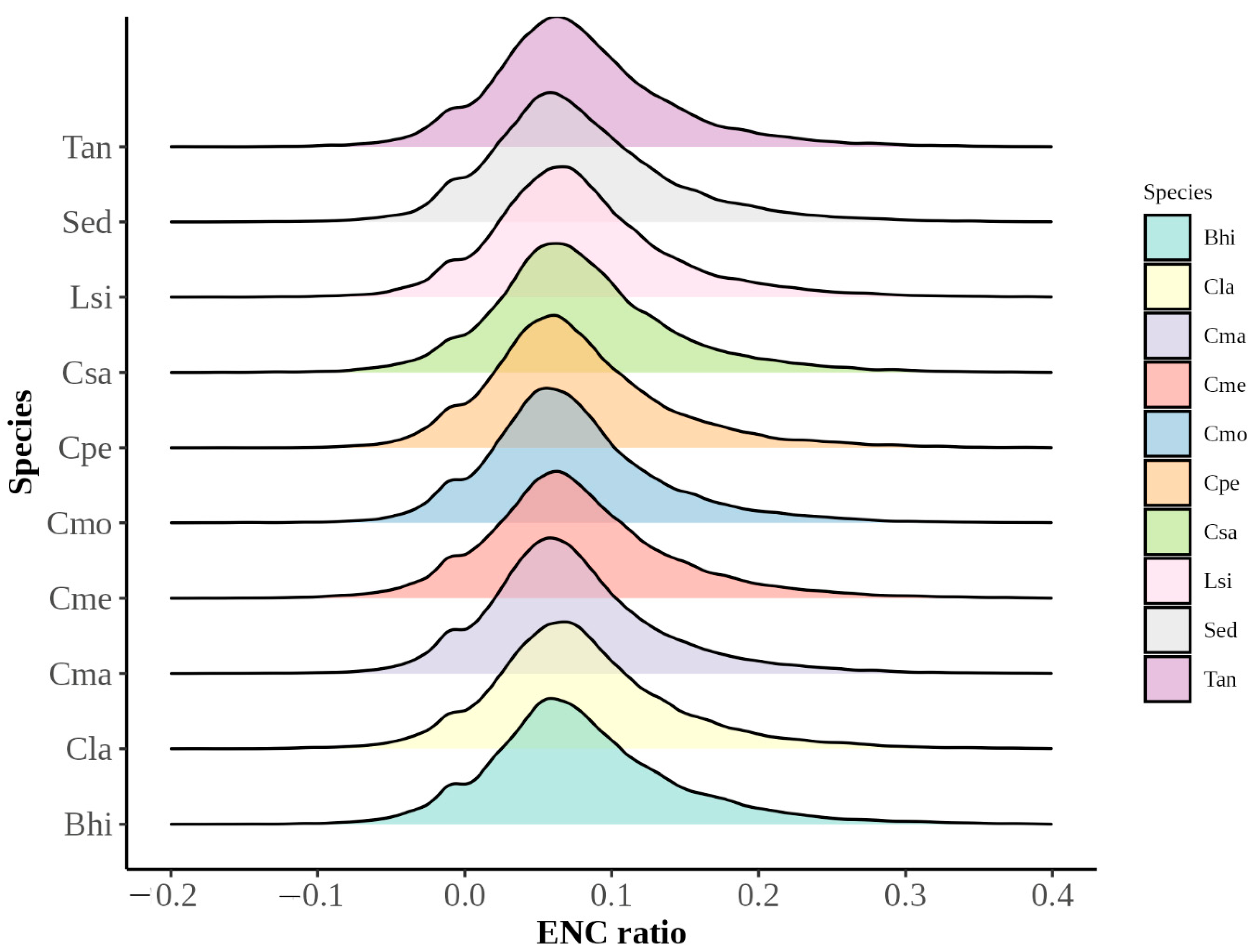
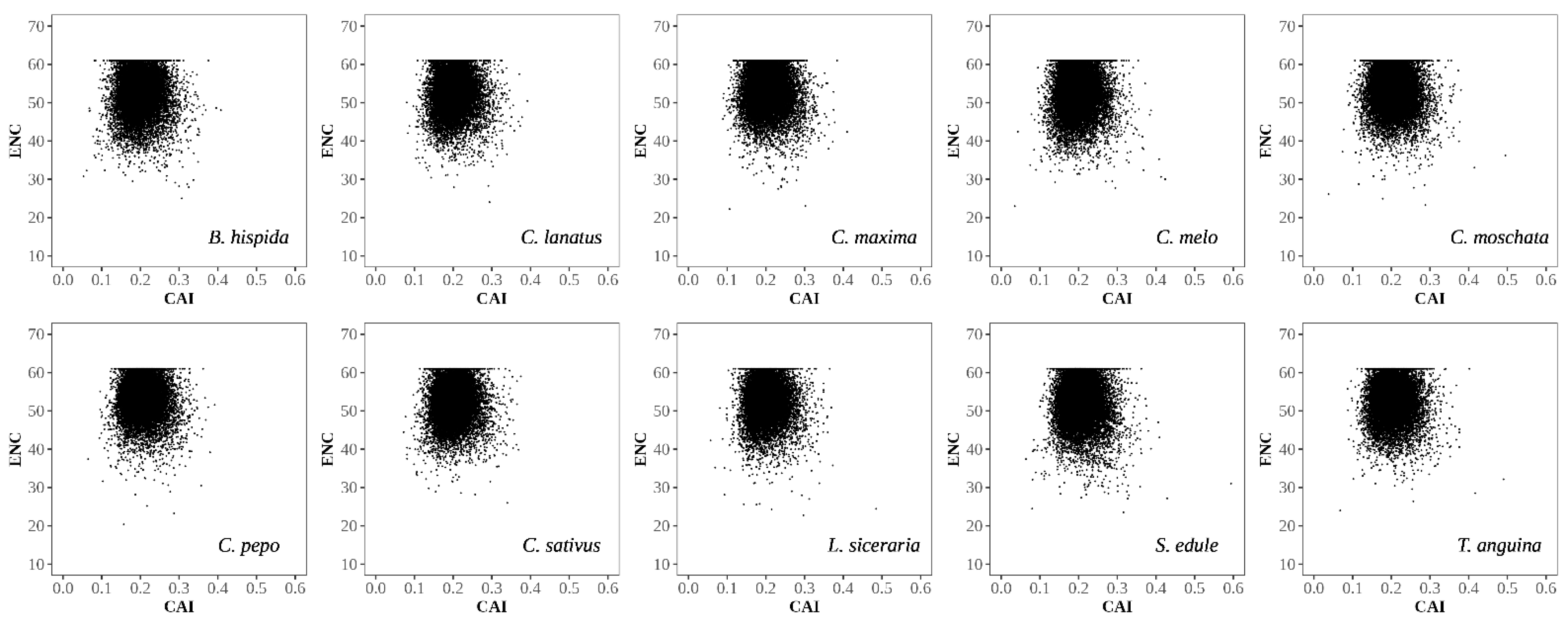
3.2.3. ENC Plot Analysis
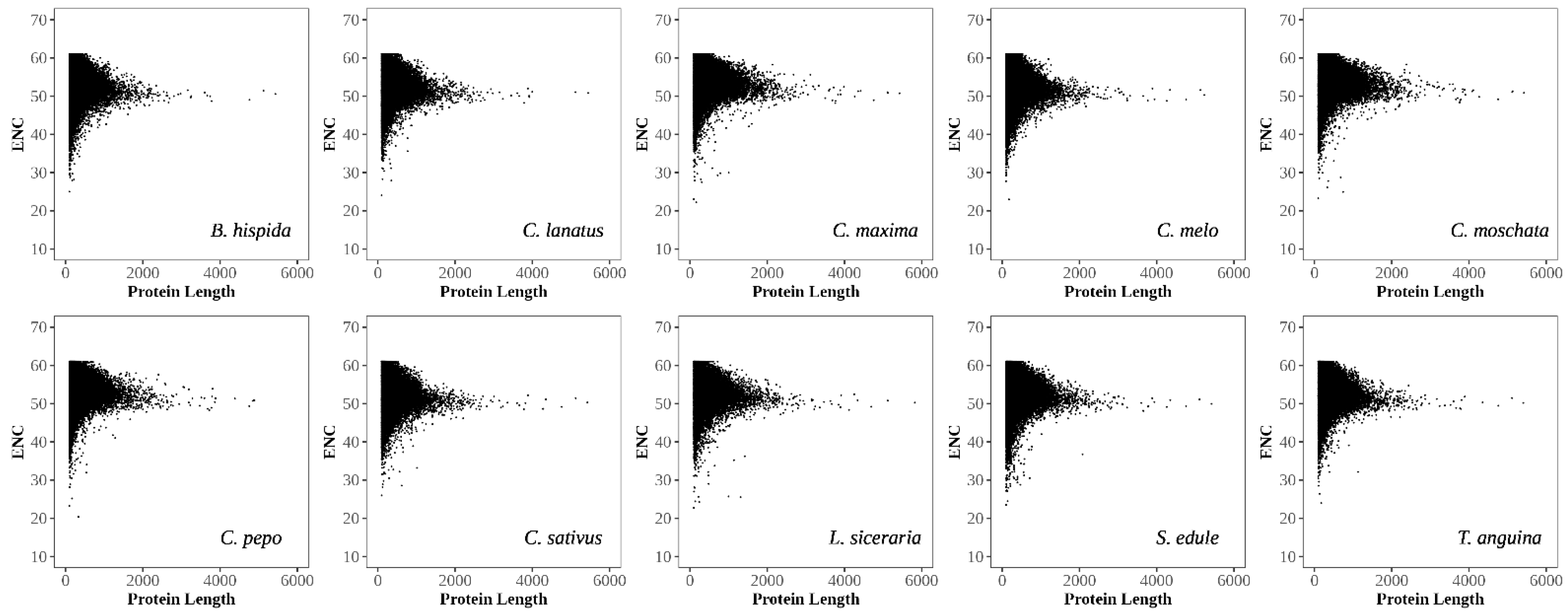
3.2.4. Analysis of Gene Expression Level, Protein Length, and Translational Selection
3.2.5. Correlation Analysis
3.3. Application of CUB
3.3.1. Identification of Optimal Codons
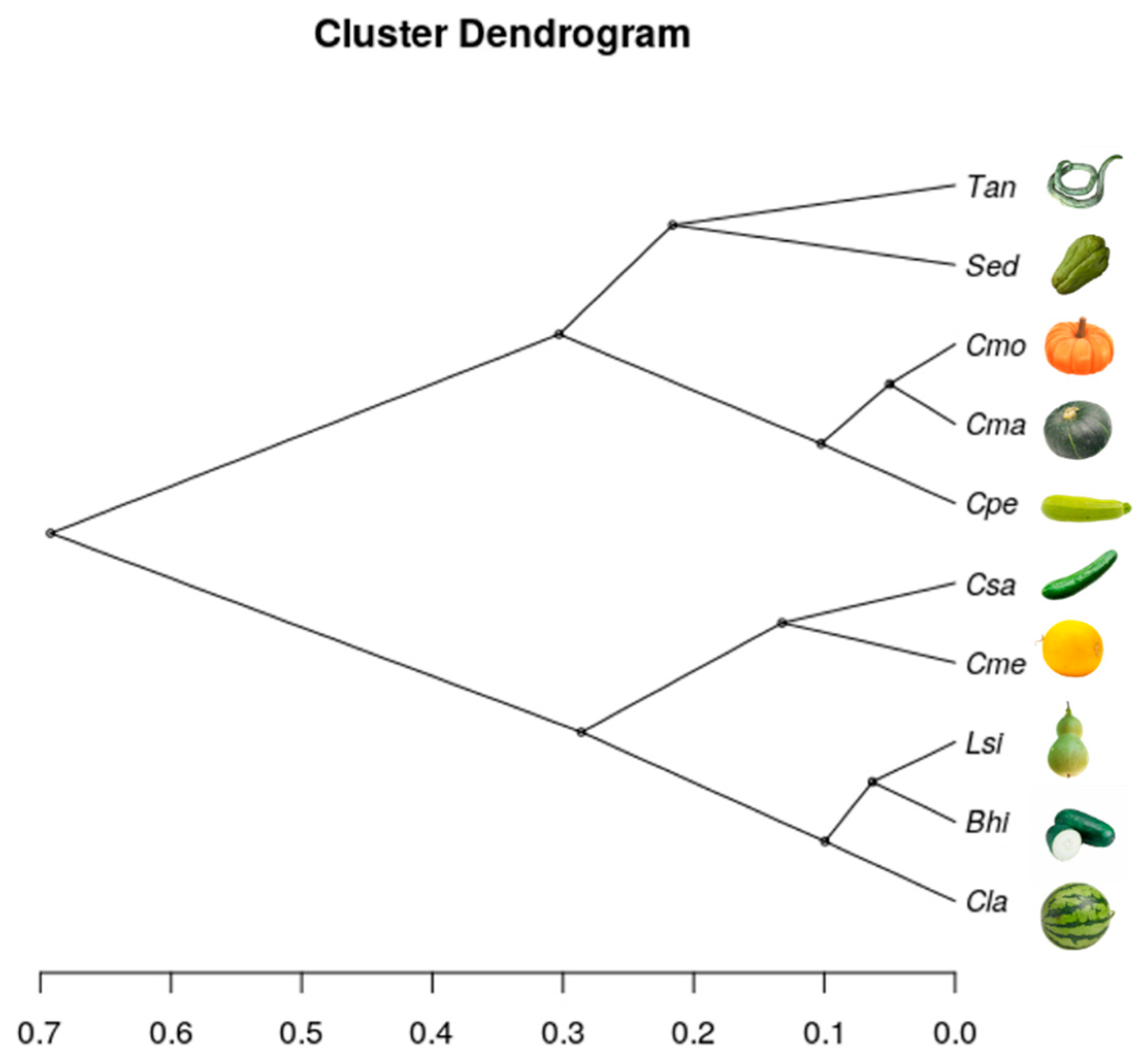
3.3.2. RSCU-Based Cluster Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The Genome of the Cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Zhang, J.; Mu, Z.; Wang, Y.; Wen, C.; Wu, T.; Yu, C.; Li, Z.; Wang, H. Recent Progress on the Molecular Breeding of Cucumis sativus L. in China. Theor. Appl. Genet. 2020, 133, 1777–1790. [Google Scholar] [CrossRef]
- Pawełkowicz, M.; Zieliński, K.; Zielińska, D.; Pląder, W.; Yagi, K.; Wojcieszek, M.; Siedlecka, E.; Bartoszewski, G.; Skarzyńska, A.; Przybecki, Z. Next Generation Sequencing and Omics in Cucumber (Cucumis sativus L.) Breeding Directed Research. Plant Sci. 2016, 242, 77–88. [Google Scholar] [CrossRef]
- Geng, Y.; Jiang, L.; Zhang, Y.; He, Z.; Wang, L.; Peng, Y.; Wang, Y.; Liu, X.; Xu, Y. Assessment of the Dissipation, Pre-Harvest Interval and Dietary Risk of Carbosulfan, Dimethoate, and Their Relevant Metabolites in Greenhouse Cucumber (Cucumis sativus L.). Pest. Manag. Sci. 2018, 74, 1654–1663. [Google Scholar] [CrossRef]
- Tang, L.; Hamid, Y.; Chen, Z.; Lin, Q.; Shohag, M.J.I.; He, Z.; Yang, X. A Phytoremediation Coupled with Agro-Production Mode Suppresses Fusarium Wilt Disease and Alleviates Cadmium Phytotoxicity of Cucumber (Cucumis sativus L.) in Continuous Cropping Greenhouse Soil. Chemosphere 2021, 270, 128634. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, L.; Golparvar, A.R.; Nasr-Esfahani, M.; Golabadi, M. Expression Analysis of Defense-Related Genes in Cucumber (Cucumis sativus L.) against Phytophthora Melonis. Mol. Biol. Rep. 2020, 47, 4933–4944. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, Q.; Wang, X.; Meng, X.; Yu, Y.; Fan, H.; Cui, N. Mildew Resistance Locus O Genes CsMLO1 and CsMLO2 Are Negative Modulators of the Cucumis sativus Defense Response to Corynespora Cassiicola. Int. J. Mol. Sci. 2019, 20, 4793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Guo, S.; Wang, Y.; Wang, L.; Shu, S.; Sun, J. Systematic Identification and Analysis of Heat-Stress-Responsive LncRNAs, CircRNAs and MiRNAs with Associated Co-Expression and CeRNA Networks in Cucumber (Cucumis sativus L.). Physiol. Plant 2020, 168, 736–754. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Ahmed, S.; Ali, A.; Yasin, N.A. 2-Hydroxymelatonin Mitigates Cadmium Stress in Cucumis sativus Seedlings: Modulation of Antioxidant Enzymes and Polyamines. Chemosphere 2020, 243, 125308. [Google Scholar] [CrossRef] [PubMed]
- Borlotti, A.; Vigani, G.; Zocchi, G. Iron Deficiency Affects Nitrogen Metabolism in Cucumber (Cucumis sativus L.) Plants. BMC Plant Biol. 2012, 12, 189. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Zhao, H.; Shi, J.; Li, J.; Nie, X.; Yang, G. Effects of 2,4-Dichlorophenoxyacetic Acid on Cucumber Fruit Development and Metabolism. Int. J. Mol. Sci. 2019, 20, 1126. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, H.; Huang, W.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A Chromosome-Scale Genome Assembly of Cucumber (Cucumis sativus L.). Gigascience 2019, 8, giz072. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Zhang, Z.; Lou, Q.; Xia, L.; Li, J.; Li, M.; Zhou, J.; Zhao, X.; Xu, Y.; Li, Q.; et al. Chromosome-Scale Genome Assembly of Cucumis Hystrix-a Wild Species Interspecifically Cross-Compatible with Cultivated Cucumber. Hortic. Res. 2021, 8, 40. [Google Scholar] [CrossRef]
- Yu, X.; Wang, P.; Li, J.; Zhao, Q.; Ji, C.; Zhu, Z.; Zhai, Y.; Qin, X.; Zhou, J.; Yu, H.; et al. Whole-Genome Sequence of Synthesized Allopolyploids in Cucumis Reveals Insights into the Genome Evolution of Allopolyploidization. Adv. Sci. (Weinh) 2021, 8, 2004222. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mas, J.; Benjak, A.; Sanseverino, W.; Bourgeois, M.; González, V.; Henaff, E.; Camara, F.; Cozzuto, L.; Lowy, E.; Alioto, T.; et al. The Genome of Melon (Cucumis melo L.). Proc. Natl. Acad. Sci. USA 2012, 109, 11872–11877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z.; et al. The Draft Genome of Watermelon (Citrullus lanatus) and Resequencing of 20 Diverse Accessions. Nat. Genet. 2013, 45, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Urasaki, N.; Takagi, H.; Natsume, S.; Uemura, A.; Taniai, N.; Miyagi, N.; Fukushima, M.; Suzuki, S.; Tarora, K.; Tamaki, M.; et al. Draft Genome Sequence of Bitter Gourd (Momordica charantia), a Vegetable and Medicinal Plant in Tropical and Subtropical Regions. DNA Res. 2016, 24, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Shamimuzzaman, M.; Sun, H.; Salse, J.; Sui, X.; Wilder, A.; Wu, Z.; Levi, A.; Xu, Y.; Ling, K.; et al. The Bottle Gourd Genome Provides Insights into Cucurbitaceae Evolution and Facilitates Mapping of a Papaya Ring-spot Virus Resistance Locus. Plant J. 2017, 92, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Wu, S.; Zhang, G.; Jiao, C.; Guo, S.; Ren, Y.; Zhang, J.; Zhang, H.; Gong, G.; Jia, Z.; et al. Karyotype Stability and Unbiased Fractionation in the Paleo-Allotetraploid Cucurbita Genomes. Mol. Plant 2017, 10, 1293–1306. [Google Scholar] [CrossRef] [Green Version]
- Montero-Pau, J.; Blanca, J.; Bombarely, A.; Ziarsolo, P.; Esteras, C.; Martí-Gómez, C.; Ferriol, M.; Gómez, P.; Jamilena, M.; Mueller, L.; et al. De Novo Assembly of the Zucchini Genome Reveals a Whole-Genome Duplication Associated with the Origin of the Cucurbita Genus. Plant Biotechnol. J. 2018, 16, 1161–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera-Redondo, J.; Ibarra-Laclette, E.; Vázquez-Lobo, A.; Gutiérrez-Guerrero, Y.T.; Sánchez de la Vega, G.; Piñero, D.; Montes-Hernández, S.; Lira-Saade, R.; Eguiarte, L.E. The Genome of Cucurbita argyrosperma (Silver-Seed Gourd) Reveals Faster Rates of Protein-Coding Gene and Long Noncoding RNA Turnover and Neofunctionalization within Cucurbita. Mol. Plant 2019, 12, 506–520. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Wang, Q.; Mu, J.; Fu, A.; Wen, C.; Zhao, X.; Gao, L.; Li, J.; Shi, K.; Wang, Y.; et al. The Genome and Transcriptome Analysis of Snake Gourd Provide Insights into Its Evolution and Fruit Development and Ripening. Hortic. Res. 2020, 7, 199. [Google Scholar] [CrossRef]
- Fu, A. Combined Genomic, Transcriptomic, and Metabolomic Analyses Provide Insights into Chayote (Sechium edule) Evolution and Fruit Development. Hortic. Res. 2021, 8, 1–15. [Google Scholar] [CrossRef]
- Camiolo, S.; Melito, S.; Porceddu, A. New Insights into the Interplay between Codon Bias Determinants in Plants. DNA Res. 2015, 22, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Zenan, S.; Gan, Z.; Zhang, F.; Yi, X.; Zhang, J.; Wan, X. Analysis of Codon Usage Patterns in Citrus Based on Coding Sequence Data. BMC Genom. 2020, 21, 234. [Google Scholar] [CrossRef]
- Chenkang, Y.; Zhao, Q.; Wang, Y.; Zhao, J.; Qiao, L.; Wu, B.; Yan, S.; Zheng, J.; Zheng, X. Comparative Analysis of Genomic and Transcriptome Sequences Reveals Divergent Patterns of Codon Bias in Wheat and Its Ancestor Species. Front. Genet. 2021, 12, 732432. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. Codon Usage in Regulatory Genes in Escherichia Coli Does Not Reflect Selection for “rare” Codons. Nucleic. Acids. Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Li, W.H. The Codon Adaptation Index--a Measure of Directional Synonymous Codon Usage Bias, and Its Potential Applications. Nucleic. Acids. Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Carbone, A.; Zinovyev, A.; Képès, F. Codon Adaptation Index as a Measure of Dominating Codon Bias. Bioinformatics 2003, 19, 2005–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, F. The ‘Effective Number of Codons’ Used in a Gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Ikemura, T. Codon Usage and TRNA Content in Unicellular and Multicellular Organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Hall, B.D. Codon Selection in Yeast. J. Biol. Chem. 1982, 257, 3026–3031. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulmer, M. The Selection-Mutation-Drift Theory of Synonymous Codon Usage. Genetics 1991, 129, 897–907. [Google Scholar] [CrossRef]
- Wang, H.-C.; Hickey, D. Rapid Divergence of Codon Usage Patterns within the Rice Genome. BMC Evol. Biol. 2007, 7 (Suppl. S1), S6. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; He, R.; Zhang, H.; Huang, Y.; Tian, M.; Junjie, Z. Analysis of Synonymous Codon Usage in Zea Mays. Mol. Biol. Rep. 2009, 37, 677–684. [Google Scholar] [CrossRef]
- Li, N.; Sun, M.; Jiang, Z.; Shu, H.; Zhang, S. Genome-Wide Analysis of the Synonymous Codon Usage Patterns in Apple. J. Integr. Agric. 2016, 15, 983–991. [Google Scholar] [CrossRef]
- Clepet, C.; Joobeur, T.; Zheng, Y.; Jublot, D.; Huang, M.; Truniger, V.; Boualem, A.; Hernandez-Gonzalez, M.E.; Dolcet-Sanjuan, R.; Portnoy, V.; et al. Analysis of Expressed Sequence Tags Generated from Full-Length Enriched CDNA Libraries of Melon. BMC Genom. 2011, 12, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 1999. [Google Scholar]
- Fuglsang, A. The “effective Number of Codons” Revisited. Biochem. Biophys. Res. Commun 2004, 317, 957–964. [Google Scholar] [CrossRef]
- Duret, L. TRNA Gene Number and Codon Usage in the C. Elegans Genome Are Co-Adapted for Optimal Translation of Highly Expressed Genes. Trends. Genet. 2000, 16, 287–289. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.-H. An Evolutionary Perspective on Synonymous Codon Usage in Unicellular Organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- McInerney, J.O. GCUA: General Codon Usage Analysis. Bioinformatics 1998, 14, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Directional Mutation Pressure and Neutral Molecular Evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueoka, N. Directional Mutation Pressure, Mutator Mutations, and Dynamics of Molecular Evolution. J. Mol. Evol. 1993, 37, 137–153. [Google Scholar] [CrossRef]
- Novembre, J.A. Accounting for Background Nucleotide Composition When Measuring Codon Usage Bias. Mol. Biol. Evol. 2002, 19, 1390–1394. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, S.; Zhang, B.; Wei, W. Analysis of Synonymous Codon Usage Bias of Zika Virus and Its Adaption to the Hosts. PLoS ONE 2016, 11, e0166260. [Google Scholar] [CrossRef] [PubMed]
- Mcewan, N.; Gatherer, D. Codon Indices as a Predictor of Gene Functionality in a Frankia Operon. Can. J. Bot. 2011, 77, 1287–1292. [Google Scholar] [CrossRef]
- Gatherer, D.; McEwan, N. Small Regions of Preferential Codon Usage and Their Effect on Overall Codon Bias—The Case of the Plp Gene. Tenn. Baptist. Mission. Board 1997, 43, 107–114. [Google Scholar] [CrossRef]
- Wu, Y.; Zhao, D.; Tao, J. Analysis of Codon Usage Patterns in Herbaceous Peony (Paeonia lactiflora Pall.) Based on Transcriptome Data. Genes 2015, 6, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Qiao, Z.; Wang, X.; Zeng, H.; Li, Y.; Cai, N.; Chen, Y. Analysis of Codon Usage Patterns in “Lonicerae Flos” (Lonicera macranthoides Hand. -Mazz.) Based on Transcriptome Data. Gene 2019, 705, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, D.; Terrazas, T.; Grego, D.; Arias, S. Phylogeny in Echinocereus (Cactaceae) Based on Combined Morphological and Molecular Evidence: Taxonomic Implications. Syst. Biodivers. 2017, 16, 28–44. [Google Scholar] [CrossRef]
- Horiike, T. An introduction to molecular phylogenetic analysis. Rev. Agric. Sci. 2016, 4, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xing, H.; Yuan, Y.; Wang, X.; Saeed, M.; Tao, J.; Feng, W.; Zhang, G.; Song, X.; Sun, X. Genome-Wide Analysis of Codon Usage Bias in Four Sequenced Cotton Species. PLoS ONE 2018, 13, e0194372. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Common Names | Abbreviations | CDS Numbers | Sequence Source | |
|---|---|---|---|---|---|
| Before Selection | After Selection | ||||
| Benincasa hispida | Wax gourd | Bhi | 27467 | 19865 | CuGenDB |
| Citrullus lanatus | Watermelon | Cla | 22596 | 19904 | CuGenDB |
| Cucurbita maxima | Rimu | Cma | 32076 | 27769 | CuGenDB |
| Cucumis melo | Melon | Cme | 29980 | 21959 | CuGenDB |
| Cucurbita moschata | Rifu | Cmo | 32205 | 28423 | CuGenDB |
| Cucurbita pepo | Zucchini | Cpe | 27868 | 22990 | CuGenDB |
| Cucumis sativus | Cucumber | Csa | 24317 | 20274 | CuGenDB |
| Lagenaria siceraria | Bottle gourd | Lsi | 22472 | 19307 | CuGenDB |
| Sechium edule | Chayote | Sed | 28237 | 26761 | CuGenDB |
| Trichosanthes anguina | Snake gourd | Tan | 22874 | 21541 | CuGenDB |
| Species | SSU | WWU | SSC | WWC | P2 |
|---|---|---|---|---|---|
| B. hispida | 4.92 | 5.08 | 2.67 | 3.21 | 0.5120 |
| C. lanatus | 4.90 | 5.06 | 2.71 | 3.24 | 0.5116 |
| C. maxima | 4.81 | 4.85 | 2.83 | 3.46 | 0.5185 |
| C. melo | 5.06 | 5.10 | 2.57 | 3.17 | 0.5176 |
| C. moschata | 4.81 | 4.83 | 2.83 | 3.47 | 0.5194 |
| C. pepo | 4.88 | 4.86 | 2.79 | 3.46 | 0.5216 |
| C. sativus | 5.11 | 5.11 | 2.53 | 3.17 | 0.5201 |
| L. siceraria | 4.92 | 5.08 | 2.70 | 3.21 | 0.5110 |
| S. edule | 4.68 | 4.96 | 2.93 | 3.36 | 0.5047 |
| T. anguina | 4.76 | 5.00 | 2.81 | 3.30 | 0.5079 |
| Amino Acids | Optimal Codons | ||||
|---|---|---|---|---|---|
| U-Ending | A-Ending | G-Ending | |||
| Ala | GCU | GCA | |||
| Arg | (CGU) | AGA | (CGA) | AGG | |
| Asn | AAU | ||||
| Asp | GAU | ||||
| Cys | UGU | ||||
| Gln | CAA | ||||
| Glu | GAA | ||||
| Gly | GGU | GGA | |||
| His | CAU | ||||
| Ile | AUU | AUA | |||
| Leu | CUU | UUA | CUA | UUG | |
| Lys | AAA | ||||
| Phe | UUU | ||||
| Pro | CCU | CCA | |||
| Ser | UCU | AGU | UCA | ||
| Thr | ACU | ACA | |||
| Tyr | UAU | ||||
| Val | GUU | GUA | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, Y.; Luo, Y.; Wang, C.; Liao, W. Deciphering Codon Usage Patterns in Genome of Cucumis sativus in Comparison with Nine Species of Cucurbitaceae. Agronomy 2021, 11, 2289. https://doi.org/10.3390/agronomy11112289
Niu Y, Luo Y, Wang C, Liao W. Deciphering Codon Usage Patterns in Genome of Cucumis sativus in Comparison with Nine Species of Cucurbitaceae. Agronomy. 2021; 11(11):2289. https://doi.org/10.3390/agronomy11112289
Chicago/Turabian StyleNiu, Yuan, Yanyan Luo, Chunlei Wang, and Weibiao Liao. 2021. "Deciphering Codon Usage Patterns in Genome of Cucumis sativus in Comparison with Nine Species of Cucurbitaceae" Agronomy 11, no. 11: 2289. https://doi.org/10.3390/agronomy11112289
APA StyleNiu, Y., Luo, Y., Wang, C., & Liao, W. (2021). Deciphering Codon Usage Patterns in Genome of Cucumis sativus in Comparison with Nine Species of Cucurbitaceae. Agronomy, 11(11), 2289. https://doi.org/10.3390/agronomy11112289