Comparison of Soil Bacterial Communities from Juvenile Maize Plants of a Long-Term Monoculture and a Natural Grassland

 , ,
, ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sites and Sampling
2.2. Physical and Chemical Analysis of the Soil Samples
2.3. MicroResp™ Substrate induced Catabolic Activity Measurements
2.4. Bacterial Diversity Analysis by Next-Generation DNA Sequencing
2.5. Statistical Analysis of the Data
3. Results
3.1. Physical and Chemical Properties of the Soil Samples
3.2. Substrate Induced Catabolic Activities of Soil Microbial Communities
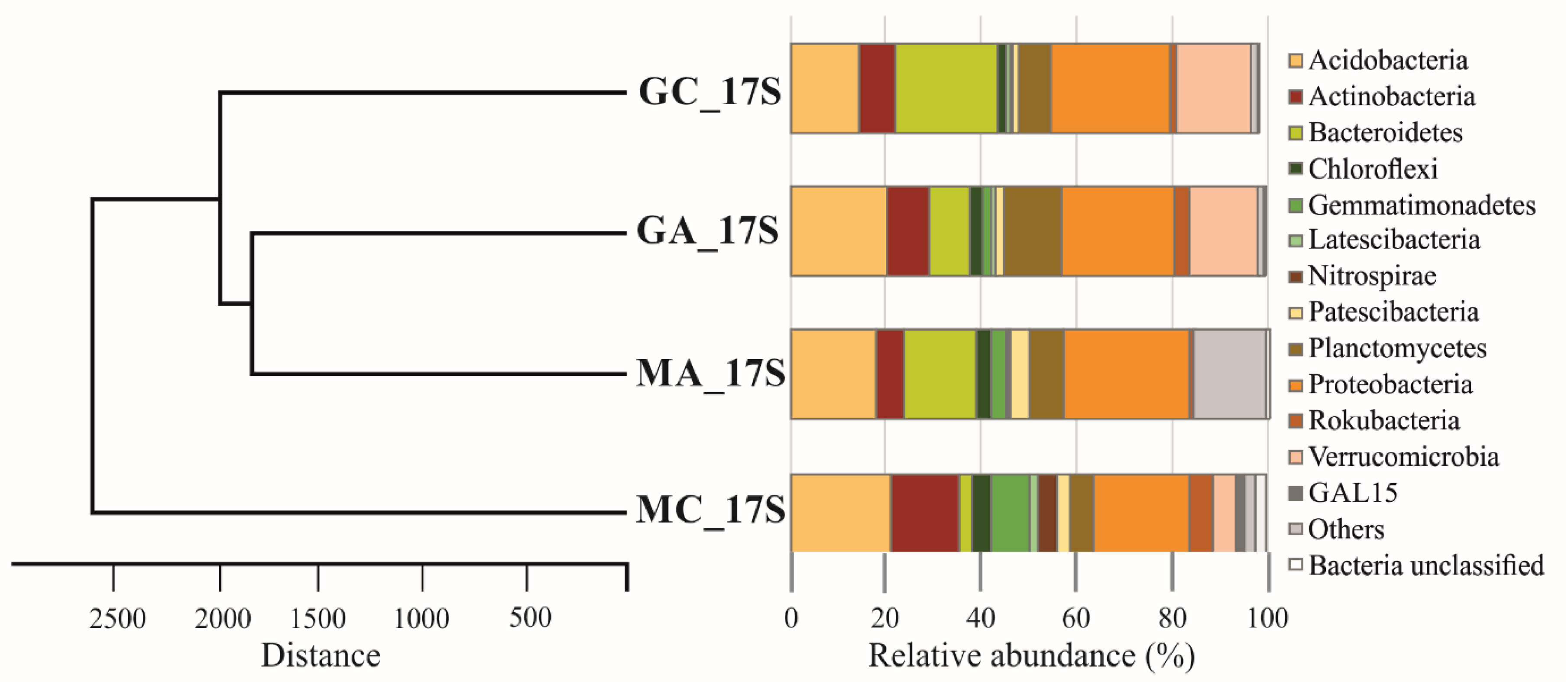
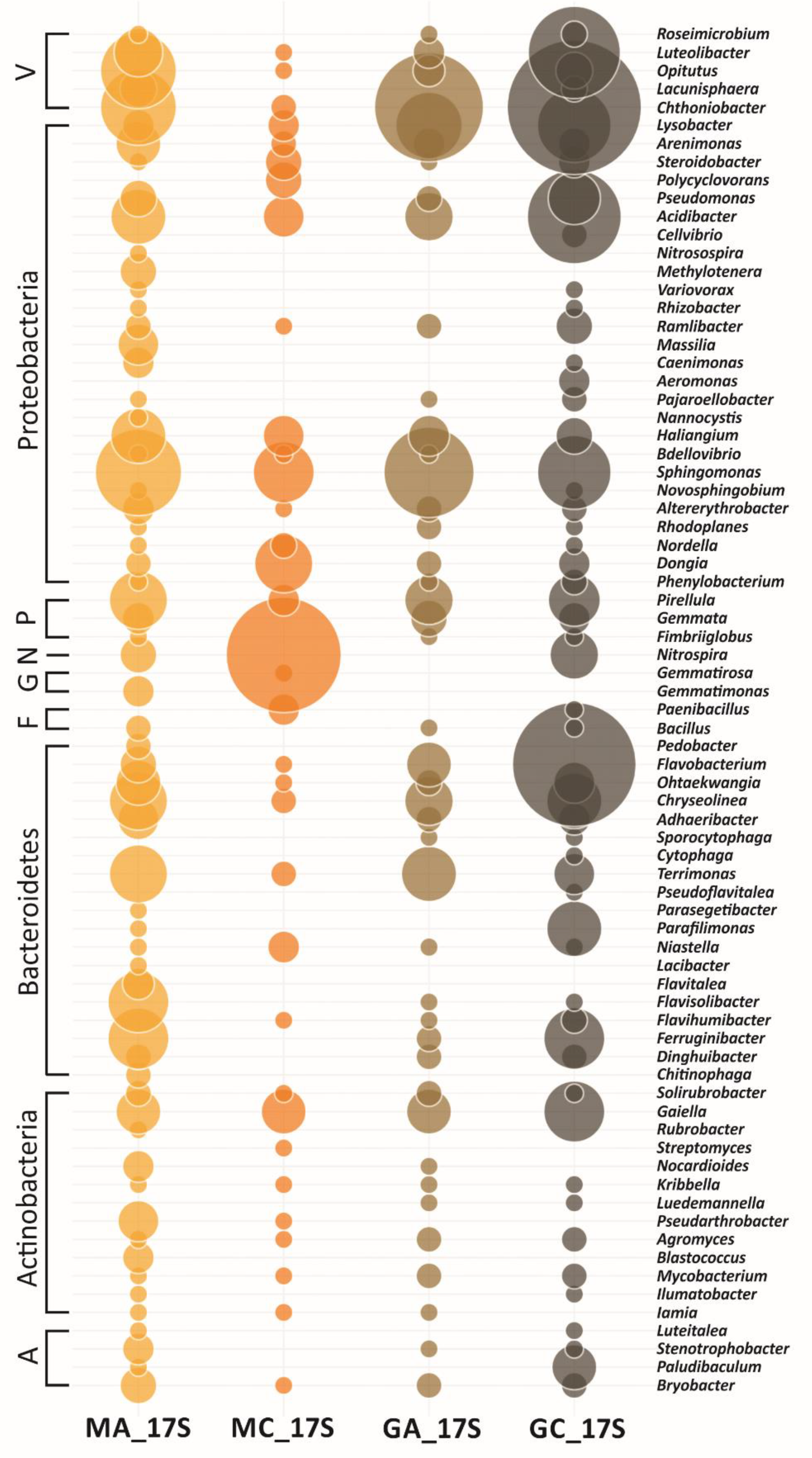
3.3. Metagenomic Diversity of Soil Bacterial Communities
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diacono, M.; Montemurro, F. Long-term effects of organic amendments on soil fertility. In Sustainable Agriculture Volume 2; Lichtfouse, E., Hamelin, M., Navarrete, M., Debaeke, P., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 30, pp. 761–786. [Google Scholar]
- Gouda, S.; Kerry, R.G.; Das, G.; Paramithiotis, S.; Shin, H.-S.; Patra, J.K. Revitalization of plant growth promoting rhizobacteria for sustainable development in agriculture. Microbiol. Res. 2018, 206, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Bandick, A.K.; Dick, R.P. Field management effects on soil enzyme activities. Soil Biol. Biochem. 1999, 31, 1471–1479. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Hayat, R.; Ali, S.; Amara, U.; Khalid, R.; Ahmed, I. Soil beneficial bacteria and their role in plant growth promotion: A review. Ann. Microbiol. 2010, 60, 579–598. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Mendes, R.; Raaijmakers, J.M. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol. Biol. 2016, 90, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef]
- Visioli, G.; Sanangelantoni, A.M.; Vamerali, T.; Dal Cortivo, C.; Blandino, M. 16S rDNA profiling to reveal the influence of seed-applied biostimulants on the rhizosphere of young maize plants. Molecules 2018, 23, 1461. [Google Scholar] [CrossRef]
- Sradnick, A.; Murugan, R.; Oltmanns, M.; Raupp, J.; Joergensen, R.G. Changes in functional diversity of the soil microbial community in a heterogeneous sandy soil after long-term fertilization with cattle manure and mineral fertilizer. Appl. Soil Ecol. 2013, 63, 23–28. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Zhao, B.; Zhou, G.; Ruan, L. Bacterial community structure in maize stubble-amended soils with different moisture levels estimated by bar-coded pyrosequencing. Appl. Soil Ecol. 2015, 86, 62–70. [Google Scholar] [CrossRef]
- Ramirez-Villanueva, D.A.; Bello-López, J.M.; Navarro-Noya, Y.E.; Luna-Guido, M.; Verhulst, N.; Govaerts, B.; Dendooven, L. Bacterial community structure in maize residue amended soil with contrasting management practices. Appl. Soil Ecol. 2015, 90, 49–59. [Google Scholar] [CrossRef]
- van Wyk, D.A.B.; Adeleke, R.; Rhode, O.H.J.; Bezuidenhout, C.C.; Mienie, C. Ecological guild and enzyme activities of rhizosphere soil microbial communities associated with Bt-maize cultivation under field conditions in North West Province of South Africa. J. Basic Microbiol. 2017, 57, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Liu, X.; Liang, Y.; Niu, J.; Xiao, Y.; Gu, Y.; Ma, L.; Meng, D.; Zhang, Y.; Huang, W.; et al. Maize growth responses to soil microbes and soil properties after fertilization with different green manures. Appl. Microbiol. Biotechnol. 2017, 101, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, X.; Li, X.; Wang, J.; Li, X.; Guo, Q.; Yu, Z.; Yang, T.; Zhang, H. Long-term no-tillage and different residue amounts alter soil microbial community composition and increase the risk of maize root rot in northeast China. Soil Tillage Res. 2020, 196, 104452. [Google Scholar] [CrossRef]
- Chelius, M.K.; Triplett, E.W. The Diversity of Archaea and Bacteria in association with the roots of Zea mays L. Microb. Ecol. 2001, 41, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Correa-Galeote, D.; Bedmar, E.J.; Arone, G.J. Maize endophytic bacterial diversity as affected by soil cultivation history. Front. Microbiol. 2018, 9, 484. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, E.; Benizri, E.; Guckert, A. Impact of growth stage on the bacterial community structure along maize roots, as determined by metabolic and genetic fingerprinting. Appl. Soil Ecol. 2002, 19, 135–145. [Google Scholar] [CrossRef]
- Li, C.H.; Ma, B.L.; Zhang, T.Q. Soil bulk density effects on soil microbial populations and enzyme activities during the growth of maize (Zea mays L.) planted in large pots under field exposure. Can. J. Soil Sci. 2002, 82, 147–154. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef]
- Garbeva, P.; van Veen, J.A.; van Elsas, J.D. Microbial diversity in soil: Selection of microbial populations by plant and soil type and implications for disease suppressiveness. Annu. Rev. Phytopathol. 2004, 42, 243–270. [Google Scholar] [CrossRef]
- Berzsenyi, Z. Significance of the 50-year-old long-term experiments in Martonvásár in improving crop production. Acta Agron. Hungarica 2010, 58, 23–34. [Google Scholar] [CrossRef]
- Csitári, G.; Hoffmann, S. Comparative study on soil biological parameters at a long-term field experiment. Arch. Agron. Soil. Sci. 2005, 51, 563–569. [Google Scholar] [CrossRef]
- Magurno, F.; Sasvári, Z.; Barchi, L.; Posta, K. From monoculture to Norfolk system: How the number of crops in rotation can influence the biodiversity of arbuscular mycorrhiza assemblages in the soil. Open J. Ecol. 2014, 4, 1080–1088. [Google Scholar] [CrossRef][Green Version]
- Mayer, Z.; Sasvári, Z.; Szentpéteri, V.; Pethőné Rétháti, B.; Vajna, B.; Posta, K. Effect of long-term cropping systems on the diversity of the soil bacterial communities. Agronomy 2019, 9, 878. [Google Scholar] [CrossRef]
- Centeri, C.; Jakab, G.; Szabo, S.; Farsang, A.; Barta, K.; Szalai, Z.; Biro, Z. Comparison of particle-size analyzing laboratory methods. Environ. Eng. Manag. J. 2015, 14, 1125–1135. [Google Scholar] [CrossRef]
- Rieder, Á.; Madarász, B.; Szabó, J.; Zacháry, D.; Vancsik, A.; Ringer, M.; Szalai, Z.; Jakab, G. Soil organic matter alteration velocity due to land-use change: A case study under conservation agriculture. Sustainability 2018, 10, 943. [Google Scholar] [CrossRef]
- Jakab, G.; Filep, T.; Király, C.; Madarász, B.; Zacháry, D.; Ringer, M.; Vancsik, A.; Gáspár, L.; Szalai, Z. Differences in mineral phase associated soil organic matter composition due to varying tillage intensity. Agronomy 2019, 9, 700. [Google Scholar] [CrossRef]
- Buurman, P.; van Lagen, B.; Velthorst, E.J. (Eds.) Manual for Soil and Water Analysis; Backhuys Publishers: Leiden, The Netherlands, 1996; ISBN 9789073348585. [Google Scholar]
- Campbell, C.D.; Chapman, S.J.; Cameron, C.M.; Davidson, M.S.; Potts, J.M. A Rapid microtiter plate method to measure carbon dioxide evolved from carbon substrate amendments so as to determine the physiological profiles of soil microbial communities by using whole soil. Appl. Environ. Microbiol. 2003, 69, 3593–3599. [Google Scholar] [CrossRef]
- Mucsi, M.; Csontos, P.; Borsodi, A.; Krett, G.; Gazdag, O.; Szili-Kovács, T. Use of the microrespiration method to analyse the metabolic activity patterns in the soil of four characteristic sodic plant associations. Agrokémia és Talajt. 2017, 66, 165–179. [Google Scholar] [CrossRef]
- Szili-Kovács, T.; Bárány, Á.; Füzy, A.; Takács, T.; Krett, G.; Kovács, R.; Borsodi, A. Analysis of the microbial metabolic activity patterns and mycorrhizal fungal colonisation in the rhizosphere of three soils neighbouring sodic lakes. Agrokémia és Talajt. 2017, 66, 149–164. [Google Scholar] [CrossRef][Green Version]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Engelbrektson, A.; Ochman, H.; Hugenholtz, P. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 2010, 12, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Busse, H.-J.; Tindall, B.J.; Ludwig, W.; Rosselló-Móra, R.; Kämpfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 2010, 60, 249–266. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Package “Vegan”: Community Ecology Package 2018. Available online: https://github.com/vegandevs/vegan (accessed on 1 February 2020).
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Jakab, G.; Madarász, B.; Szabó, J.; Tóth, A.; Zacháry, D.; Szalai, Z.; Kertész, Á.; Dyson, J. Infiltration and soil loss changes during the growing season under ploughing and conservation tillage. Sustainability 2017, 9, 1726. [Google Scholar] [CrossRef]
- Schad, P.; van Huyssten, C.; Michéli, E. International soil classification system for naming soils and creating legends for soil maps. In World Reference Base for Soil Resources; FAO: Rome, Italy, 2015; ISBN 9789251083703. [Google Scholar]
- Creamer, R.E.; Stone, D.; Berry, P.; Kuiper, I. Measuring respiration profiles of soil microbial communities across Europe using MicroRespTM method. Appl. Soil Ecol. 2016, 97, 36–43. [Google Scholar] [CrossRef]
- Zhang, Z.; Liang, S.; Wang, J.; Zhang, X.; Mahamood, M.; Yu, J.; Zhang, X.; Liang, A.; Liang, W. Tillage and crop succession effects on soil microbial metabolic activity and carbon utilization in a clay loam soil. Eur. J. Soil Biol. 2018, 88, 97–104. [Google Scholar] [CrossRef]
- Kraffczyk, I.; Trolldenier, G.; Beringer, H. Soluble root exudates of maize: Influence of potassium supply and rhizosphere microorganisms. Soil Biol. Biochem. 1984, 16, 315–322. [Google Scholar] [CrossRef]
- Yun-Choi, H.S.; Kim, J.H.; Lee, J.R. Potential inhibitors of platelet aggregation from plant sources, III. J. Nat. Prod. 1987, 50, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Blume, E.; Bischoff, M.; Reichert, J.M.; Moorman, T.; Konopka, A.; Turco, R.F. Surface and subsurface microbial biomass, community structure and metabolic activity as a function of soil depth and season. Appl. Soil Ecol. 2002, 20, 171–181. [Google Scholar] [CrossRef]
- Gartzia-Bengoetxea, N.; Kandeler, E.; Martínez de Arano, I.; Arias-González, A. Soil microbial functional activity is governed by a combination of tree species composition and soil properties in temperate forests. Appl. Soil Ecol. 2016, 100, 57–64. [Google Scholar] [CrossRef]
- Turrini, A.; Caruso, G.; Avio, L.; Gennai, C.; Palla, M.; Agnolucci, M.; Tomei, P.E.; Giovannetti, M.; Gucci, R. Protective green cover enhances soil respiration and native mycorrhizal potential compared with soil tillage in a high-density olive orchard in a long term study. Appl. Soil Ecol. 2017, 116, 70–78. [Google Scholar] [CrossRef]
- Chaudhry, V.; Rehman, A.; Mishra, A.; Chauhan, P.S.; Nautiyal, C.S. Changes in bacterial community structure of agricultural land due to long-term organic and chemical amendments. Microb. Ecol. 2012, 64, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Steenwerth, K.L.; Jackson, L.E.; Calderon, F.J.; Scow, K.M.; Rolsotn, D.E. Response of microbial community composition and activity in agricultural and grassland soils after a simulated rainfall. Soil Biol. Biochem. 2005, 37, 2249–2262. [Google Scholar] [CrossRef]
- Buckley, D.H.; Schmidt, T.M. The structure of microbial communities in soil and the lasting impact of cultivation. Microb. Ecol. 2001, 42, 11–21. [Google Scholar] [CrossRef]
- Li, X.; Rui, J.; Mao, Y.; Yannarell, A.; Mackie, R. Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar. Soil Biol. Biochem. 2014, 68, 392–401. [Google Scholar] [CrossRef]
- Aguirre-von-Wobeser, E.; Rocha-Estrada, J.; Shapiro, L.R.; de la Torre, M. Enrichment of Verrucomicrobia, Actinobacteria and Burkholderiales drives selection of bacterial community from soil by maize roots in a traditional milpa agroecosystem. PLoS ONE 2018, 13, e0208852. [Google Scholar] [CrossRef]
- Johnston-Monje, D.; Lundberg, D.S.; Lazarovits, G.; Reis, V.M.; Raizada, M.N. Bacterial populations in juvenile maize rhizospheres originate from both seed and soil. Plant Soil 2016, 405, 337–355. [Google Scholar] [CrossRef]
- Yin, A.; Jia, Y.; Qiu, T.; Gao, M.; Cheng, S.; Wang, X.; Sun, Y. Poly-γ-glutamic acid improves the drought resistance of maize seedlings by adjusting the soil moisture and microbial community structure. Appl. Soil Ecol. 2018, 129, 128–135. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Ladau, J.; Clemente, J.C.; Leff, J.W.; Owens, S.M.; Pollard, K.S.; Knight, R.; Gilbert, J.A.; McCulley, R.L. Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 2013, 342, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.-L.; Zhang, X.-S.; Lu, X.-H.; Qin, R.; Bi, Y.-M.; Gao, W.-W. Effects of maize rotation on the physicochemical properties and microbial communities of American ginseng cultivated soil. Sci. Rep. 2019, 9, 8615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zuo, S.; Xu, L.; Zou, Y.; Song, W. Study on diversity of endophytic bacterial communities in seeds of hybrid maize and their parental lines. Arch. Microbiol. 2012, 194, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Lv, F.; Wang, X.; Yuan, M.; Li, J.; Wu, Q.; Sun, J. Flavobacterium endophyticum sp. nov., a nifH gene-harbouring endophytic bacterium isolated from maize root. Int. J. Syst. Evol. Microbiol. 2015, 65, 3900–3904. [Google Scholar] [CrossRef]
- Sun, J.-G.; Zhang, Y.-C.; Xu, J.; Hu, H.-Y. Isolation, identification and inoculation effect of nitrogen-fixing bacteria Sphingomonas GD542 from maize rhizosphere. Chinese J. Eco-Agriculture 2010, 18, 89–93. [Google Scholar] [CrossRef]
- Attard, E.; Poly, F.; Commeaux, C.; Laurent, F.; Terada, A.; Smets, B.F.; Recous, S.; Roux, X. Le Shifts between Nitrospira-and Nitrobacter-like nitrite oxidizers underlie the response of soil potential nitrite oxidation to changes in tillage practices. Environ. Microbiol. 2010, 12, 315–326. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Maize Monoculture | Natural Grassland | |||
|---|---|---|---|---|
| Horizon | A | C | A | C |
| Sample designations | MA_17S | MC_17S | GA_17S | GC_17S |
| clay (%, v/v) 1 | 24.9 ± 3.3 | 24.5 ± 0.7 | 20.7 ± 1.3 | 18.8 ± 0.3 |
| silt (%, v/v) 1 | 45.6 ± 1.9 | 44.9 ± 0.9 | 56.3 ± 3.3 | 30.7 ± 3.1 |
| sand (%, v/v) 1 | 29.4 ± 2.2 | 30.6 ± 1.5 | 22.9 ± 2.1 | 50.5 ± 3.4 |
| pH(dw)1 | 7.7 ± 0.2 ab | 8.1 ± 0.1 a | 7.4 ± 0.2 b | 8.2 ± 0.1 a |
| pH(KCl)1 | 7.3 ± 0.1 b | 7.6 ± 0.0 a | 7.1 ± 0.2 b | 7.6 ± 0.0 a |
| CaCO3 (%, m/m) 1 | 2.9 ± 2.3 a | 19.6 ± 1.6 a | 9.1 ± 0.9 a | 15.0 ± 1.4 a |
| SOC (g/kg) 1 | 8.3 ± 0.3 a | 1.2 ± 0.2 a | 26.1 ± 0.6 b | 6.5 ± 0.9 a |
| TN (g/kg) 1 | 0.6 ± 0.1 a | 0.1 ± 0.0 b | 1.4 ± 0.3 c | NM |
| C:N1 | 18.8 ± 4.0 | ND | 20.7 ± 1.3 | ND |
| Maize Monoculture | Natural Grassland | |||
|---|---|---|---|---|
| Horizon | A | C | A | C |
| Sample sign | MA_17S | MC_17S | GA_17S | GC_17S |
| Good’s coverage (%) | 100.0 | 98.7 | 98.0 | 97.8 |
| Number of OTUs | 2803 | 2063 | 2888 | 2469 |
| Chao 1 | 2803 | 2224 | 3070 | 2828 |
| ACE | 2804 | 2304 | 3277 | 2989 |
| Inverse Simpson’s (1/D) | 360 | 172 | 184 | 198 |
| Shannon’s diversity (H’) | 6.9 | 6.2 | 6.6 | 6.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ujvári, G.; Borsodi, A.K.; Megyes, M.; Mucsi, M.; Szili-Kovács, T.; Szabó, A.; Szalai, Z.; Jakab, G.; Márialigeti, K. Comparison of Soil Bacterial Communities from Juvenile Maize Plants of a Long-Term Monoculture and a Natural Grassland. Agronomy 2020, 10, 341. https://doi.org/10.3390/agronomy10030341
Ujvári G, Borsodi AK, Megyes M, Mucsi M, Szili-Kovács T, Szabó A, Szalai Z, Jakab G, Márialigeti K. Comparison of Soil Bacterial Communities from Juvenile Maize Plants of a Long-Term Monoculture and a Natural Grassland. Agronomy. 2020; 10(3):341. https://doi.org/10.3390/agronomy10030341
Chicago/Turabian StyleUjvári, Gergely, Andrea K. Borsodi, Melinda Megyes, Márton Mucsi, Tibor Szili-Kovács, Attila Szabó, Zoltán Szalai, Gergely Jakab, and Károly Márialigeti. 2020. "Comparison of Soil Bacterial Communities from Juvenile Maize Plants of a Long-Term Monoculture and a Natural Grassland" Agronomy 10, no. 3: 341. https://doi.org/10.3390/agronomy10030341
APA StyleUjvári, G., Borsodi, A. K., Megyes, M., Mucsi, M., Szili-Kovács, T., Szabó, A., Szalai, Z., Jakab, G., & Márialigeti, K. (2020). Comparison of Soil Bacterial Communities from Juvenile Maize Plants of a Long-Term Monoculture and a Natural Grassland. Agronomy, 10(3), 341. https://doi.org/10.3390/agronomy10030341