Divergent Genomic Selection for Herbage Accumulation and Days-To-Heading in Perennial Ryegrass

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Genotyping and Determination of GEBVs
2.2. Selection and Polycross Isolations
2.3. Evalaution Trials
3. Results and Discussion
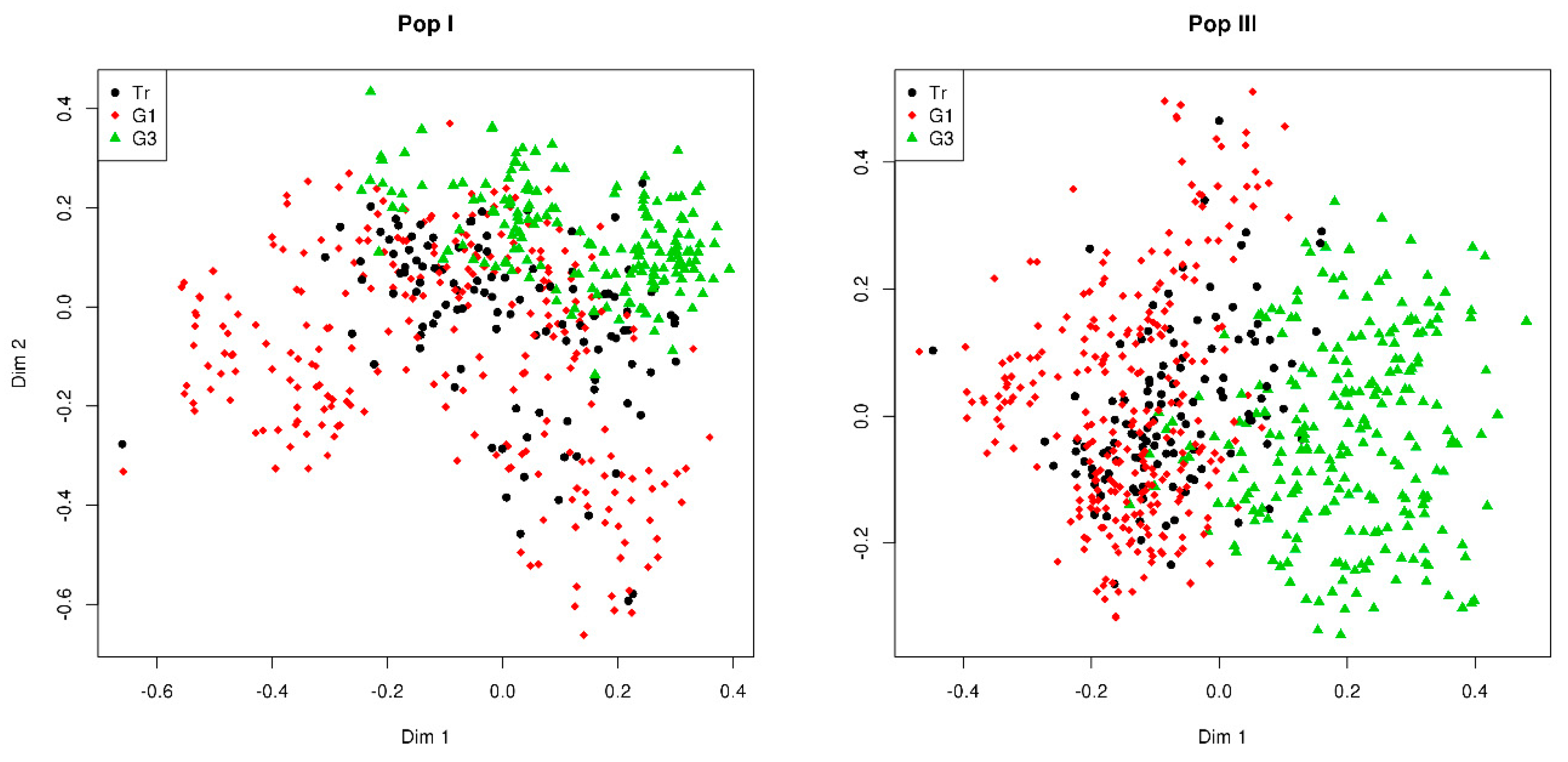
3.1. GBS Data and GEBVs
3.2. Phenotypic Evaluation of Divergent Genomic Selections
3.2.1. Genomic Selection for DTH
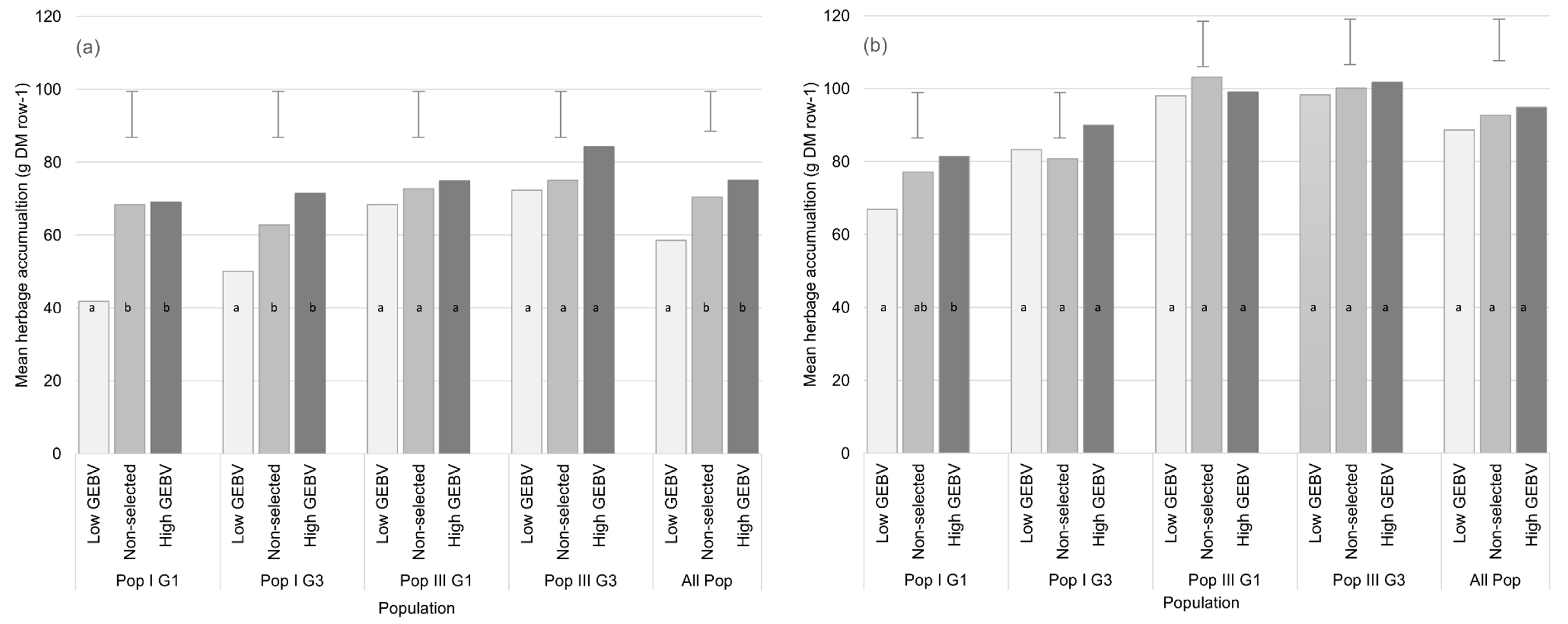
3.2.2. Genomic Selection for HA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Williams, W.; Easton, H.; Jones, C. Future options and targets for pasture plant breeding in New Zealand. N. Z. Agric. Res. 2007, 50, 223–248. [Google Scholar] [CrossRef]
- Thorrold, B.S.; Doyle, P.T. Nature or nurture–forces shaping the current and future state of dairy farming in New Zealand and Australia. In Meeting the Challenges for Pasture Based Dairying, Proceedings of the 3rd Australasian Dairy Science Symposium; University of Melbourne: Melbourne, VIC, Australia, 18–20 September 2007. [Google Scholar]
- McEvoy, M.; O’Donovan, M.; Shalloo, L. Development and application of an economic ranking index for perennial ryegrass cultivars. J. Dairy Sci. 2011, 94, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, M.; McHugh, N.; McEvoy, M.; Grogan, D.; Shalloo, L. Combining seasonal yield, silage dry matter yield, quality and persistency in an economic index to assist perennial ryegrass variety selection. J. Agric. Sci. 2017, 155, 556–568. [Google Scholar] [CrossRef]
- Chapman, D.; Bryant, J.; Olayemi, M.; Edwards, G.; Thorrold, B.; McMillan, W.; Kerr, G.A.; Judson, G.; Cookson, T.; Moorhead, A.; et al. An economically based evaluation index for perennial and short-term ryegrasses in New Zealand dairy farm systems. Grass Forage Sci. 2017, 72, 1–21. [Google Scholar] [CrossRef]
- Leddin, C.; Jacobs, J.; Smith, K.; Giri, K.; Malcolm, B.; Ho, C. Development of a system to rank perennial ryegrass cultivars according to their economic value to dairy farm businesses in south-eastern Australia. Anim. Prod. Sci. 2018, 58, 1552–1558. [Google Scholar] [CrossRef]
- Wilkins, P.W.; Humphreys, M.O. Progress in breeding perennial forage grasses for temperate agriculture. J. Agric. Sci. 2003, 140, 129–150. [Google Scholar] [CrossRef]
- Easton, H.S.; Amyes, J.M.; Cameron, N.E.; Green, R.B.; Kerr, G.A.; Norriss, M.G.; Stewart, A.V. Pasture plant breeding in New Zealand: Where to from here? N. Z. Grassl. Assoc. 2002, 64, 173–179. [Google Scholar]
- McDonagh, J.; O’Donovan, M.; McEvoy, M.; Gilliland, T.J. Genetic gain in perennial ryegrass (Lolium perenne) varieties 1973 to 2013. Euphytica 2016, 212, 187–199. [Google Scholar] [CrossRef]
- Sampoux, J.-P.; Baudouin, P.; Bayle, B.; Béguier, V.; Bourdon, P.; Chosson, J.-F.; Deneufbourg, F.; Galbrun, C.; Ghesquière, M.; Noël, D.; et al. Breeding perennial grasses for forage usage: An experimental assessment of trait changes in diploid perennial ryegrass (Lolium perenne L.) cultivars released in the last four decades. Field Crop. Res. 2011, 123, 117–129. [Google Scholar] [CrossRef]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef]
- Humphreys, M.O. The contribution of conventional plant breeding to forage crop improvement. In Proceedings of the 18th International Grassland Congress, Winnipeg, and Saskatoon, SK, Canada, 8–17 June 1997. [Google Scholar]
- Casler, M.D.; Brummer, E.C. Theoretical expected genetic gains for among-and-within-family selection methods in perennial forage crops. Crop Sci. 2008, 48, 890–902. [Google Scholar] [CrossRef]
- Conaghan, P.; Casler, M.; McGilloway, D.; O’Kiely, P.; Dowley, L. Genotype × environment interactions for herbage yield of perennial ryegrass sward plots in Ireland. Grass Forage Sci. 2008, 63, 107–120. [Google Scholar] [CrossRef]
- Casler, M.D. Genetic variation within eight populations of perennial forage grasses. Plant Breed. 1998, 117, 243–249. [Google Scholar] [CrossRef]
- Hayward, M.D.; Vivero, J.L. Selection for yield in Lolium perenne. II. performance of spaced plant selections under competitive conditions. Euphytica 1984, 33, 787–800. [Google Scholar] [CrossRef]
- Waldron, B.L.; Robins, J.G.; Peel, M.D.; Jensen, K.B. Predicted efficiency of spaced-plant selection to indirectly improve tall fescue sward yield and quality. Crop Sci. 2008, 48, 443–449. [Google Scholar] [CrossRef]
- Hayes, B.J.; Cogan, N.O.; Pembleton, L.W.; Goddard, M.E.; Wang, J.; Spangenberg, G.C.; Forster, J.W. Prospects for genomic selection in forage plant species. Plant Breed. 2013, 132, 133–143. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Faville, M.J.; Ganesh, S.; Cao, M.; Jahufer, M.Z.Z.; Bilton, T.P.; Easton, H.S.; Ryan, D.L.; Trethewey, J.A.K.; Rolston, M.P.; Griffiths, A.G.; et al. Predictive ability of genomic selection models in a multi-population perennial ryegrass training set using genotyping-by-sequencing. Theor. Appl. Genet. 2018, 131, 703–720. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, J.; Cogan, N.O.I.; Pembleton, L.W.; Badenhorst, P.; Forster, J.W.; Spangenberg, G.C.; Hayes, B.J.; Daetwyler, H.D. Optimizing resource allocation in a genomic breeding program for perennial ryegrass to balance genetic gain, cost, and inbreeding. Crop Sci. 2017, 57, 243–252. [Google Scholar] [CrossRef]
- Grinberg, N.F.; Lovatt, A.; Hegarty, M.; Lovatt, A.; Skøt, K.P.; Kelly, R.; Blackmore, T.; Thorogood, D.; King, R.D.; Armstead, I.; et al. Implementation of genomic prediction in Lolium perenne (L.) breeding populations. Front. Plant Sci. 2016, 7, 133. [Google Scholar] [CrossRef]
- Arojju, S.K.; Cao, M.; Zulfi Jahufer, M.Z.; Barrett, B.A.; Faville, M.J. Genomic predictive ability for foliar nutritive traits in perennial ryegrass. G3 Genes Genomes Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fè, D.; Ashraf, B.H.; Pedersen, M.G.; Janss, L.; Byrne, S.; Roulund, N.; Lenk, I.; Didion, T.; Asp, T.; Jensen, C.S.; et al. Accuracy of genomic prediction in a commercial perennial ryegrass breeding program. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Fè, D.; Cericola, F.; Byrne, S.; Lenk, I.; Ashraf, B.H.; Pedersen, M.G.; Roulund, N.; Asp, T.; Janss, L.; Jensen, C.S.; et al. Genomic dissection and prediction of heading date in perennial ryegrass. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pembleton, L.W.; Inch, C.; Baillie, R.C.; Drayton, M.C.; Thakur, P.; Ogaji, Y.O.; Spangenberg, G.C.; Forster, J.W.; Daetwyler, H.D.; Cogan, N.O.I. Exploitation of data from breeding programs supports rapid implementation of genomic selection for key agronomic traits in perennial ryegrass. Theor. Appl. Genet. 2018, 131, 1891–1902. [Google Scholar] [CrossRef]
- Anderson, C.B.; Franzmayr, B.K.; Hong, S.W.; Larking, A.C.; van Stijn, T.C.; Tan, R.; Moraga, R.; Faville, M.J.; Faville, A.G. Protocol: A versatile, inexpensive, high-throughput plant genomic DNA extraction method suitable for genotyping-by-sequencing. Plant Methods 2018, 14, 75. [Google Scholar] [CrossRef]
- Hahn, H.; Huth, W.; Schöberlein, W.; Diepenbrock, W.; Weber, W.E. Detection of endophytic fungi in Festuca spp. by means of tissue print immunoassay. Plant Breed. 2003, 122, 217–222. [Google Scholar] [CrossRef]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Byrne, S.L.; Nagy, I.; Pfeifer, M.; Armstead, I.; Swain, S.; Studer, B.; Mayer, K.; Campbell, J.D.; Czaban, A.; Hentrup, S.; et al. A synteny-based draft genome sequence of the forage grass Lolium perenne. Plant J. 2015, 84, 816–826. [Google Scholar] [CrossRef]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; van Stijn, T.C.; Kristjánsson, T.; Clarke, S.M. Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genom. 2015, 16, 1047. [Google Scholar] [CrossRef]
- Lee, J.M.; Thom, E.R.; Chapman, D.F.; Wynn, K.; Waugh, D.; Rossi, L.; Faville, M.J.; Bell, N.L.; McNeill, M.R. High perennial ryegrass seeding rates do not negatively impact pasture sward persistence. Grass Forage Sci. 2017. [Google Scholar] [CrossRef]
- Lee, J.M.; Thom, E.R.; Waugh, C.D.; Bell, N.L.; McNeill, M.R.; Wilson, D.J.; Chapman, D.F. Trajectory and causes of decline in the botanical composition of dairy-grazed pasture in the Waikato. J. N. Z. Grassl. 2017, 79, 83–88. [Google Scholar]
- Jahufer, M.Z.Z.; Luo, D. DeltaGen: A comprehensive decision support tool for plant breeders. Crop Sci. 2018, 58, 1118–1131. [Google Scholar] [CrossRef]
- Galwey, N.W. Introduction to Mixed Modelling: Beyond Regression and Analysis of Variance; John Wiley & Sons, Ltd.: West Sussex, UK, 2006. [Google Scholar]
- Gallagher, J.A.; Turner, L.B.; Cairns, A.J.; Farrell, M.; Lovatt, J.A.; Skøt, K.; Armstead, I.P.; Humphreys, M.O.; Roldan-Ruiz, I. Genetic differentiation in response to selection for water-soluble carbohydrate content inperennial ryegrass (Lolium perenne L.). Bioenergy Res. 2015, 8, 77–90. [Google Scholar] [CrossRef]
- Byrne, S.L.; Conaghan, P.; Barth, S.; Arojju, S.K.; Casler, M.; Michel, T.; Velmurugan, J.; Milbourne, D. Using variable importance measures to identify a small set of SNPs to predict heading date in perennial ryegrass. Sci. Rep. 2017, 7, 3566. [Google Scholar] [CrossRef]
- Frandsen, K.J. Variability and inheritance of digestibility in perennial ryegrass (Lolium perenne), meadow fescue (Festuca pratensis) and cocksfoot (Dactylis glomerata). Acta Agric. Scand. 1986, 36, 241–263. [Google Scholar] [CrossRef]
- Fè, D.; Pedersen, M.G.; Jensen, C.S.; Jensen, J. Genetic and environmental variation in a commercial breeding program of perennial ryegrass. Crop Sci. 2015, 55, 631–640. [Google Scholar] [CrossRef]
- Clark, S.A.; Hickey, J.M.; Daetwyler, H.D.; van der Werf, J.H.J. The importance of information on relatives for the prediction of genomic breeding values and the implications for the makeup of reference data sets in livestock breeding schemes. Genet. Sel. Evol. 2012, 44, 4. [Google Scholar] [CrossRef]
- Scutari, M.; Mackay, I.; Balding, D. Using genetic distance to infer the accuracy of genomic prediction. PLoS Genet. 2016, 12, e1006288. [Google Scholar] [CrossRef]
- Wientjes, Y.C.J.; Veerkamp, R.F.; Calus, M.P.L. The effect of linkage disequilibrium and family relationships on the reliability of genomic prediction. Genetics 2013, 193, 621. [Google Scholar] [CrossRef]
- Habier, D.; Tetens, J.; Seefried, F.-R.; Lichtner, P.; Thaller, G. The impact of genetic relationship information on genomic breeding values in German Holstein cattle. Genet. Sel. Evol. 2010, 42, 5. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. The impact of genetic relationship information on genome-assisted breeding values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Arojju, S.K.; Barth, S.; Milbourne, D.; Conaghan, P.; Velmurugan, J.; Hodkinson, T.R.; Byrne, S.L. Markers associated with heading and aftermath heading in perennial ryegrass full-sib families. BMC Plant Biol. 2016, 16, 160. [Google Scholar] [CrossRef] [PubMed]
- Heslot, N.; Jannink, J.-L.; Sorrells, M.E. Perspectives for genomic selection applications and research in plants. Crop Sci. 2015, 55, 1–12. [Google Scholar] [CrossRef]
- Ghamkhar, K.; Irie, K.; Hagedorn, M.; Hsiao, J.; Fourie, J.; Gebbie, S.; Hoyos-Villegas, V.; George, R.; Stewart, A.; Inch, C.; et al. Real-time, non-destructive and in-field foliage yield and growth rate measurement in perennial ryegrass (Lolium perenne L.). Plant Methods 2019, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Badenhorst, P.; Phelan, A.; Pembleton, L.; Shi, F.; Cogan, N.; Spangenberg, G.; Smith, K. Using sensors and unmanned aircraft systems for high-throughput phenotyping of biomass in perennial ryegrass breeding trials. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Population | Total | ||||||
|---|---|---|---|---|---|---|---|
| Pop I Training | Pop I G1 | Pop I G3 | Pop III Training | Pop III G1 | Pop III G3 | ||
| No. Plants with GEBV | - | 278 | 177 | - | 280 | 235 | 970 |
| No. Plants Available for Selection | - | 238 | 167 | - | 253 | 137 | 795 |
| Mean Coefficient of Relatedness 1 | |||||||
| Within Population | 0.175 | 0.199 | 0.234 | 0.107 | 0.129 | 0.172 | - |
| To Training Set | 0.175 | 0.163 | 0.137 | 0.107 | 0.100 | 0.084 | - |
| Location | Population | Mean HA, g DM Row−1 (± SE) | L.S.D0.05 | Selection Response, g DM Row−1 (%) | ||||
|---|---|---|---|---|---|---|---|---|
| High GEBV | Non-Selected | Low GEBV | High-Low | High-Non-Selected | Non-Selected-Low | |||
| Waikato | Pop I G1 | 69.0 (5.35) | 68.3 (5.27) | 41.8 (5.39) | 12.7 | 27.2 * (65%) | 0.7 ns (1.0%) | 26.5 * (63%) |
| Pop I G3 | 71.5 (5.32) | 62.8 (5.37) | 50.1 (5.40) | 12.7 | 21.4 * (43%) | 8.7 ns (14%) | 12.7 ns (25%) | |
| Pop III G1 | 74.9 (5.83) | 72.7 (5.32) | 68.3 (5.35) | 12.7 | 6.6 ns (10%) | 2.2 ns (3%) | 4.4 ns (6%) | |
| Pop III G3 | 84.2 (5.40) | 75.0 (5.40) | 72.3 (5.33) | 12.7 | 11.9 ns (16%) | 9.2 ns (12%) | 2.7 ns (4%) | |
| All Pop | 75.0 (2.33) | 70.4 (3.17) | 58.5 (3.22) | 11.0 | 16.5 * (28%) | 4.6 ns (7%) | 11.9 * (20%) | |
| Manawatu | Pop I G1 | 81.4 (4.65) | 77.1 (4.71) | 66.9 (4.62) | 12.5 | 14.5 * (22%) | 4.3 ns (6%) | 10.2 ns (15%) |
| Pop I G3 | 90.0 (4.74) | 80.8 (4.62) | 83.3 (4.64) | 12.5 | 6.7 ns (8%) | 9.2 ns (11%) | −2.5 ns (3%) | |
| Pop III G1 | 99.0 (4.72) | 103.2 (4.63) | 98.1 (4.69) | 12.5 | 0.9 ns (1%) | −4.2 ns (4%) | 5.1 ns (5%) | |
| Pop III G3 | 101.8 (4.64) | 100.2 (4.70) | 98.3 (4.70) | 12.5 | 3.5 ns (4%) | 1.6 ns (2%) | 1.9 ns (2%) | |
| All Pop | 94.9 (2.40) | 92.8 (2.41) | 88.7 (2.38) | 11.4 | 6.2 ns (7%) | 2.1 ns (2%) | 4.1 ns (5%) | |
| Combined | Pop I G1 | 75.6 (6.43) | 73.2 (6.45) | 52.8 (6.52) | 12.2 | 22.8 * (43%) | 2.4 ns (3%) | 20.4 * (39%) |
| Pop I G3 | 83.0 (6.49) | 72.1 (6.45) | 67.3 (6.46) | 12.2 | 15.7 * (23%) | 10.9 ns (15%) | 4.8 ns (7%) | |
| Pop III G1 | 85.9 (6.37) | 84.0 (6.39) | 82.0 (6.39) | 12.2 | 3.9 ns (5%) | 1.9 ns (2%) | 2.0 ns (2%) | |
| Pop III G3 | 91.1 (6.40) | 85.7 (6.38) | 84.3 (6.40) | 12.2 | 6.8 ns (8%) | 5.4 ns (6%) | 1.4 ns (2%) | |
| All Pop | 81.5 (5.43) | 76.8 (5.45) | 69.4 (5.43) | 11.8 | 12.1 * (17%) | 4.7 ns (6%) | 7.4 ns (11%) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faville, M.J.; Cao, M.; Schmidt, J.; Ryan, D.L.; Ganesh, S.; Jahufer, M.Z.Z.; Hong, S.W.; George, R.; Barrett, B.A. Divergent Genomic Selection for Herbage Accumulation and Days-To-Heading in Perennial Ryegrass. Agronomy 2020, 10, 340. https://doi.org/10.3390/agronomy10030340
Faville MJ, Cao M, Schmidt J, Ryan DL, Ganesh S, Jahufer MZZ, Hong SW, George R, Barrett BA. Divergent Genomic Selection for Herbage Accumulation and Days-To-Heading in Perennial Ryegrass. Agronomy. 2020; 10(3):340. https://doi.org/10.3390/agronomy10030340
Chicago/Turabian StyleFaville, Marty J., Mingshu Cao, Jana Schmidt, Douglas L. Ryan, Siva Ganesh, M. Z. Zulfi Jahufer, Soon Won Hong, Richard George, and Brent A. Barrett. 2020. "Divergent Genomic Selection for Herbage Accumulation and Days-To-Heading in Perennial Ryegrass" Agronomy 10, no. 3: 340. https://doi.org/10.3390/agronomy10030340
APA StyleFaville, M. J., Cao, M., Schmidt, J., Ryan, D. L., Ganesh, S., Jahufer, M. Z. Z., Hong, S. W., George, R., & Barrett, B. A. (2020). Divergent Genomic Selection for Herbage Accumulation and Days-To-Heading in Perennial Ryegrass. Agronomy, 10(3), 340. https://doi.org/10.3390/agronomy10030340