Identification of Non-Pleiotropic Loci in Flowering and Maturity Control in Soybean

 ,
,
Abstract
1. Introduction
2. Materials and Methods
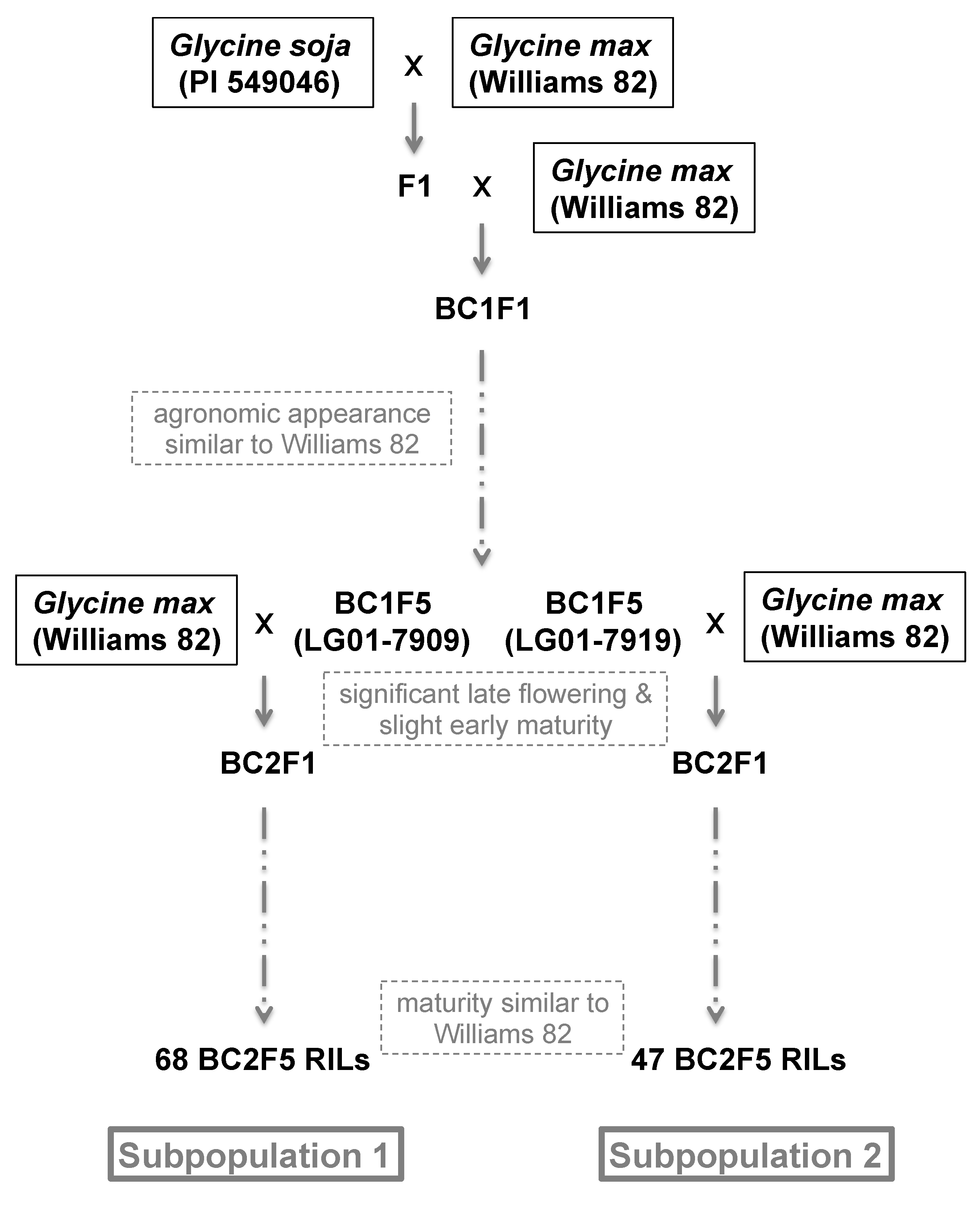
2.1. Mapping Population
2.2. Field Evaluation
2.3. Statistical Analysis of Phenotypic Data
2.4. Genotyping
2.5. SNP Selection
2.6. QTL Mapping
2.7. Candidate Gene Identification
2.8. Plant Growth Condition
3. Results
3.1. Selection for Non-Pleiotropy
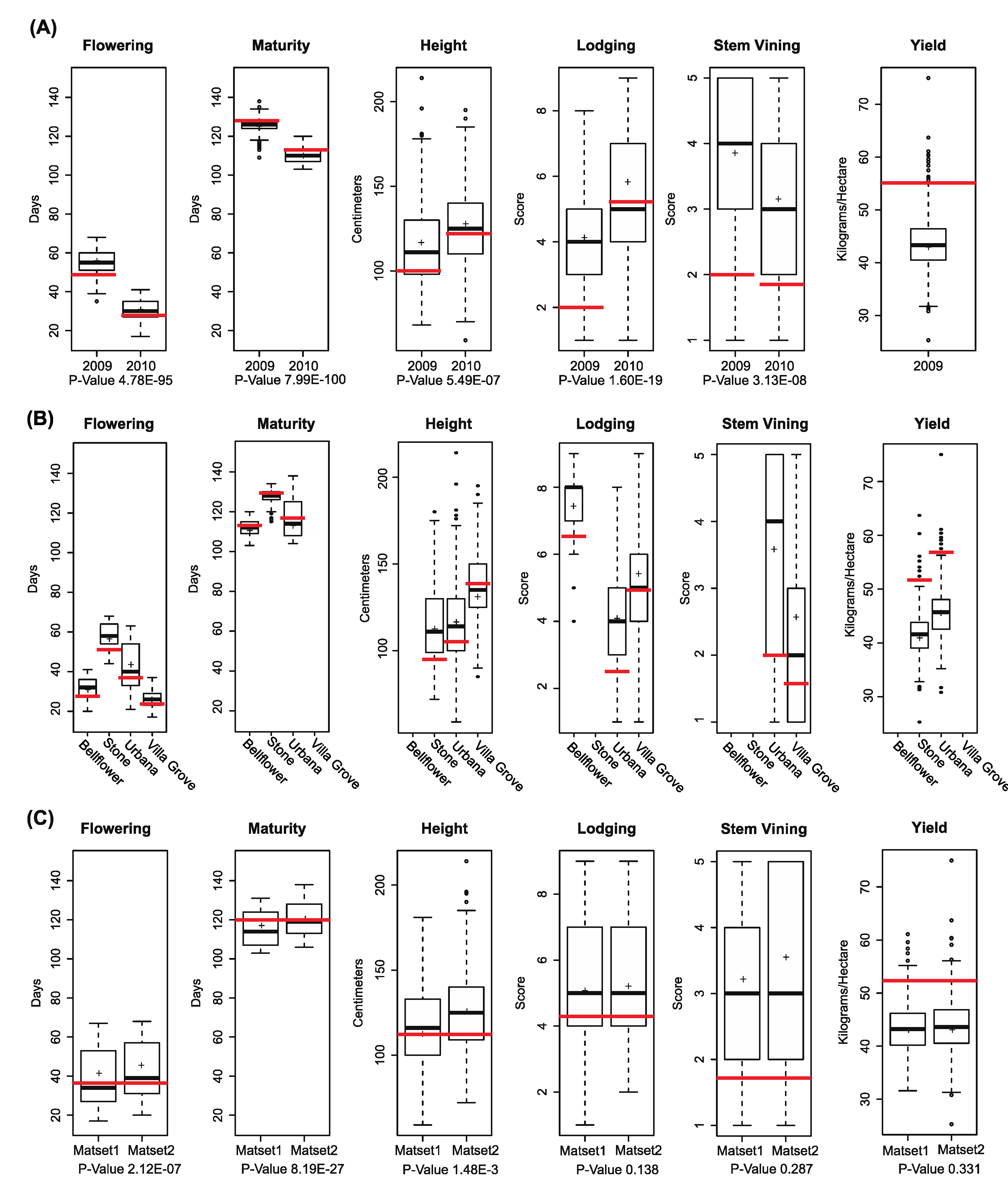
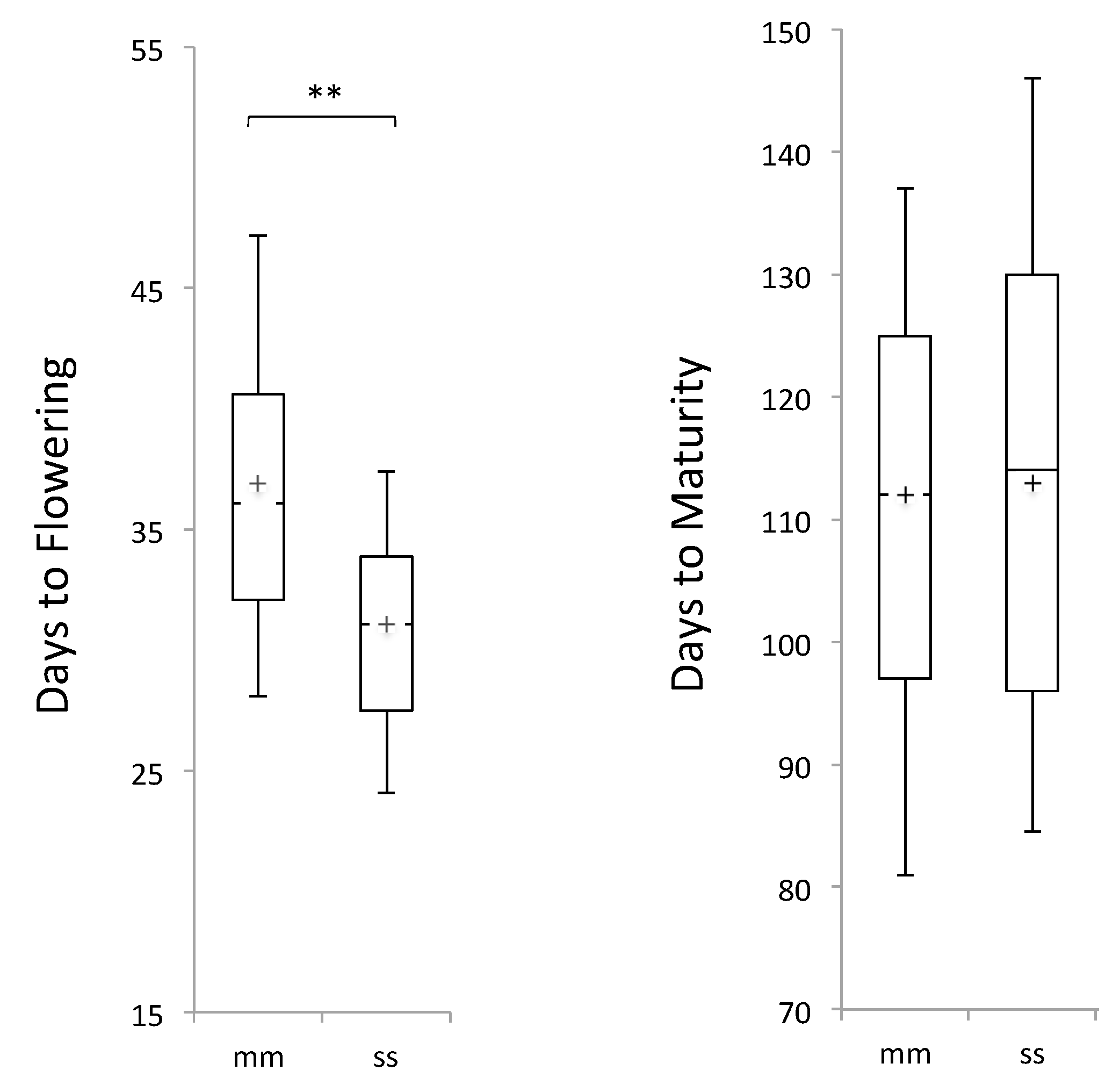
3.2. Phenotypic Distribution
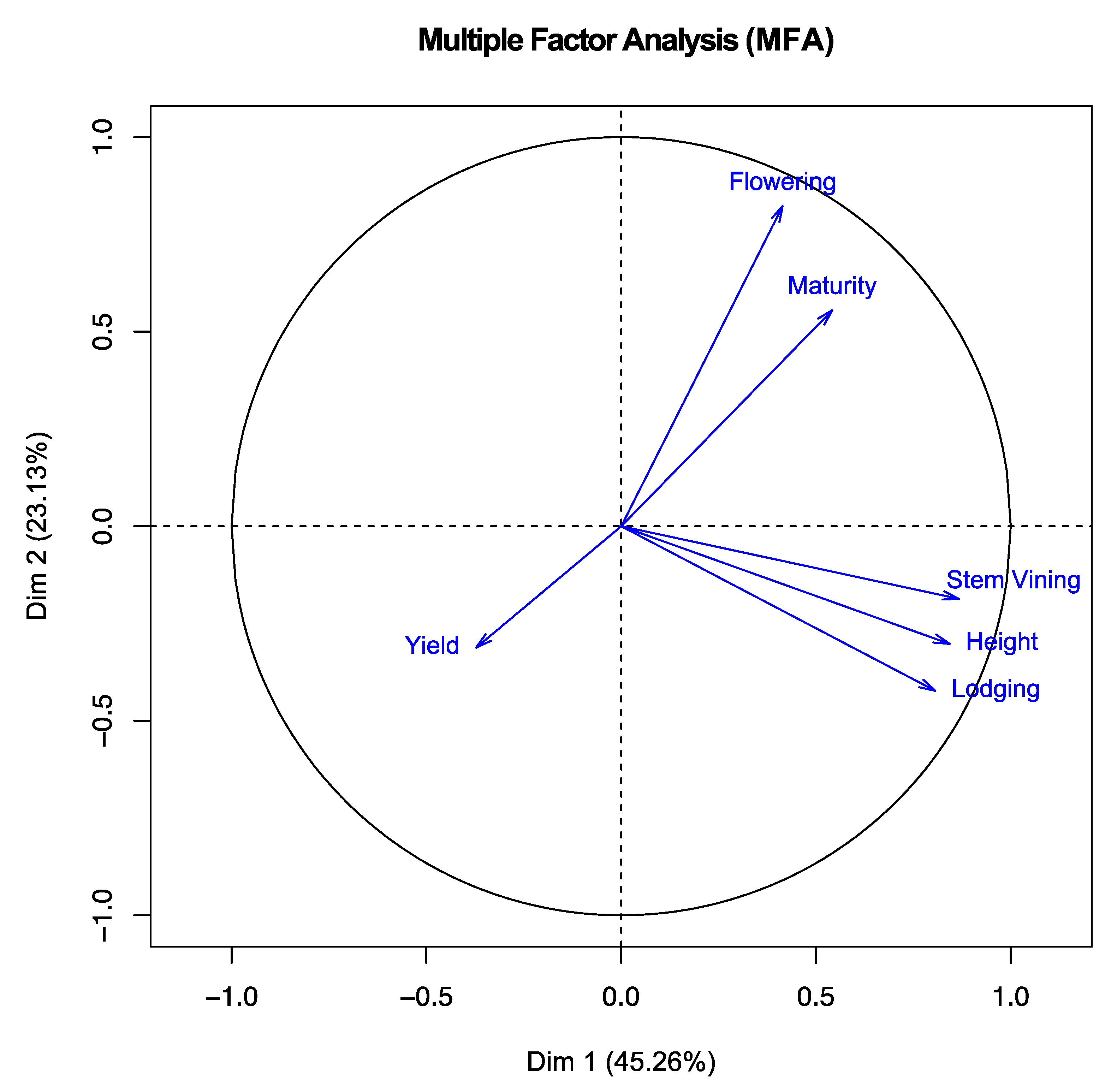
3.3. Multiple Factor Analysis (MFA)
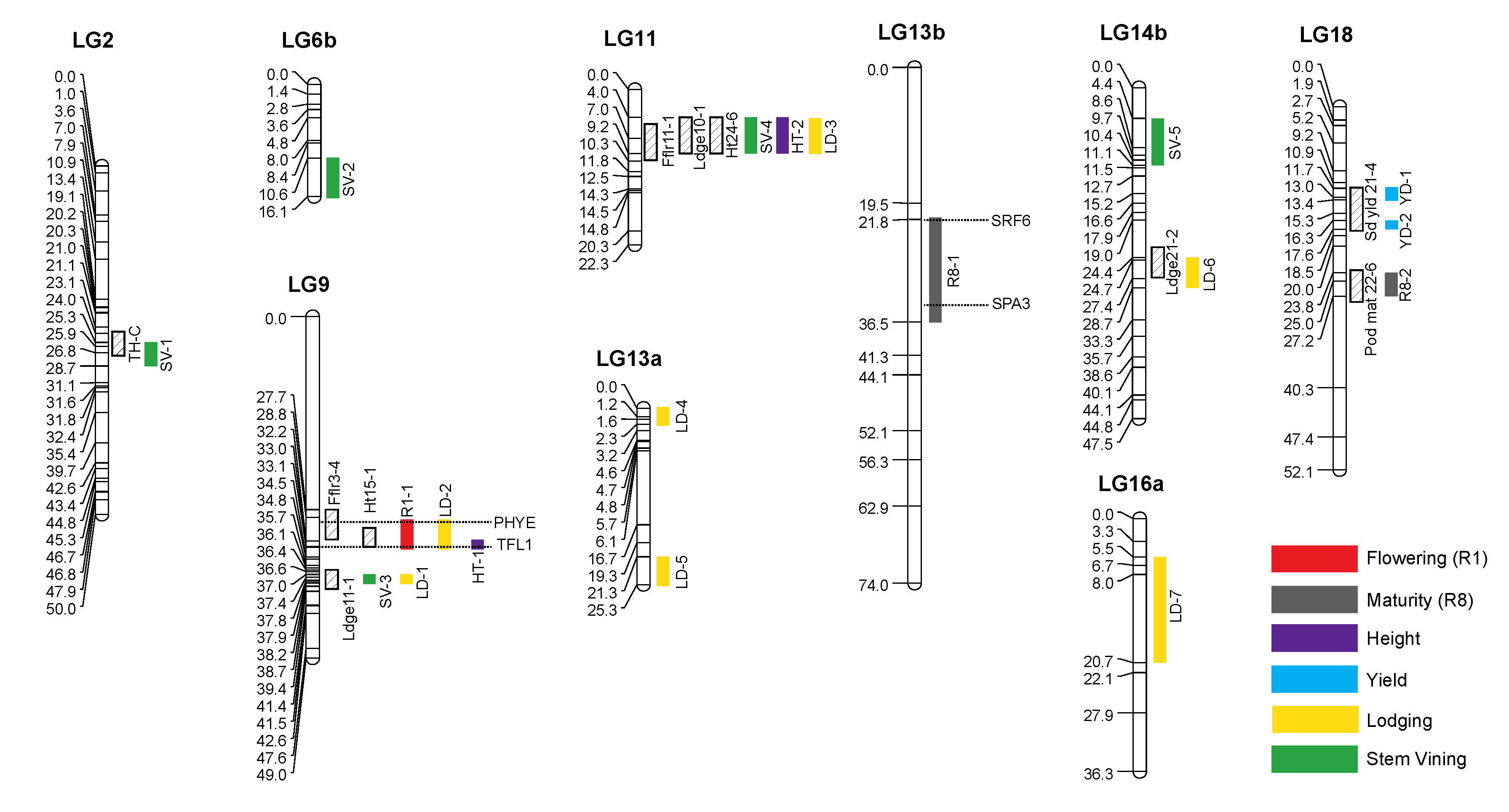
3.4. Construction of Linkage Map
3.5. QTL Mapping
3.6. Pleiotropic QTL and Verification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kimura, M.; Ohta, T. On some principles governing molecular evolution. Proc. Natl. Acad. Sci. USA 1974, 71, 2848–2852. [Google Scholar] [CrossRef]
- Ohta, T.; Gillespie, J.H. Development of neutral and nearly neutral theories. Theor. Popul. Biol. 1996, 49, 128–142. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kenney-Hunt, J.P.; Pavlicev, M.; Peck, J.R.; Waxman, D.; Cheverud, J.M. Pleiotropic scaling of gene effects and the ‘cost of complexity. Nature 2008, 27, 470–472. [Google Scholar] [CrossRef]
- Wang, Z.; Liao, B.-Y.; Zhang, J. Genomic patterns of pleiotropy and the evolution of complexity. Proc. Natl. Acad. Sci. USA 2010, 107, 18034–18039. [Google Scholar] [CrossRef] [PubMed]
- Hammer, K. Das domestikationssyndrom. Die Kult. 1984, 32, 11–34. (In German) [Google Scholar] [CrossRef]
- Carter, T., Jr.; Hymowitz, T.; Nelson, R. Biogeography, local adaptation, Vavilov, and genetic diversity in soybean. In Biological Resources and Migration; Springer: Berlin/Heidelberg, Germany, 2004; pp. 47–59. [Google Scholar]
- Kim, M.Y.; Van, K.; Kang, Y.J.; Kim, K.H.; Lee, S.-H. Tracing soybean domestication history: From nucleotide to genome. Breed. Sci. 2012, 61, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, E.J.; Wu, F.; Hanzawa, Y. Soybean domestication, the origin, genetic architecture and molecular bases. New Phytol. 2017, 214, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yang, X.; Liu, J.; Wang, B.-H.; Liu, B.-L.; Wang, Y.-Z. Pod shattering resistance associated with domestication is mediated by a NAC gene in soybean. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Funatsuki, H.; Suzuki, M.; Hirose, A.; Inaba, H.; Yamada, T.; Hajika, M.; Komatsu, K.; Katayama, T.; Sayama, T.; Ishimoto, M.; et al. Molecular basis of a shattering resistance boosting global dissemination of soybean. Proc. Natl. Acad. Sci. USA 2014, 111, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Miao, Z.; Cai, C.; Zhang, D.; Zhao, M.; Wu, Y.; Zhang, X.; Swarm, S.A.; Zhou, L.; Zhang, Z.J.; et al. GmHs1-1, encoding a calcineurin-like protein, controls hard-seededness in soybean. Nat. Genet. 2015, 47, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Sedivy, E.J.; Price, W.B.; Haider, W.; Hanzawa, Y. Evolutionary trajectories of duplicated FT homologues and their roles in soybean domestication. Plant J. 2017, 90, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Cober, E.R.; Morrison, M.J. Regulation of seed yield and agronomic characters by photoperiod sensitivity and growth habit genes in soybean. Theor. Appl. Genet. 2010, 120, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Shin, J.H.; Kang, Y.J.; Shim, S.R.; Lee, S.-H. Divergence of flowering genes in soybean. J. Biosci. 2012, 37, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Takeshima, R.; Zhao, C.; Liu, B.; Jun, A.; Kong, F. Molecular mechanisms of flowering under long days and stem growth habit in soybean. J. Exp. Bot. 2017, 68, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Samanfar, B.; Molnar, S.J.; Charette, M.; Schoenrock, A.; Dehne, F.; Golshani, A. Mapping and identification of a potential candidate gene for a novel maturity locus, E10, in soybean. Theor. Appl. Genet. 2017, 130, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Watanabe, S.; Yamada, T.; Tsubokura, Y.; Nakashima, H.; Zhai, H.; Anai, T.; Sato, S.; Yamazaki, T.; Lu, S.; et al. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc. Natl. Acad. Sci. USA 2012, 109, E2155–E2164. [Google Scholar] [CrossRef]
- Watanabe, S.; Xia, Z.; Hideshima, R.; Tsubokura, Y.; Sato, S.; Yamanaka, N.; Takahashi, R.; Anai, T.; Tabata, S.; Kitamura, K. A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 2011, 188, 395–407. [Google Scholar] [CrossRef]
- Liu, B.; Kanazawa, A.; Matsumura, H.; Takahashi, R.; Harada, K.; Abe, J. Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome A gene. Genetics 2008, 180, 995–1007. [Google Scholar] [CrossRef]
- Watanabe, S.; Hideshima, R.; Xia, Z.; Tsubokura, Y.; Sato, S.; Nakamoto, Y.; Yamanaka, N.; Takahashi, R.; Ishimoto, M.; Anai, T. Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 2009, 182, 1251–1262. [Google Scholar] [CrossRef]
- Zhao, C.; Takeshima, R.; Zhu, J.; Xu, M.; Sato, M.; Watanabe, S.; Kanazawa, A.; Liu, B.; Kong, F.; Yamada, T.; et al. A recessive allele for delayed flowering at the soybean maturity locus E9 is a leaky allele of FT2a, a FLOWERING LOCUS T ortholog. BMC Plant Biol. 2016, 16, 20. [Google Scholar] [CrossRef]
- Lu, S.; Zhao, X.; Hu, Y.; Liu, S.; Nan, H.; Li, X.; Fang, C.; Cao, D.; Shi, X.; Kong, L.; et al. Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat. Genet. 2017, 49, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.L. Response to Selection for Time of Flowering in Soybean. Crop Sci. 1988, 623–626. [Google Scholar] [CrossRef]
- Smith, J.R.; Nelson, R.L. Selection for Seed-Filling Period in Soybean. Crop Sci. 1986, 26, 466–469. [Google Scholar] [CrossRef]
- Fehr, W.R.; Caviness, C.E.; Burmood, D.T.; Pennington, J.S. Stage of development descriptions for soybeans, Glycine max (L.) Merrill. Crop Sci. 1971, 11, 929–931. [Google Scholar] [CrossRef]
- Revelle, W. An Overview of the Psych Package. Department of Psychology, Northwestern University. 2017. Available online: http://personality-project.org/r/overview.pdf (accessed on 1 July 2017).
- Pagès, J. Multiple Factor Analysis, Main Features and Application to Sensory Data. Agrocampus Ouest, Rennes, France. Available online: http://factominer.free.fr/more/PagesAFM.pdf (accessed on 1 April 2016).
- Hyten, D.L.; Cannon, S.B.; Song, Q.; Weeks, N.; Fickus, E.W.; Shoemaker, R.C.; Specht, J.E.; Farmer, A.D.; May, G.D.; Cregan, P.B. High-throughput SNP discovery through deep resequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genom. 2010, 11, 38. [Google Scholar] [CrossRef]
- Hyten, D.L.; Song, Q.; Choi, I.Y.; Yoon, M.S.; Specht, J.E.; Matukumalli, L.K.; Nelson, R.L.; Shoemaker, R.C.; Young, N.D.; Cregan, P.B. High-throughput genotyping with the GoldenGate assay in the complex genome of soybean. Theor. Appl. Genet. 2008, 116, 945–952. [Google Scholar] [CrossRef]
- Wu, F.; Kang, X.; Wang, M.; Haider, W.; Price, W.B.; Hajek, B.; Hanzawa, Y. Transcriptome-enabled network inference revealed the GmCOL1 feed-forward look and its roles in photoperiodic flowering of soybean. Front. Plant Sci. 2019. [Google Scholar] [CrossRef]
- Wang, S.C.; Basten, C.J.; Zeng, Z.-B. Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. 2012. Available online: http://statgen.ncsu.edu/qtlcart/WQTLCart.htm (accessed on 1 April 2016).
- Li, Z.; Nelson, R.L. RAPD Marker Diversity among Cultivated and Wild Soybean Accessions from Four Chinese Provinces. Crop Sci. 2002, 42, 1737–1744. [Google Scholar] [CrossRef]
- McBlain, B.A.; Bernard, R.L. A new gene affecting the time of flowering and maturity in soybeans. J. Hered. 1987, 78, 160–162. [Google Scholar] [CrossRef]
- Buzzell, R.V.H. Inheritance of insensitivity to long day length. Soybean Genet. Newsl. 1980, 7, 26–29. [Google Scholar]
- Liu, B.; Fujita, T.; Yan, Z.H.; Sakamoto, S.; Xu, D.; Abe, J. QTL mapping of domestication-related traits in soybean (Glycine max). Ann. Bot. 2007, 100, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.K.; Wang, Y.J.; Luo, G.Z.; Zhang, J.S.; He, C.Y.; Wu, X.L.; Gai, J.Y.; Chen, S.Y. QTL mapping of ten agronomic traits on the soybean (Glycine max, L. Merr.) genetic map and their association with EST markers. Theor. Appl. Genet. 2004, 108, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Guzman, P.S.; Diers, B.W.; Neece, D.J.; St Martin, S.K.; LeRoy, A.R.; Grau, C.R.; Hughes, T.J.; Nelson, R.L. QTL associated with yield in three backcross-derived populations of soybean. Crop Sci. 2007, 12, 111–122. [Google Scholar] [CrossRef]
- Doust, A.N.; Lukens, L.; Olsen, K.M.; Mauro-Herrera, M.; Meyer, A.; Rogers, K. Beyond the single gene, How epistasis and gene-by-environment effects influence crop domestication. Proc. Natl. Acad. Sci. USA 2014, 111, 6178–6183. [Google Scholar] [CrossRef] [PubMed]
- Kryazhimskiy, S.; Rice, D.P.; Jerison, E.R.; Desai, M.M. Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science 2014, 344, 1519–1522. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Dimension 1 (45.3%) | Dimension 2 (23.1%) | Dimension 3 (16.7%) |
|---|---|---|---|
| Flowering | 0.414 | 0.822 | 0.019 |
| Maturity | 0.541 | 0.555 | 0.500 |
| Height | 0.842 | −0.302 | 0.167 |
| Lodging | 0.806 | −0.423 | 0.018 |
| Yield | −0.372 | −0.312 | 0.838 |
| Stem Vining | 0.866 | −0.186 | −0.141 |
| Trait | QTL | Chromosome | Year/Location | Peak Marker | Position (cM) | Position (Mbp) | LOD | R2 | Additive Effect |
|---|---|---|---|---|---|---|---|---|---|
| Flowering | |||||||||
| R1-1 | 9 | matset 2 | BARC-056323-14257 | 32.24 | 20.99 | 4.10 | 0.26 | 6.41 | |
| Maturity | |||||||||
| R8-1 | 13 | pop2(7919) | BARC-041649-08056 | 21.76 | 33.28 | 5.88 | 0.32 | −1.31 | |
| R8-2 | 18 | matset2 | BARC-015067-02556 | 25.04 | 2.15 | 3.14 | 0.17 | −1.18 | |
| Height | |||||||||
| HT-1 | 9 | All years | BARC-056323-14257 | 32.24 | 20.99 | 3.15 | 0.10 | 8.29 | |
| 9 | 2009 | BARC-056323-14257 | 32.24 | 20.99 | 3.15 | 0.10 | 8.29 | ||
| HT-2 | 11 | pop1(7909) | BARC-040851-07854 | 7.00 | 7.61 | 5.48 | 0.26 | 13.87 | |
| 11 | Urbana09 | BARC-040851-07854 | 7.00 | 7.61 | 4.92 | 0.15 | 15.13 | ||
| Yield | |||||||||
| YD-1 | 18 | Stone09 | BARC-014395-01348 | 13.02 | 3.44 | 4.71 | 0.15 | 3.83 | |
| YD-2 | 18 | Stone09 | BARC-060195-16470 | 16.32 | 0.47 | 3.77 | 0.13 | 3.60 | |
| Lodging | |||||||||
| LD-1 | 9 | pop1(7909) | BARC-028249-05804 | 38.15 | 7.47 | 3.18 | 0.16 | 1.08 | |
| 9 | Urbana09 | BARC-028249-05804 | 38.15 | 7.47 | 3.60 | 0.13 | 1.39 | ||
| LD-2 | 9 | Urbana10 | BARC-056323-14257 | 32.24 | 20.99 | 3.59 | 0.11 | 0.66 | |
| LD-3 | 11 | 2010 | BARC-040851-07854 | 6.98 | 7.61 | 5.36 | 0.17 | 0.94 | |
| LD-4 | 13 | matset2 | BARC-062009-17616 | 0.00 | 19.33 | 3.64 | 0.16 | −0.52 | |
| LD-5 | 13 | matset2 | BARC-065851-19789 | 25.32 | 2.44 | 3.72 | 0.18 | 0.51 | |
| LD-6 | 14 | matset2 | BARC-017933-02457 | 24.71 | 46.13 | 3.39 | 0.14 | −0.43 | |
| LD-7 | 16 | pop2(7919) | BARC-054099-12340 | 6.74 | 3.56 | 3.18 | 0.20 | 0.43 | |
| Stem vining | |||||||||
| SV-1 | 2 | Matset2 | BARC-021647-04164 | 25.85 | 46.04 | 4.54 | 0.23 | 1.36 | |
| SV-2 | 6 | VG10 | BARC-025179-06455 | 10.62 | 50.32 | 4.37 | 0.16 | 0.97 | |
| SV-3 | 9 | All years | BARC-028249-05804 | 38.15 | 7.47 | 2.90 | 0.09 | 1.02 | |
| 9 | 2009 | BARC-028249-05804 | 38.15 | 7.47 | 2.90 | 0.09 | 1.02 | ||
| SV-4 | 11 | pop1(7909) | BARC-040851-07854 | 6.98 | 7.61 | 4.96 | 0.24 | 1.09 | |
| 11 | Urbana09 | BARC-040851-07854 | 6.98 | 7.61 | 4.38 | 0.14 | 0.99 | ||
| 11 | Urbana10 | BARC-040851-07854 | 6.98 | 7.61 | 4.21 | 0.13 | 0.92 | ||
| 11 | 2010 | BARC-040851-07854 | 6.98 | 7.61 | 3.34 | 0.10 | 0.85 | ||
| SV-5 | 14 | Matset2 | BARC-052759-11611 | 4.40 | 10.01 | 3.05 | 0.14 | −0.42 |
| Trait | QTL | Chromosome | Peak Marker | Previously Identified QTL |
|---|---|---|---|---|
| R1 | R1-1 | 9 | BARC-056323-14257 | Fflr 3-4 |
| R8 | R8-2 | 18 | BARC-015067-02556 | Pod mat 22-6 |
| Height | HT-1 | 9 | BARC-056323-14257 | Pl ht 15-1 |
| HT-2 | 11 | BARC-040851-07854 | Pl ht 24-6 | |
| Yield | YD-1 | 18 | BARC-014395-01348 | Sd yld 21-4 |
| YD-2 | 18 | BARC-060195-16470 | Sd yld 21-4 | |
| Lodging | LD-1 | 9 | BARC-028249-05804 | Ldge 11-1 |
| LD-3 | 11 | BARC-040851-07854 | Ldge 10-1 | |
| LD-6 | 14 | BARC-017933-02457 | Ldge 21-2 | |
| Stem vining | SV-1 | 2 | BARC-021647-04164 | TH-C |
| QTL | Chromosome | Year/Location | Peak Marker | Gene | ID Number | Start Position |
|---|---|---|---|---|---|---|
| R8-1 | 13 | pop2(7919) | BARC-041649-08056 | SPA3 | Glyma.13g276700 | 37212524 |
| SRF6 | Glyma.13g241100 | 34503402 | ||||
| R1-1 | 9 | matset 2 | BARC-056323-14257 | PHYE | Glyma.09g088500 | 11657930 |
| TFL1 | Glyma.09g143500 | 36559409 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedivy, E.J.; Akpertey, A.; Vela, A.; Abadir, S.; Khan, A.; Hanzawa, Y. Identification of Non-Pleiotropic Loci in Flowering and Maturity Control in Soybean. Agronomy 2020, 10, 1204. https://doi.org/10.3390/agronomy10081204
Sedivy EJ, Akpertey A, Vela A, Abadir S, Khan A, Hanzawa Y. Identification of Non-Pleiotropic Loci in Flowering and Maturity Control in Soybean. Agronomy. 2020; 10(8):1204. https://doi.org/10.3390/agronomy10081204
Chicago/Turabian StyleSedivy, Eric J., Abraham Akpertey, Angela Vela, Sandra Abadir, Awais Khan, and Yoshie Hanzawa. 2020. "Identification of Non-Pleiotropic Loci in Flowering and Maturity Control in Soybean" Agronomy 10, no. 8: 1204. https://doi.org/10.3390/agronomy10081204
APA StyleSedivy, E. J., Akpertey, A., Vela, A., Abadir, S., Khan, A., & Hanzawa, Y. (2020). Identification of Non-Pleiotropic Loci in Flowering and Maturity Control in Soybean. Agronomy, 10(8), 1204. https://doi.org/10.3390/agronomy10081204