22KD Zein Content Coordinates Transcriptional Activity during Starch Synthesis in Maize Endosperm

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sample Collection
2.2. RNA Extraction and Library Construction
2.3. Identification of DEGs
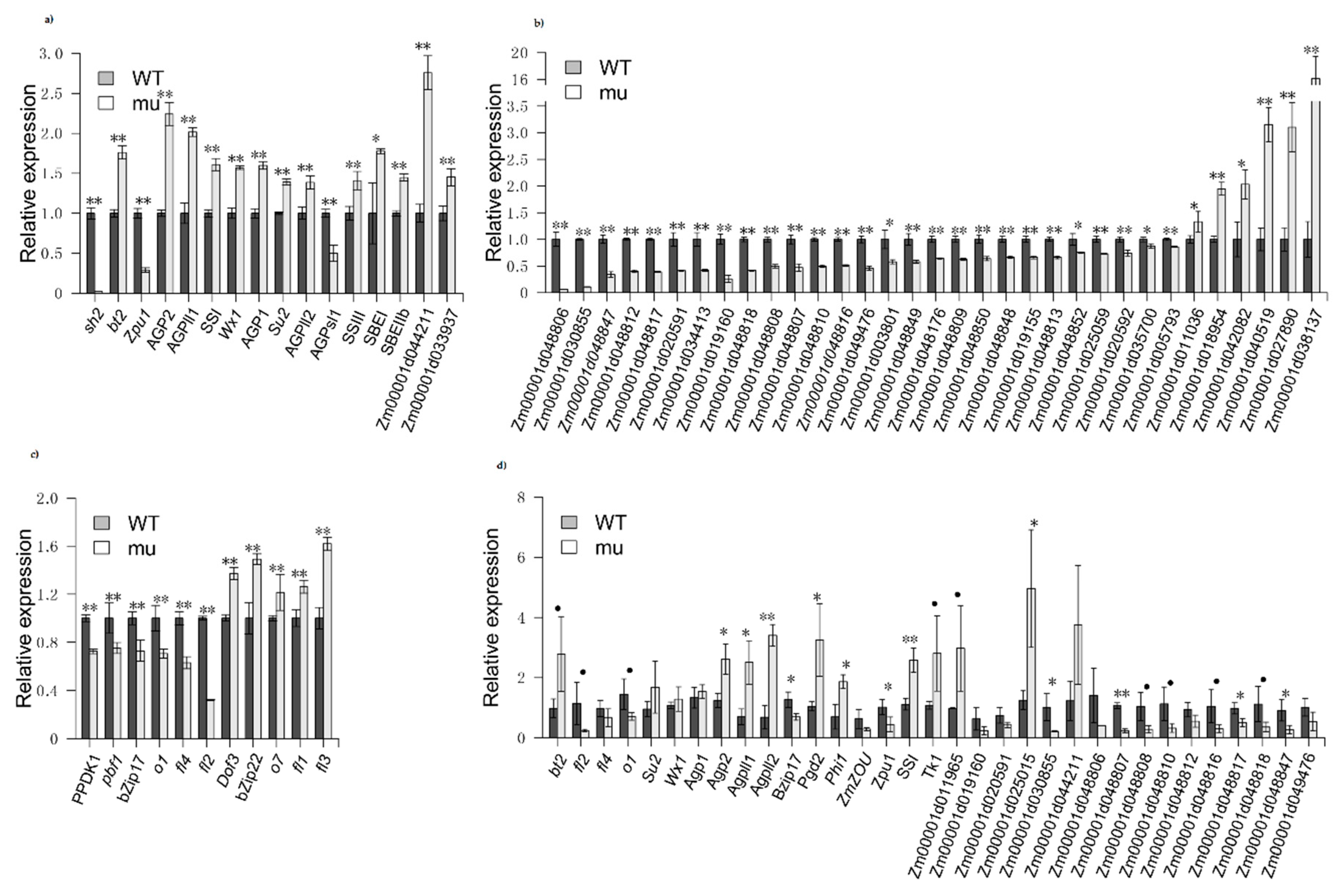
2.4. Confirmation of DEG
2.5. Co-Expression Network Analysis of Well-Known Maize Kernel Mutant Transcriptomes
3. Results
3.1. Isolation of Shrunken Mutant and DEGs
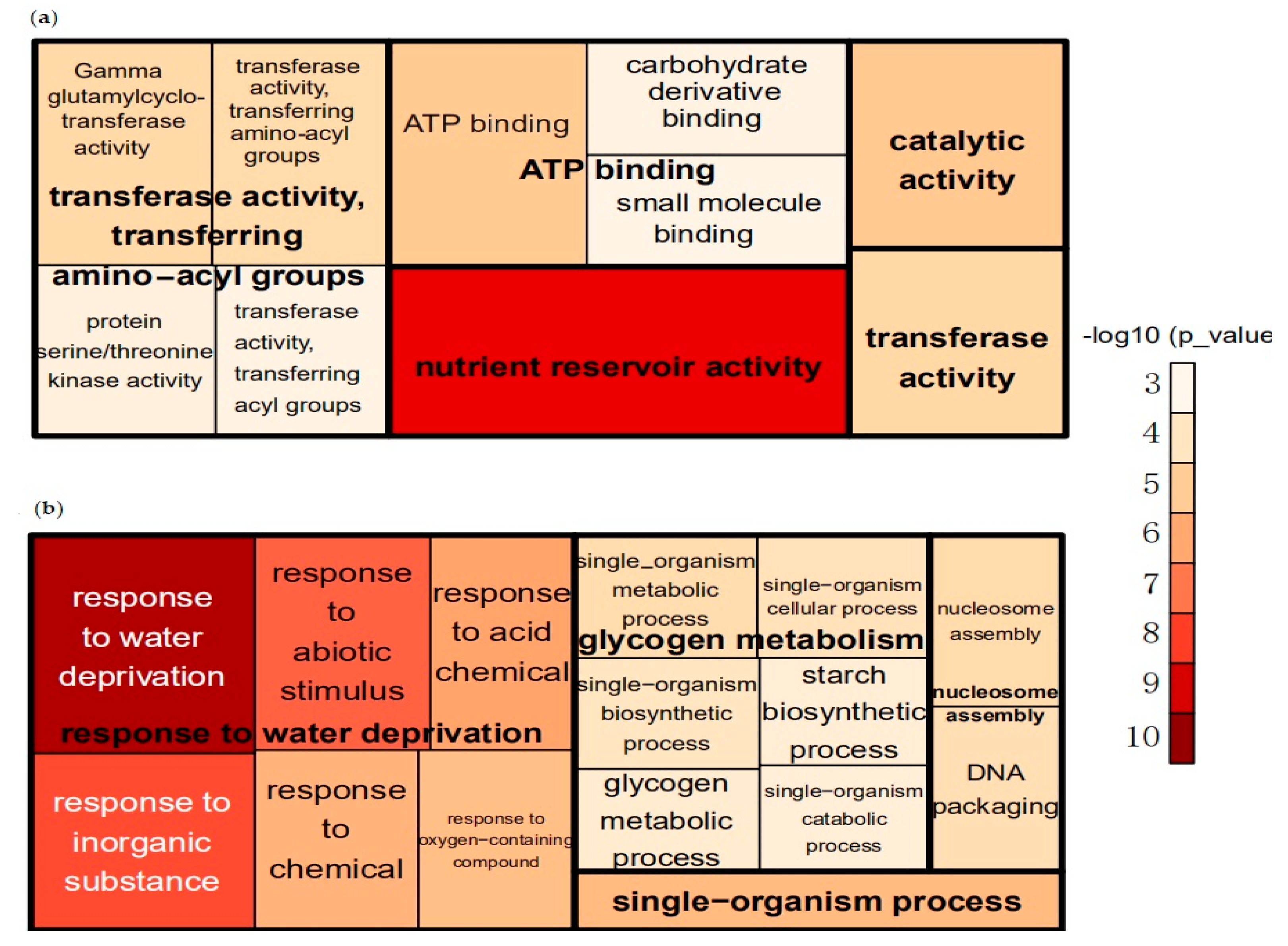
3.2. GO Enrichment Analysis of DEGs
3.3. Starch Biosynthesis and Zein-Related Genes Are Differentially Regulated in sh2 Kernels
3.4. Carbohydrate Catabolism and Electron Transport Chain Related Genes Are Differentially Regulated in sh2 Kernels
3.5. Hormone-Related DEGs
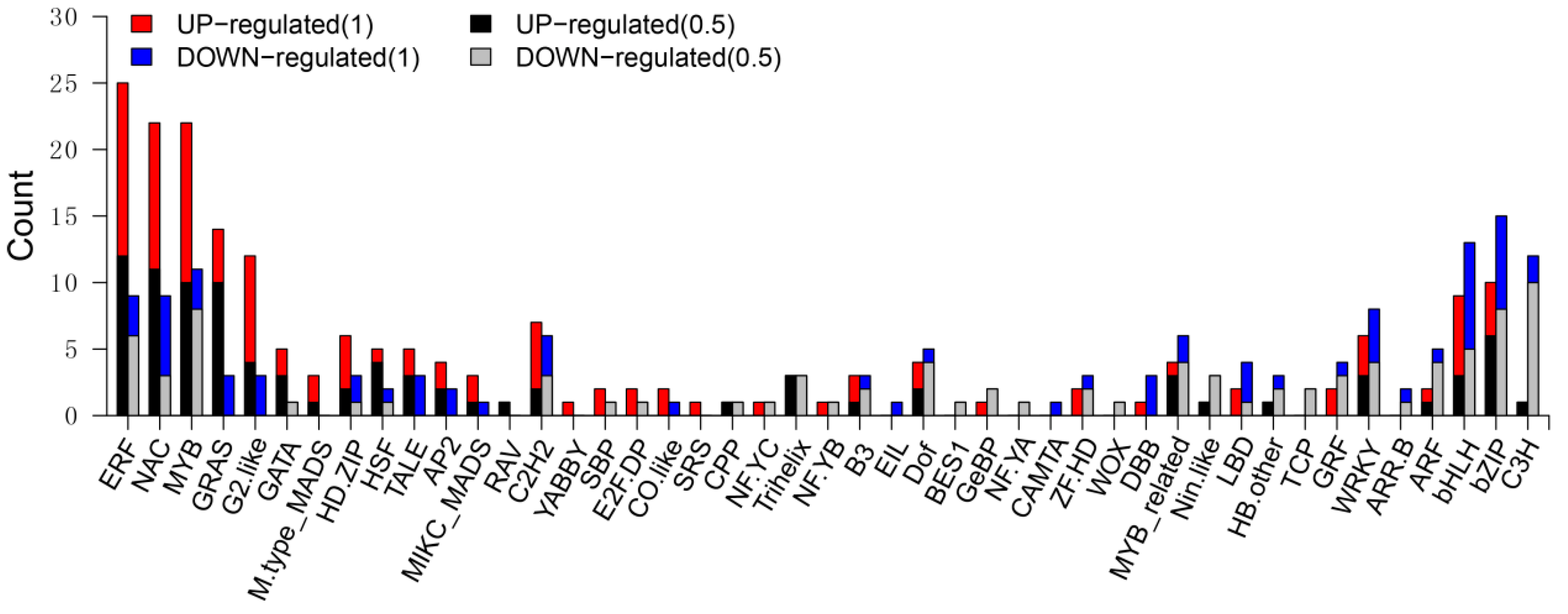
3.6. Differentially Expressed TF Genes between the Wild Type and sh2
3.7. Transcriptional Activity is a Key Regulator of Starch Synthesis at 15 DAP
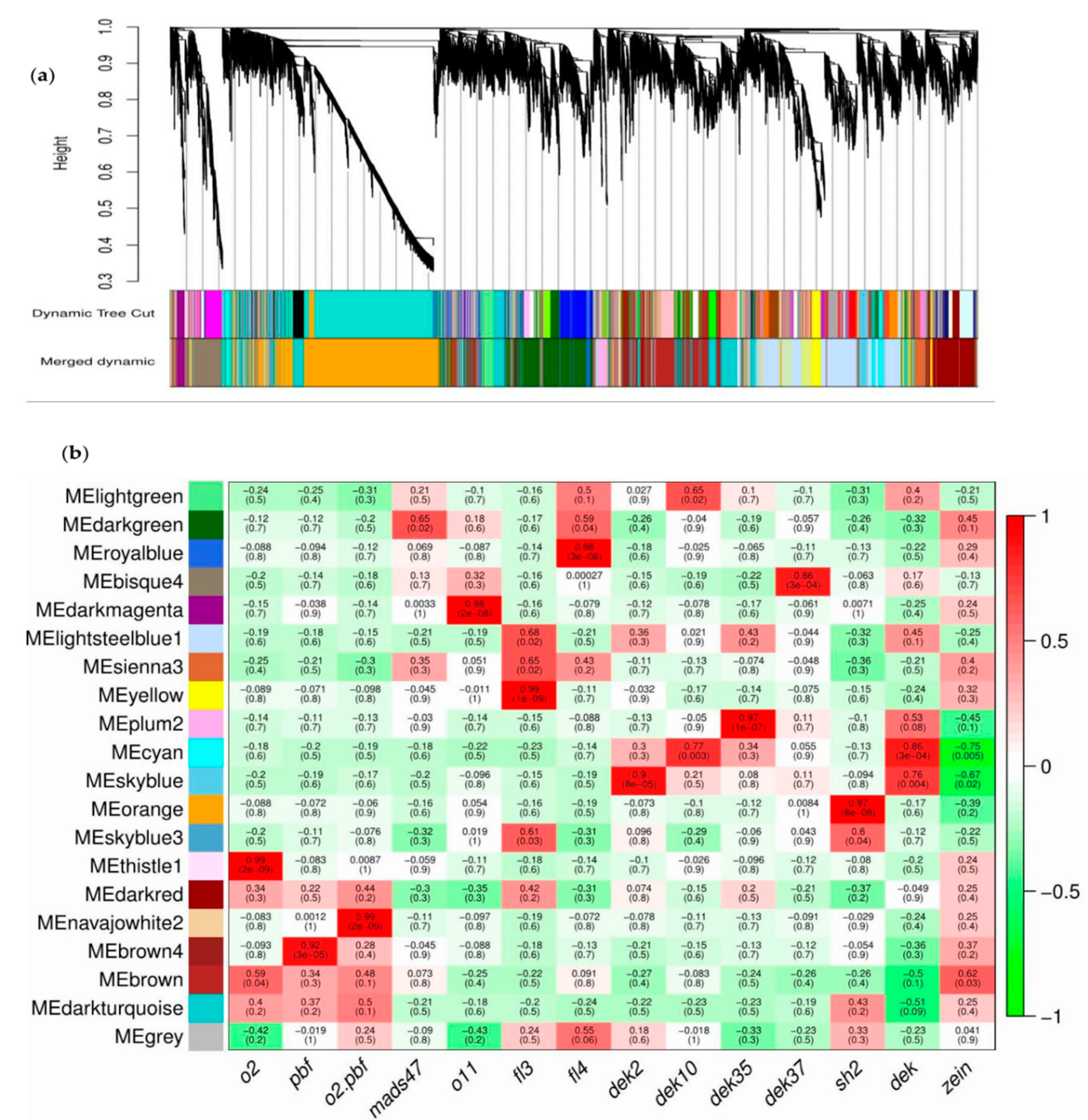
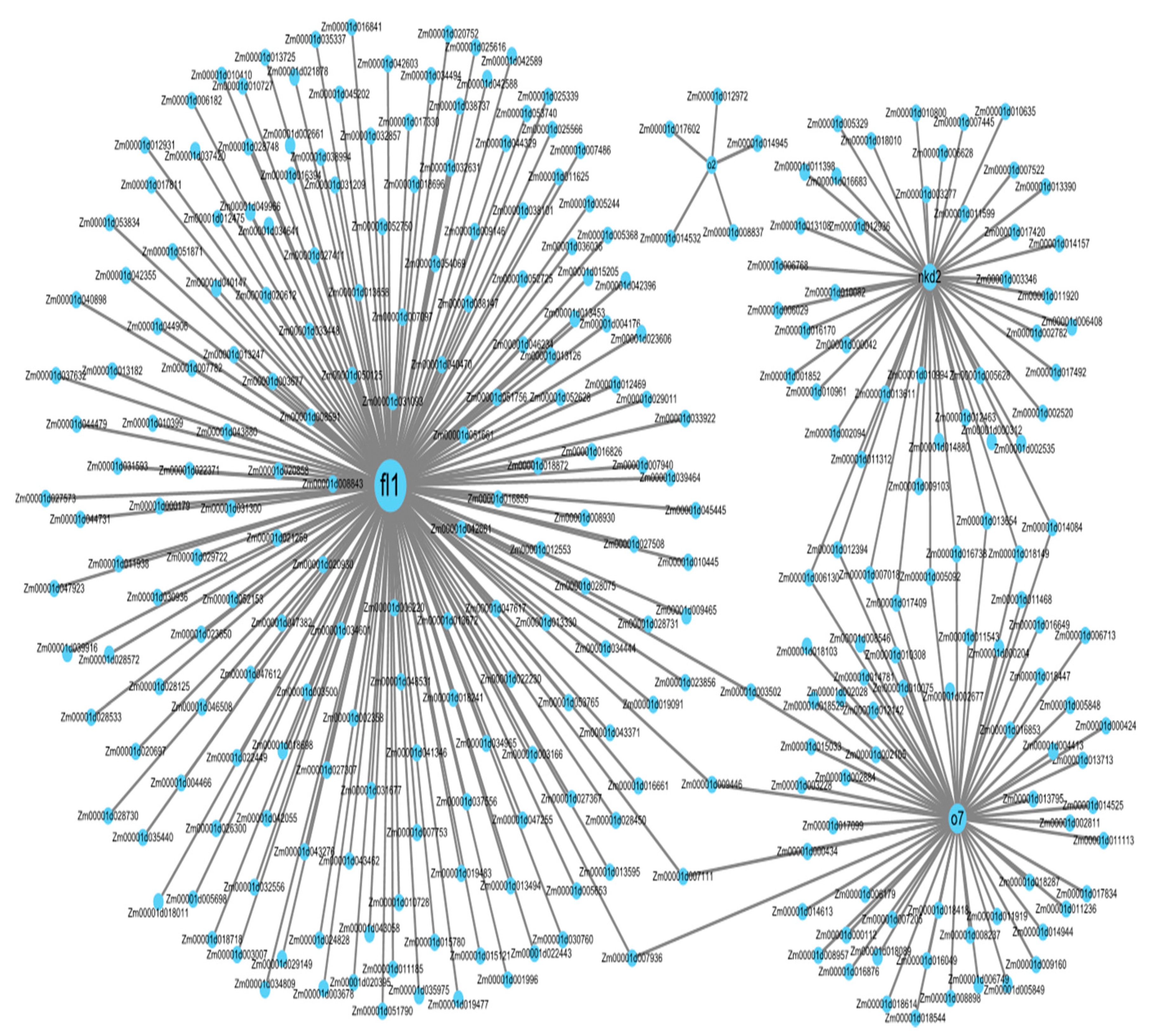
3.8. Co-Expression Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ballicora, M.A.; Iglesias, A.A.; Preiss, J. ADP-Glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol. Mol. Biol. Rev. 2003, 67, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Hannah, L.C.; James, M. The complexities of starch biosynthesis in cereal endosperms. Curr. Opin. Biotechnol. 2008, 19, 160–165. [Google Scholar] [CrossRef]
- Georgelis, N.; Braun, E.L.; Shaw, J.R.; Hannah, L.C. The two AGPase subunits evolve at different rates in angiosperms, yet they are equally sensitive to activity-altering amino acid changes when expressed in bacteria. Plant Cell 2007, 19, 1458–1472. [Google Scholar] [CrossRef] [PubMed]
- Hannah, L.C.; Giroux, M.; Boyer, C. Biotechnological modification of carbohydrates for sweet corn and maize improvement. Sci. Hortic. 1993, 55, 177–197. [Google Scholar] [CrossRef]
- Stark, D.M.; Timmerman, K.P.; Barry, G.F.; Preiss, J.; Kishore, G.M. Regulation of the amount of starch in plant tissues by ADP glucose pyrophosphorylase. Science 1992, 258, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Sakulsingharoj, C.; Choi, S.-B.; Hwang, S.-K.; Edwards, G.E.; Bork, J.; Meyer, C.R.; Preiss, J.; Okita, T.W. Engineering starch biosynthesis for increasing rice seed weight: The role of the cytoplasmic ADP-glucose pyrophosphorylase. Plant Sci. 2004, 167, 1323–1333. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Wang, J.; Liu, T.; Liu, Y.; Zhao, L.; Wang, G. Increasing maize seed weight by enhancing the cytoplasmic ADP-glucose pyrophosphorylase activity in transgenic maize plants. Plant Cell. Tissue Organ Cult. 2007, 88, 83–92. [Google Scholar] [CrossRef]
- Giroux, M.J.; Shaw, J.; Barry, G.; Cobb, B.G.; Greene, T.; Okita, T.; Hannah, L.C. A single mutation that increases maize seed weight. Proc. Natl. Acad. Sci. USA 1996, 93, 5824–5829. [Google Scholar] [CrossRef]
- Li, N.; Zhang, S.; Zhao, Y.; Li, B.; Zhang, J. Over-expression of AGPase genes enhances seed weight and starch content in transgenic maize. Planta 2011, 233, 241–250. [Google Scholar] [CrossRef]
- Smidansky, E.D.; Clancy, M.; Meyer, F.D.; Lanning, S.P.; Blake, N.K.; Talbert, L.E.; Giroux, M.J. Enhanced ADP-glucose pyrophosphorylase activity in wheat endosperm increases seed yield. Proc. Natl. Acad. Sci. USA 2002, 99, 1724–1729. [Google Scholar]
- Smidansky, E.D.; Martin, J.M.; Hannah, L.C.; Fischer, A.M.; Giroux, M.J. Seed yield and plant biomass increases in rice are conferred by deregulation of endosperm ADP-glucose pyrophosphorylase. Planta 2003, 216, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Hannah, L.C.; Futch, B.; Bing, J.; Shaw, J.R.; Boehlein, S.; Stewart, J.D.; Beiriger, R.; Georgelis, N.; Greene, T. A shrunken-2 transgene increases maize yield by acting in maternal tissues to increase the frequency of seed development. Plant Cell 2012, 24, 2352–2363. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Kunca, S.; Muc, S. Summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Holding, D.R.; Otegui, M.S.; Li, B.; Meeley, R.B.; Dam, T.; Hunter, B.G.; Jung, R.; Larkins, B.A. The maize Floury1 gene encodes a novel endoplasmic reticulum protein involved in zein protein body formation. Plant Cell 2007, 19, 2569–2582. [Google Scholar] [CrossRef]
- Coleman, C.E.; Lopes, M.A.; Gillikin, J.W.; Boston, R.S.; Larkins, B.A. A defective signal peptide in the maize high-lysine mutant floury 2. Proc. Natl. Acad. Sci. USA 1995, 92, 6828–6831. [Google Scholar] [CrossRef]
- Li, Q.; Wang, J.; Ye, J.; Zheng, X.; Xiang, X.; Li, C.; Fu, M.; Wang, Q.; Zhang, Z.; Wu, Y. The maize imprinted gene floury3 encodes a PLATZ protein required for tRNA and 5S rRNA transcription through interaction with RNA polymerase III. Plant Cell 2017, 29, 2661–2675. [Google Scholar] [CrossRef]
- Wang, G.; Qi, W.; Wu, Q.; Yao, D.; Zhang, J.; Zhu, J.; Wang, G.; Wang, G.; Tang, Y.; Song, R. Identification and characterization of maize floury4 as a novel semidominant opaque mutant that disrupts protein body assembly. Plant Physiol. 2014, 165, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Sun, X.; Wang, G.; Wang, F.; Gao, Q.; Sun, X.; Tang, Y.; Chang, C.; Lai, J.; Zhu, L.; et al. Opaque7 encodes an acyl-activating enzyme-like protein that affects storage protein synthesis in maize endosperm. Genetics 2011, 189, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zheng, X.; Yang, J.; Messing, J.; Wu, Y. Maize endosperm-specific transcription factors O2 and PBF network the regulation of protein and starch synthesis. Proc. Natl. Acad. Sci. USA 2016, 113, 10842–10847. [Google Scholar] [CrossRef]
- Zhan, J.; Li, G.; Ryu, C.H.; Ma, C.; Zhang, S.; Lloyd, A.; Hunter, B.G.; Larkins, B.A.; Drews, G.N.; Wang, X.; et al. Opaque-2 regulates a complex gene network associated with cell differentiation and storage functions of maize endosperm. Plant Cell 2018, 30, 2425–2446. [Google Scholar] [PubMed]
- Lappe, R.R.; Baier, J.W.; Boehlein, S.K.; Huffman, R.; Lin, Q.; Wattebled, F.; Mark Settles, A.; Curtis Hannah, L.; Borisjuk, L.; Rolletschek, H.; et al. Functions of maize genes encoding pyruvate phosphate dikinase in developing endosperm. Proc. Natl. Acad. Sci. USA 2018, 115, E24–E33. [Google Scholar] [PubMed]
- Tobias, R.B.; Boyer, C.D.; Shannon, J.C. Alterations in carbohydrate intermediates in the endosperm of starch-deficient maize (Zea mays L.) genotypes. Plant Physiol. 1992, 99, 146–152. [Google Scholar] [CrossRef]
- Tobias, R.B.; Boyer, C.D.; Shannon, J.C. Enzymes catalyzing the reversible conversion of fructose-6-phosphate and fructose-I,6-bisphosphate in maize (Zea mays L.) Kernels’. Plant Physiol. 1992, 99, 140–145. [Google Scholar] [CrossRef]
- Duncan, K.A.; Hardin, S.C.; Huber, S.C. The three maize sucrose synthase isoforms siffer in sistribution, localization, and phosphorylation. Plant Cell Physiol. 2006, 47, 959–971. [Google Scholar] [CrossRef]
- Tsuwamoto, R.; Yokoi, S.; Takahata, Y. Arabidopsis embryomaker encoding an AP2 domain transcription factor plays a key role in developmental change from vegetative to embryonic phase. Plant Mol. Biol. 2010, 73, 481–492. [Google Scholar] [CrossRef]
- Sekhar, S.; Panda, B.B.; Mohapatra, T.; Das, K.; Shaw, B.P.; Kariali, E.; Mohapatra, P.K. Spikelet-specific variation in ethylene production and constitutive expression of ethylene receptors and signal transducers during grain filling of compact- and lax-panicle rice (Oryza sativa) cultivars. J. Plant Physiol. 2015, 179, 21–34. [Google Scholar] [CrossRef]
- Wang, G.-Q.; Hao, S.-S.; Gao, B.; Chen, M.-X.; Liu, Y.-G.; Yang, J.-C.; Ye, N.-H.; Zhang, J.-H. Regulation of gene expression in the remobilization of carbon reserves in rice stems during grain filling. Plant Cell Physiol. 2017, 58, 1391–1404. [Google Scholar] [CrossRef]
- Iuchi, S.; Kobayashi, M.; Taji, T.; Naramoto, M.; Seki, M.; Kato, T.; Tabata, S.; Kakubari, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis. Plant J. 2001, 27, 325–333. [Google Scholar] [CrossRef]
- Nambara, E.; Marion-Poll, A. Abscisic acid biosynthesis and catabolism. Annu. Rev. Plant Biol. 2005, 56, 165–185. [Google Scholar] [CrossRef]
- Liu, L.; Tong, H.; Xiao, Y.; Che, R.; Xu, F.; Hu, B.; Liang, C.; Chu, J.; Li, J.; Chu, C. Activation of big grain1 significantly improves grain size by regulating auxin transport in rice. Proc. Natl. Acad. Sci. USA 2015, 112, 11102–11107. [Google Scholar] [CrossRef]
- Weng, X.; Wang, L.; Wang, J.; Hu, Y.; Du, H.; Xu, C.; Xing, Y.; Li, X.; Xiao, J.; Zhang, Q. Grain number, plant height, and heading date7 is a central regulator of growth, development, and stress response. Plant Physiol. 2014, 164, 735–747. [Google Scholar] [CrossRef]
- Hakata, M.; Muramatsu, M.; Nakamura, H.; Hara, N.; Kishimoto, M.; Iida-Okada, K.; Kajikawa, M.; Imai-Toki, N.; Toki, S.; Nagamura, Y.; et al. Overexpression of TIFY genes promotes plant growth in rice through jasmonate signaling. Biosci. Biotechnol. Biochem. 2017, 81, 906–913. [Google Scholar] [CrossRef]
- Che, R.; Tong, H.; Shi, B.; Liu, Y.; Fang, S.; Liu, D.; Xiao, Y.; Hu, B.; Liu, L.; Wang, H.; et al. Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nat. Plants 2015, 2, 15195. [Google Scholar] [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Pingyong, S.; Wuhan, Z.; Yihua, W.; Qiang, H.; Fu, S.; Hai, L.; Jie, W.; Jianmin, W.; Longping, Y.; Huafeng, D. OsGRF4controls grain shape, panicle length andseed shattering in rice. J. Integr. Plant Biol. 2016, 58, 836–847. [Google Scholar]
- Schläpfer, P.; Zhang, P.; Wang, C.; Kim, T.; Banf, M.; Chae, L.; Dreher, K.; Chavali, A.K.; Nilo-Poyanco, R.; Bernard, T.; et al. Genome-Wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol. 2017, 173, 2041–2059. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Li, C.; Qiao, Z.; Qi, W.; Wang, Q.; Yuan, Y.; Yang, X.; Tang, Y.; Mei, B.; Lv, Y.; Zhao, H.; et al. Genome-wide characterization of cis-acting DNA targets reveals the transcriptional regulatory framework of opaque2 in maize. Plant Cell 2015, 27, 532–545. [Google Scholar] [CrossRef]
- Feng, F.; Qi, W.; Lv, Y.; Yan, S.; Xu, L.; Yang, W.; Yuan, Y.; Chen, Y.; Zhao, H.; Song, R. OPAQUE11 Is a central hub of the regulatory network for maize endosperm development and nutrient metabolism. Plant Cell 2018, 30, 375–396. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, Y.; Du, J.; Li, H.; Wei, B.; Wang, Y.; Li, Y.; Yu, G.; Liu, H.; Zhang, J.; et al. ZmMYB14 is an important transcription factor involved in the regulation of the activity of the ZmBT1 promoter in starch biosynthesis in maize. FEBS J. 2017, 284, 3079–3099. [Google Scholar]
- Janick, J. Plant Breeding Reviews; John Wiley & Sons: Hoboken, NJ, USA, 1996; Volume 14, ISBN 9780470650073. [Google Scholar]
- Dodson-Swenson, H.G.; Tracy, W.F. Endosperm carbohydrate composition and kernel characteristics of shrunken2-intermediate (Sh2-i/sh2-i su1/su1) and shrunken2-intermediate–sugary1- reference (sh2-i/sh2-i su1-r/su1-r) in sweet corn. Crop Sci. 2015, 55, 2647–2656. [Google Scholar] [CrossRef]
- Greene, T.; Hannah, L. Maize endosperm ADP-glucose pyrophosphorylase SHRUNKEN2 and BRITTLE2 subunit interactions. Plant Cell 1998, 10, 1295–1306. [Google Scholar]
- Giroux, M.J.; Boyer, C.; Feix, G.; Hannah, L.C. Coordinated transcriptional regulation of storage product genes in the maize endosperm. Plant Physiol. 1994, 106, 713–722. [Google Scholar] [CrossRef]
- Bhave, M.R.; Lawrence, S.; Barton, C.; Curtis, L. ldentification and molecular characterization of shrunken-2 cDNA clones of maize. Plant Cell 1990, 6, 581–588. [Google Scholar]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Wang, G.; Hauck, A.; Lai, J. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, J.; Ji, C.; Wu, Y.; Messing, J. NAC-type transcription factors regulate accumulation of starch and protein in maize seeds. Proc. Natl. Acad. Sci. USA 2019, 166, 11223–11228. [Google Scholar] [CrossRef]
- Keeling, P.L.; Myers, A.M. Biochemistry and genetics of starch synthesis. Annu. Rev. Food Sci. Technol. 2010, 1, 271–303. [Google Scholar] [CrossRef] [PubMed]
- Prioul, J.L.; Méchin, V.; Lessard, P.; Thévenot, C.; Grimmer, M.; Chateau-Joubert, S.; Coates, S.; Hartings, H.; Kloiber-Maitz, M.; Murigneux, A.; et al. A joint transcriptomic, proteomic and metabolic analysis of maize endosperm development and starch filling. Plant Biotechnol. J. 2008, 6, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Ma, C.; Feng, J.; Xu, S.; Wang, L.; Li, F.; Li, Y.; Zhang, R.; Zhang, X.; Xue, J.; et al. Transcriptome dynamics during maize endosperm development. PLoS ONE 2016, 11, e0163814. [Google Scholar] [CrossRef]
- Miclaus, M.; Wu, Y.; Xu, J.H.; Dooner, H.K.; Messing, J. The maize high-lysine mutant opaque7 is defective in an Acyl-CoA synthetase-like protein. Genetics 2011, 189, 1271–1280. [Google Scholar] [CrossRef]
- Bemer, M.; Heijmans, K.; Airoldi, C.; Davies, B.; Angenent, G.C. An atlas of type I MADS box gene expression during female gametophyte and seed development in Arabidopsis. Plant Physiol. 2010, 154, 287–300. [Google Scholar] [CrossRef]
- Gontarek, B.C.; Neelakandan, A.K.; Wu, H.; Becraft, P.W. NKD Transcription factors are central regulators of maize endosperm development. Plant Cell 2016, 28, 2916–2936. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Gene Number | Module | Gene Number | Module | Gene Number |
|---|---|---|---|---|---|
| Bisque4 | 527 | Dark turquoise | 438 | Royal blue | 86 |
| Brown | 429 | Light green | 3 | Sienna3 | 26 |
| Brown4 | 77 | Light steel blue1 | 1230 | Sky blue | 71 |
| Cyan | 62 | Navajo white2 | 21 | Skyblue3 | 14 |
| Dark green | 622 | Orange | 2465 | Thistle1 | 21 |
| Dark magenta | 104 | Plum2 | 89 | ||
| Dark red | 115 | Yellow | 327 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelly Sandrine, A.M.; Zhao, H.; Qin, Y.; Sun, Q.; Gong, D.; Pan, Z.; Qiu, F. 22KD Zein Content Coordinates Transcriptional Activity during Starch Synthesis in Maize Endosperm. Agronomy 2020, 10, 624. https://doi.org/10.3390/agronomy10050624
Nelly Sandrine AM, Zhao H, Qin Y, Sun Q, Gong D, Pan Z, Qiu F. 22KD Zein Content Coordinates Transcriptional Activity during Starch Synthesis in Maize Endosperm. Agronomy. 2020; 10(5):624. https://doi.org/10.3390/agronomy10050624
Chicago/Turabian StyleNelly Sandrine, Ada Menie, Hailiang Zhao, Yao Qin, Qin Sun, Dianming Gong, Zhenyuan Pan, and Fazhan Qiu. 2020. "22KD Zein Content Coordinates Transcriptional Activity during Starch Synthesis in Maize Endosperm" Agronomy 10, no. 5: 624. https://doi.org/10.3390/agronomy10050624
APA StyleNelly Sandrine, A. M., Zhao, H., Qin, Y., Sun, Q., Gong, D., Pan, Z., & Qiu, F. (2020). 22KD Zein Content Coordinates Transcriptional Activity during Starch Synthesis in Maize Endosperm. Agronomy, 10(5), 624. https://doi.org/10.3390/agronomy10050624