
De novo QTL-seq Identifies Loci Linked to Blanchability in Peanut (Arachis hypogaea) and Refines Previously Identified QTL with Low Coverage Sequence

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Blanching QTL-seq and Validation
2.2. Development of RIL Population and Marker Validation
2.3. Analysis of Published Datasets
2.4. Post-Khufu Filtering
3. Results and Discussion
3.1. Blanchability QTL-seq
3.2. Linkage Drag from Wild Introgressions
3.3. Improving QTL-seq Analysis in Polyploid Crops
3.4. Potential for Genotyping Using Skim Sequencing
3.5. Future Considerations for QTL-seq Using Low Coverage Sequence Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giovannoni, J.J.; Wing, R.A.; Ganal, M.W.; Tanksley, S.D. Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res. 1991, 19, 6553–6558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelmore, R.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [Green Version]
- Borevitz, J.O.; Liang, D.; Plouffe, D.; Chang, H.S.; Zhu, T.; Weigel, D.; Berry, C.C.; Winzeler, E.; Chory, J. Large-scale identification of single-feature polymorphisms in complex genomes. Genome Res. 2003, 13, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenreich, I.M.; Torabi, N.; Jia, Y.; Kent, J.; Martis, S.; Shapiro, J.A.; Gresham, D.; Caudy, A.A.; Kruglyak, L. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature 2010, 464, 1039–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossowski, S.; Schneeberger, K.; Clark, R.M.; Lanz, C.; Warthmann, N.; Weigel, D. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008, 18, 2024–2033. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, K.; Ossowski, S.; Lanz, C.; Juul, T.; Petersen, A.H.; Nielsen, K.L.; Jørgensen, J.E.; Weigel, D.; Andersen, S.U. SHOREmap: Simultaneous mapping and mutation identification by deep sequencing. Nat. Methods 2009, 6, 550–551. [Google Scholar] [CrossRef] [PubMed]
- Galvão, V.C.; Nordström, K.J.; Lanz, C.; Sulz, P.; Mathieu, J.; Posé, D.; Schmid, M.; Weigel, D.; Schneeberger, K. Synteny-based mapping-by-sequencing enabled by targeted enrichment. Plant J. Cell Mol. Biol. 2012, 71, 517–526. [Google Scholar] [CrossRef]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [Green Version]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap+: Genetic mapping and mutant identification without crossing in rice. PLoS ONE 2013, 8, e68529. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Illa-Berenguer, E.; Van Houten, J.; Huang, Z.; van der Knaap, E. Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 2015, 128, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Upadhyaya, H.D.; Bajaj, D.; Kujur, A.; Badoni, S.; Laxmi Kumar, V.; Tripathi, S.; Gowda, C.L.; Sharma, S.; Singh, S.; et al. Deploying QTL-seq for rapid delineation of a potential candidate gene underlying major trait-associated QTL in chickpea. DNA Res. 2015, 22, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Lin, T.; Klein, J.; Wang, S.; Qi, J.; Zhou, Q.; Sun, J.; Zhang, Z.; Weng, Y.; Huang, S. QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber. TAG Theor. Appl. Genet. 2014, 127, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hai, J.; Yang, J.; Tian, J.; Chen, W.; Chen, T.; Luo, H.; Wang, H. Influence of leaf and silique photosynthesis on seeds yield and seeds oil quality of oilseed rape (Brassica napus L.). Eur. J. Agron. 2016, 74, 112–118. [Google Scholar] [CrossRef]
- Tamiru, M.; Natsume, S.; Takagi, H.; White, B.; Yaegashi, H.; Shimizu, M.; Yoshida, K.; Uemura, A.; Oikawa, K.; Abe, A.; et al. Genome sequencing of the staple food crop white Guinea yam enables the development of a molecular marker for sex determination. BMC Biol. 2017, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Gimode, W.; Clevenger, J.; McGregor, C. Fine-mapping of a major quantitative trait locus Qdff3-1 controlling flowering time in watermelon. Mol. Breed. 2020, 40, 3. [Google Scholar] [CrossRef]
- Pandey, M.K.; Khan, A.W.; Singh, V.K.; Vishwakarma, M.K.; Shasidhar, Y.; Kumar, V.; Garg, V.; Bhat, R.S.; Chitikineni, A.; Janila, P.; et al. QTL-seq approach identified genomic regions and diagnostic markers for rust and late leaf spot resistance in groundnut (Arachis hypogaea L.). Plant Biotechnol. J. 2017, 15, 927–941. [Google Scholar] [CrossRef] [Green Version]
- Clevenger, J.; Chu, Y.; Chavarro, C.; Botton, S.; Culbreath, A.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Mapping Late Leaf Spot Resistance in Peanut (Arachis hypogaea) Using QTL-seq Reveals Markers for Marker-Assisted Selection. Front. Plant Sci. 2018, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Janila, P.; Vishwakarma, M.K.; Khan, A.W.; Manohar, S.S.; Gangurde, S.S.; Variath, M.T.; Shasidhar, Y.; Pandey, M.K.; Varshney, R.K. Whole-genome resequencing-based QTL-seq identified candidate genes and molecular markers for fresh seed dormancy in groundnut. Plant Biotechnol. J. 2020, 18, 992–1003. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, J.; Li, M.; Deng, L.; Li, G.; Xia, H.; Zhao, S.; Hou, L.; Li, P.; Ma, C.; et al. Whole-genome resequencing-based QTL-seq identified AhTc1 gene encoding a R2R3-MYB transcription factor controlling peanut purple testa colour. Plant Biotechnol. J. 2020, 18, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Schiessl, S.-V.; Katche, E.; Ihien, E.; Chawla, H.S.; Mason, A.S. The role of genomic structural variation in the genetic improvement of polyploid crops. Crop. J. 2019, 7, 127–140. [Google Scholar] [CrossRef]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 2020, 182, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Nettelblad, C. The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS Genet. 2019, 15, e1008302. [Google Scholar] [CrossRef] [Green Version]
- Glover, N.M.; Redestig, H.; Dessimoz, C. Homoeologs: What Are They and How Do We Infer Them? Trends Plant Sci. 2016, 21, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevenger, J.; Chavarro, C.; Pearl, S.A.; Ozias-Akins, P.; Jackson, S.A. Single Nucleotide Polymorphism Identification in Polyploids: A Review, Example, and Recommendations. Mol. Plant 2015, 8, 831–846. [Google Scholar] [CrossRef] [Green Version]
- Cui, R.; Clevenger, J.; Chu, Y.; Brenneman, T.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Quantitative trait loci sequencing—Derived molecular markers for selection of stem rot resistance in peanut. Crop. Sci. 2020, 60, 2008–2018. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Segawa, T.; Tamiru, M.; Abe, A.; Sakamoto, S.; Uemura, A.; Oikawa, K.; Kutsuzawa, H.; Koga, H.; Imamura, T.; et al. Next-generation sequencing-based bulked segregant analysis for QTL mapping in the heterozygous species Brassica rapa. Theor. Appl. Genet. 2019, 132, 2913–2925. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.C.; Borgognone, M.G.; OConnor, D.J.; Rachaputi, R.C.N.; Henry, R.J.; Furtado, A.; Anglin, N.L.; Freischfresser, D.B. Breeding for improved blanchability in peanut: Phenotyping, genotype × environment interaction and selection. Crop. Pasture Sci. 2018, 69, 1237–1250. [Google Scholar] [CrossRef]
- Cruickshank, A.W.; Tonks, J.W.; Kelly, A.K. Blanchability of peanut (Arachis hypogaea L.) kernels: Early generation selection and genotype stability over three environments. Aust. J. Agric. Res. 2003, 54, 885–888. [Google Scholar] [CrossRef] [Green Version]
- Kovach, M.J.; McCouch, S.R. Leveraging natural diversity: Back through the bottleneck. Curr. Opin. Plant Biol. 2008, 11, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Leal-Bertioli, S.C.; Cavalcante, U.; Gouvea, E.G.; Ballén-Taborda, C.; Shirasawa, K.; Guimarães, P.M.; Jackson, S.A.; Bertioli, D.J.; Moretzsohn, M.C. Identification of QTLs for Rust Resistance in the Peanut Wild Species Arachis magna and the Development of KASP Markers for Marker-Assisted Selection. G3 2015, 5, 1403–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballén-Taborda, C.; Chu, Y.; Ozias-Akins, P.; Timper, P.; Holbrook, C.C.; Jackson, S.A.; Bertioli, D.J.; Leal-Bertioli, S.C.M. A new source of root-knot nematode resistance from Arachis stenosperma incorporated into allotetraploid peanut (Arachis hypogaea). Sci. Rep. 2019, 9, 17702. [Google Scholar] [CrossRef] [PubMed]
- Stalker, H. Utilizing Wild Species for Peanut Improvement. Crop. Sci. 2017, 57, 1102–1120. [Google Scholar] [CrossRef]
- Lamon, S.; Chu, Y.; Guimaraes, L.A.; Bertioli, D.J.; Leal-Bertioli, S.C.; Santos, J.F.; Godoy, I.J.; Culbreath, A.K.; Holbrook, C.C.; Ozias-Akins, P. Characterization of peanut lines with interspecific introgressions conferring late leaf spot resistance. Crop. Sci. 2021, 61, 1724–1738. [Google Scholar] [CrossRef]
- Bertioli, D.J.; Clevenger, J.; Godoy, I.; Stalker, T.; Santos, J.F.; Wood, S.; Abernathy, B.; Azevedo, V.; Campbell, J.; Chu, Y.; et al. Legacy genetics of Arachis cardenasii in the peanut crop. Proc. Natl. Acad. Sci. USA 2021, 118, e2104899118. [Google Scholar] [CrossRef] [PubMed]
- Khedikar, Y.P.; Gowda, M.V.; Sarvamangala, C.; Patgar, K.V.; Upadhyaya, H.D.; Varshney, R.K. A QTL study on late leaf spot and rust revealed one major QTL for molecular breeding for rust resistance in groundnut (Arachis hypogaea L.). Theor. Appl. Genet. 2010, 121, 971–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Cui, R.; Chavarro, C.; Tseng, Y.-C.; Zhou, H.; Peng, Z.; Chu, Y.; Yang, X.; Lopez, Y.; Tillman, B.; et al. Mapping quantitative trait loci (QTLs) and estimating the epistasis controlling stem rot resistance in cultivated peanut (Arachis hypogaea). Theor. Appl. Genet. 2020, 133, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Chee, P.; Culbreath, A.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Major QTLs for Resistance to Early and Late Leaf Spot Diseases Are Identified on Chromosomes 3 and 5 in Peanut (Arachis hypogaea). Front. Plant Sci. 2019, 10, 883. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Pandey, A.K.; Kumar, R.; Nwosu, C.V.; Guo, B.; Wright, G.C.; Bhat, R.S.; Chen, X.; Bera, S.K.; Yuan, M.; et al. Translational genomics for achieving higher genetic gains in groundnut. Theor. Appl. Genet. 2020, 133, 1679–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertioli, D.J.; Abernathy, B.; Seijo, G.; Clevenger, J.; Cannon, S. Evaluating two different models of peanut’s origin. Nat. Genet. 2020, 52, 557–559. [Google Scholar] [CrossRef]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Dudchenko, O.; Gao, D.; Seijo, G.; Leal-Bertioli, S.; Ren, L.; Farmer, A.D.; Pandey, M.K.; et al. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.C.; Zhang, L.; Zhang, X.; Tang, R.; et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, G.; Clevenger, J.; Kale, S.M.; Wang, H.; Pandey, M.K.; Choudhary, D.; Yuan, M.; Wang, X.; Culbreath, A.K.; Holbrook, C.C.; et al. A recombination bin-map identified a major QTL for resistance to Tomato Spotted Wilt Virus in peanut (Arachis hypogaea). Sci. Rep. 2019, 9, 18246. [Google Scholar] [CrossRef] [PubMed]
- Korani, W.; Clevenger, J.P.; Chu, Y.; Ozias-Akins, P. Machine Learning as an Effective Method for Identifying True Single Nucleotide Polymorphisms in Polyploid Plants. Plant Genome 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
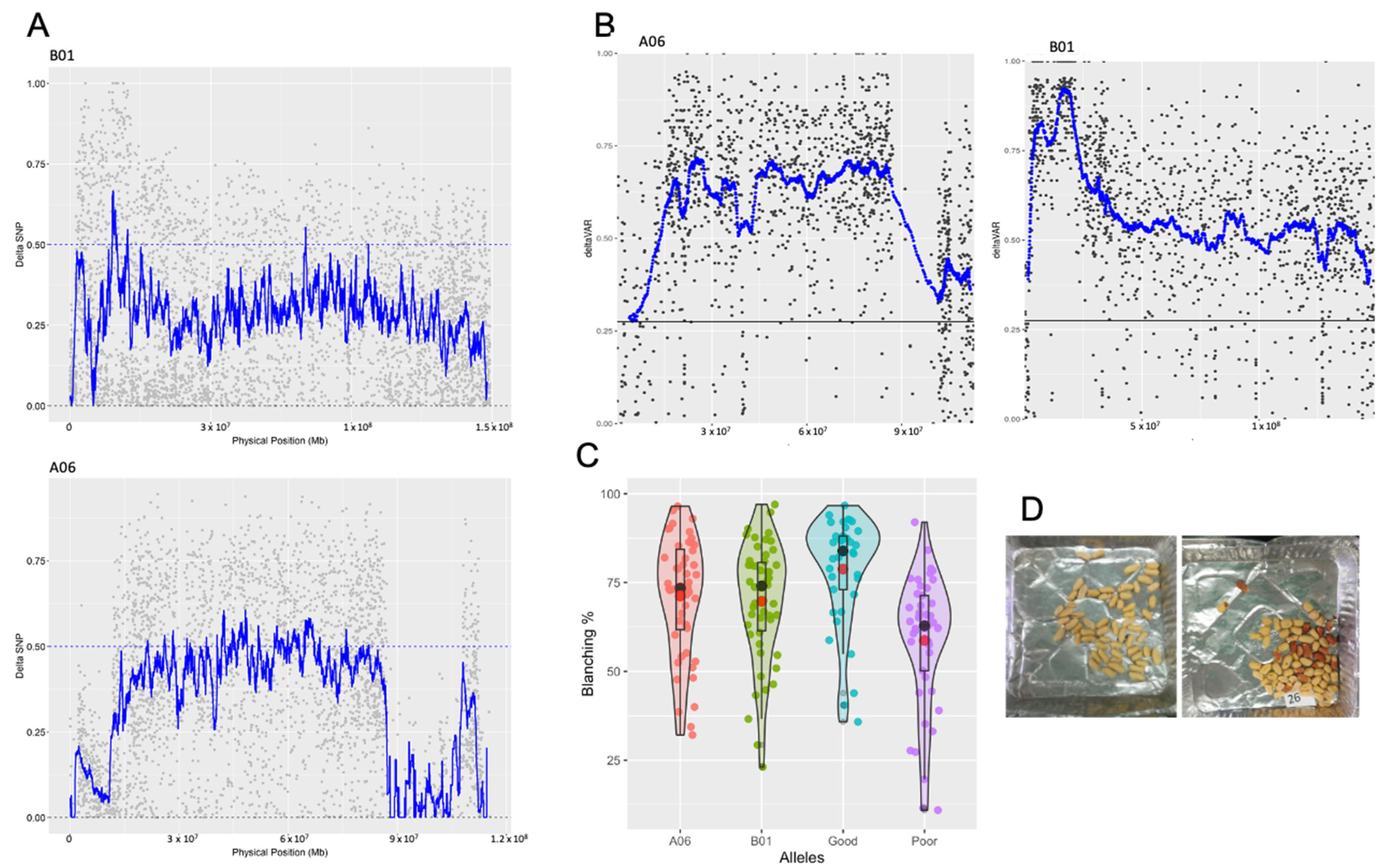


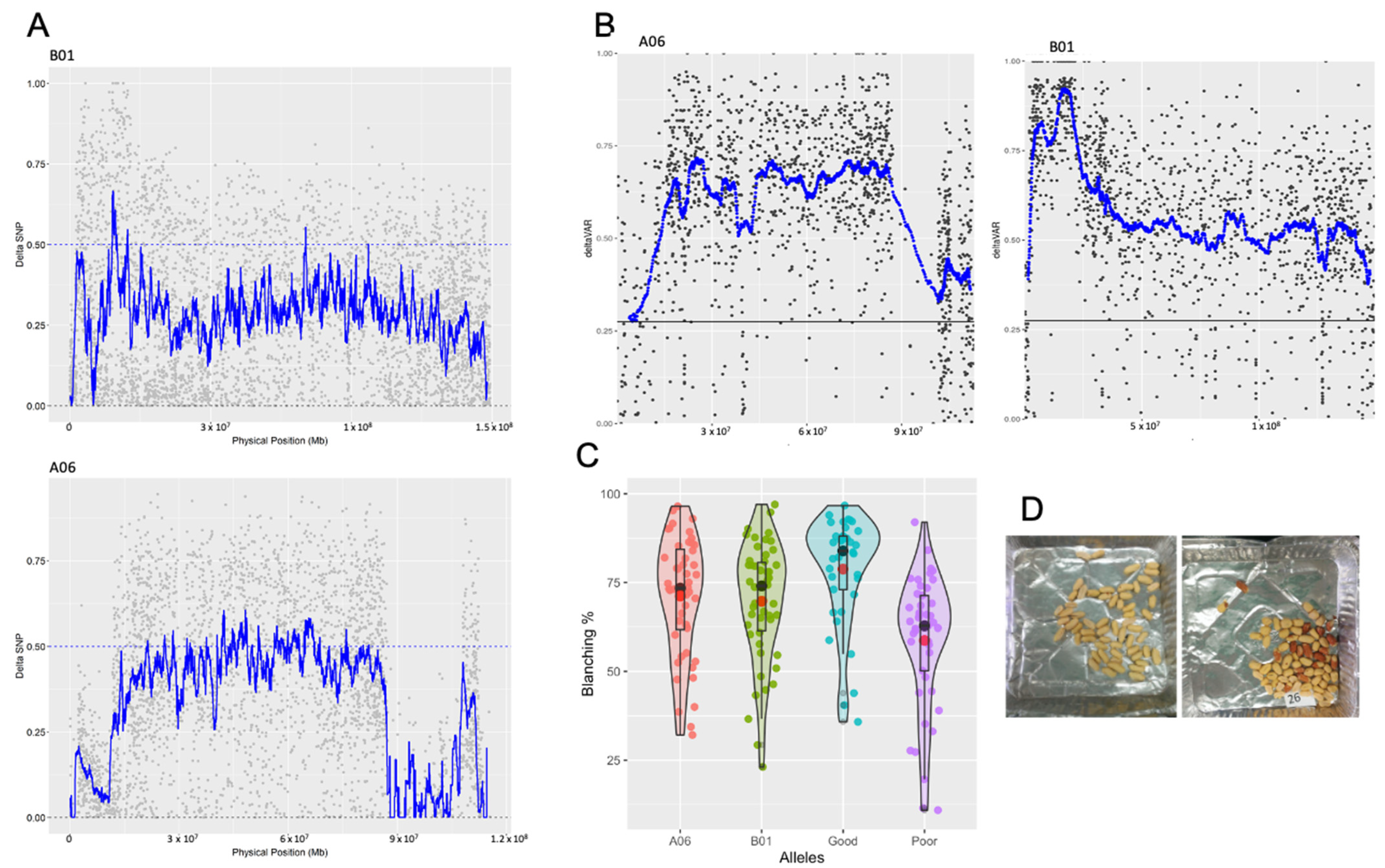{kind=link}
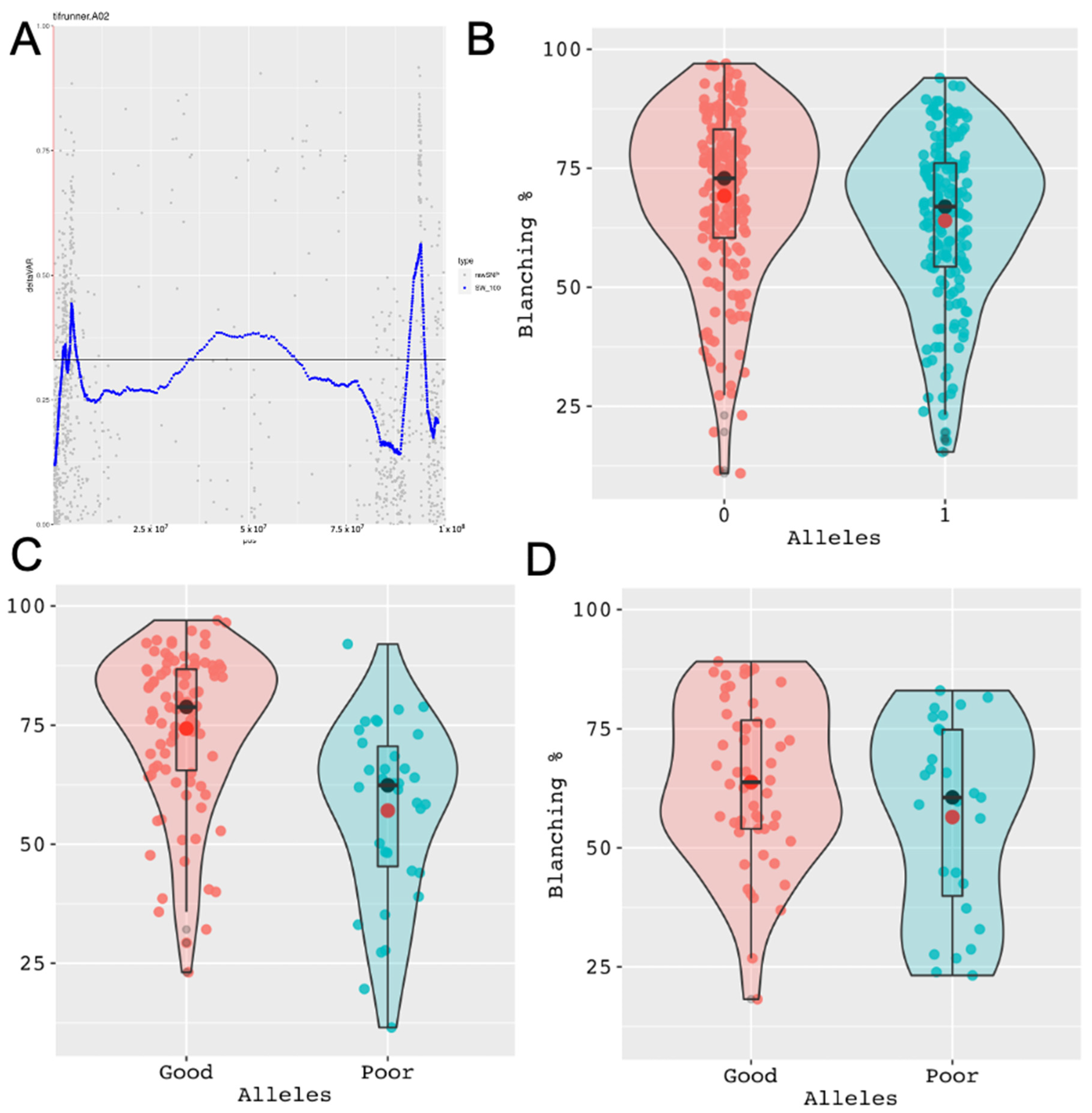
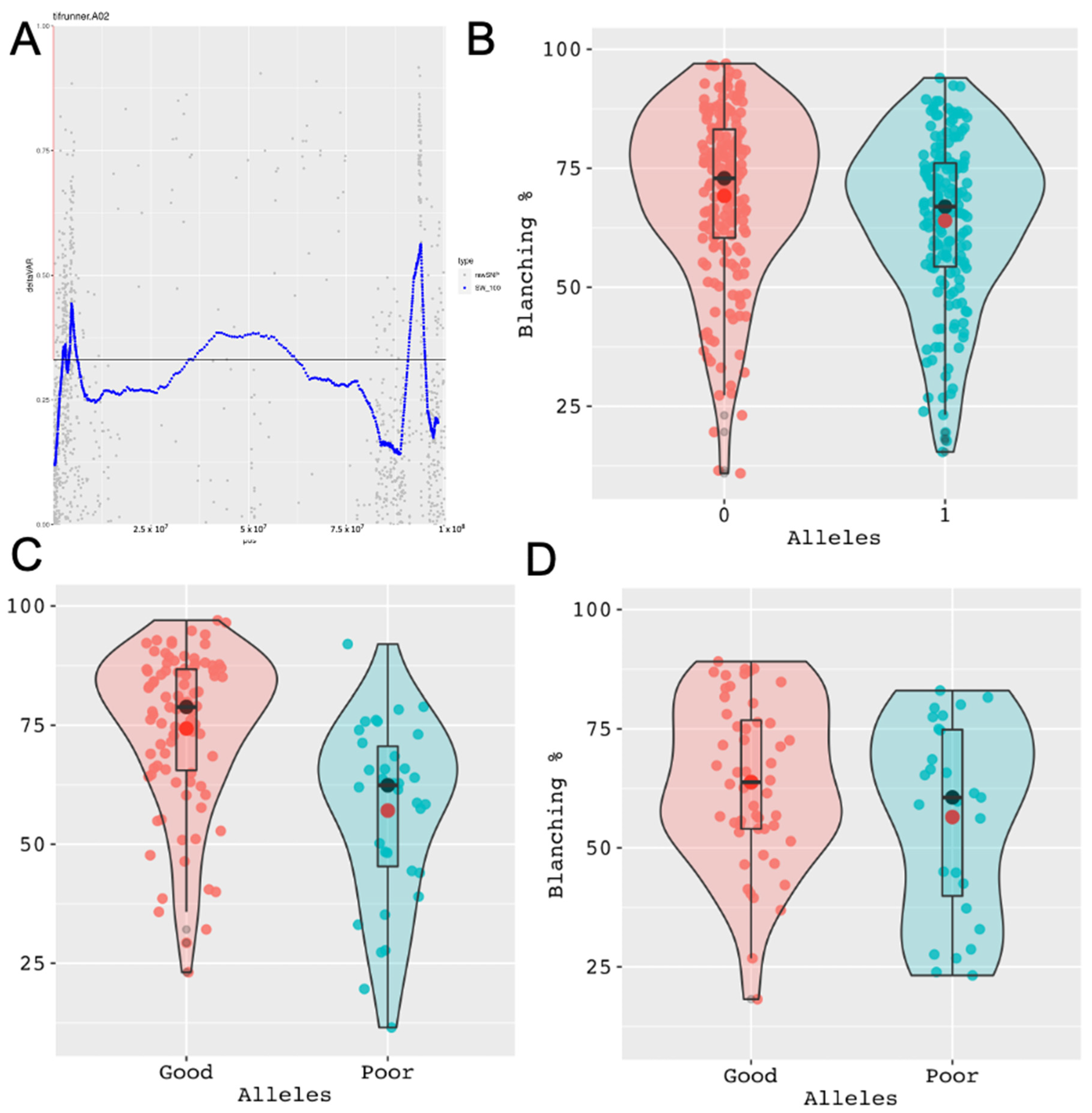{kind=link}
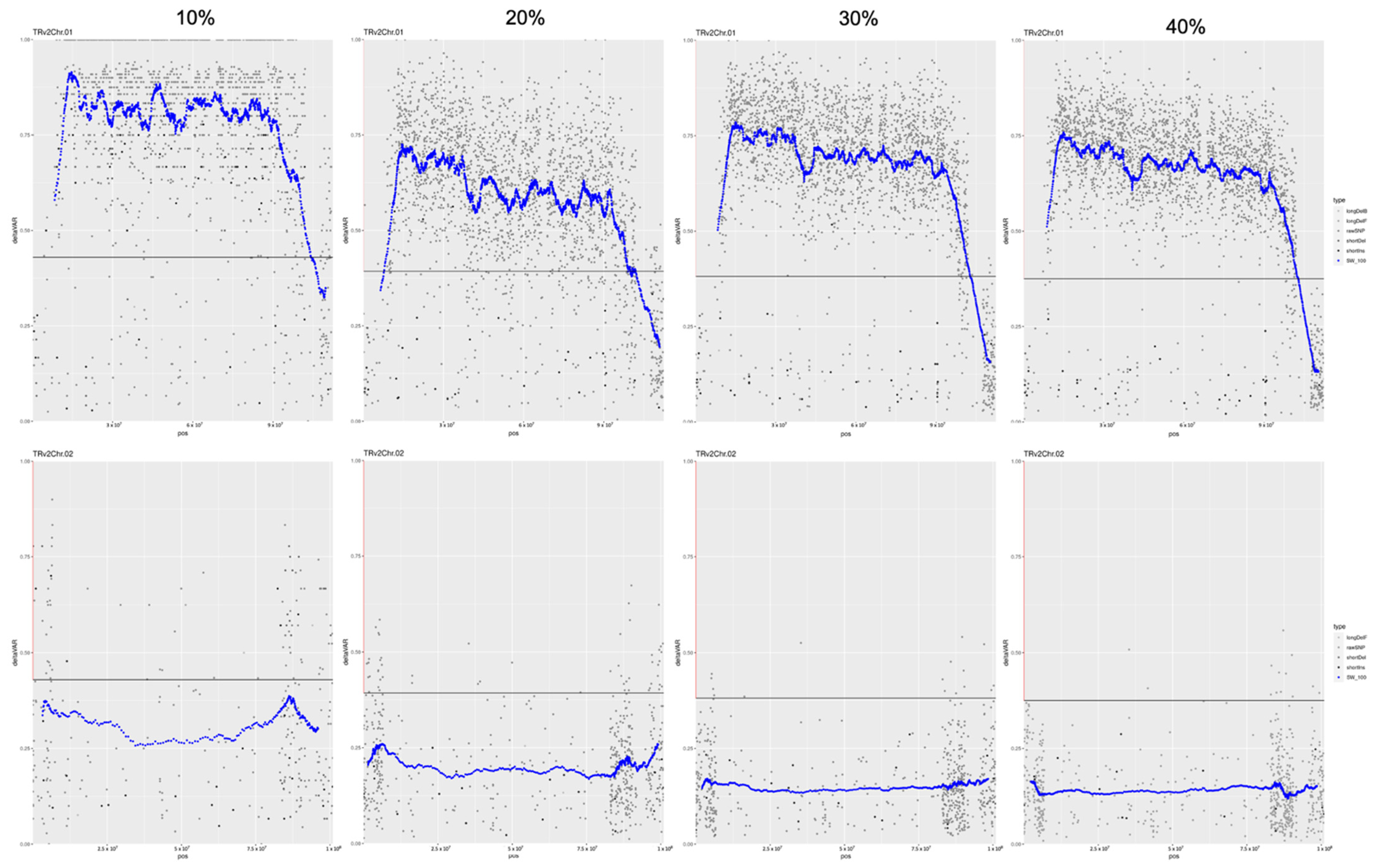{kind=link}
| Genotype | Blanching | Pedigree | Bulk |
|---|---|---|---|
| P24-p188-93 | 55.4 | Tingoora × D147-p3-115 | Poor Blancher |
| P13-p07-219 | 59.6 | Sutherland × Walter | Poor Blancher |
| P24-p187/205-97 | 71.5 | Tingoora × D147-p3-115 | Poor Blancher |
| P23-p157-65 | 74.1 | Redvale × D147-p3-115 | Poor Blancher |
| P13-p45-235 | 76.7 | Sutherland × Walter | Poor Blancher |
| P23-p157-64 | 84.0 | Redvale × D147-p3-115 | Good Blancher |
| P13-p23-233 | 85.2 | Sutherland × Walter | Good Blancher |
| P23-p153-62 | 85.8 | Redvale × D147-p3-115 | Good Blancher |
| P13-p21-223 | 91.0 | Sutherland × Walter | Good Blancher |
| P23-p171-85 | 91.0 | Redvale × D147-p3-115 | Good Blancher |
| Walter | 92.2 | Good Blanching Parent | |
| Redvale | 93.3 | Walter × D45-p37-102 | Good Blanching Parent |
| Sutherland * | 75.4 | Poor Blanching Parent |
| A02 | A03 | A06 + B01 | Blanching (Lsmeans) | Blanching (Median) | Number of Individuals |
|---|---|---|---|---|---|
| car | car | Good | 63.4 | 63.8 | 52 |
| car | hyp | Good | 68.0 | 70.4 | 60 |
| hyp | car | Good | 69.5 | 71.1 | 38 |
| hyp | hyp | Good | 74.3 | 78.9 | 92 |
| car | car | Poor | 56.5 | 60.4 | 27 |
| car | hyp | Poor | 60.9 | 69 | 23 |
| hyp | car | Poor | 57.7 | 67.2 | 12 |
| hyp | hyp | Poor | 57.1 | 62.6 | 34 |
| car | car | - | 63.4 | 62.6 | 79 |
| car | hyp | - | 68.0 | 69.2 | 83 |
| hyp | car | - | 69.5 | 69.7 | 56 |
| hyp | hyp | - | 74.1 | 74.2 | 126 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korani, W.; O’Connor, D.; Chu, Y.; Chavarro, C.; Ballen, C.; Guo, B.; Ozias-Akins, P.; Wright, G.; Clevenger, J. De novo QTL-seq Identifies Loci Linked to Blanchability in Peanut (Arachis hypogaea) and Refines Previously Identified QTL with Low Coverage Sequence. Agronomy 2021, 11, 2201. https://doi.org/10.3390/agronomy11112201
Korani W, O’Connor D, Chu Y, Chavarro C, Ballen C, Guo B, Ozias-Akins P, Wright G, Clevenger J. De novo QTL-seq Identifies Loci Linked to Blanchability in Peanut (Arachis hypogaea) and Refines Previously Identified QTL with Low Coverage Sequence. Agronomy. 2021; 11(11):2201. https://doi.org/10.3390/agronomy11112201
Chicago/Turabian StyleKorani, Walid, Dan O’Connor, Ye Chu, Carolina Chavarro, Carolina Ballen, Baozhu Guo, Peggy Ozias-Akins, Graeme Wright, and Josh Clevenger. 2021. "De novo QTL-seq Identifies Loci Linked to Blanchability in Peanut (Arachis hypogaea) and Refines Previously Identified QTL with Low Coverage Sequence" Agronomy 11, no. 11: 2201. https://doi.org/10.3390/agronomy11112201
APA StyleKorani, W., O’Connor, D., Chu, Y., Chavarro, C., Ballen, C., Guo, B., Ozias-Akins, P., Wright, G., & Clevenger, J. (2021). De novo QTL-seq Identifies Loci Linked to Blanchability in Peanut (Arachis hypogaea) and Refines Previously Identified QTL with Low Coverage Sequence. Agronomy, 11(11), 2201. https://doi.org/10.3390/agronomy11112201