
Preparation and Characterization of Epoxy Resin Cross-Linked with High Wood Pyrolysis Bio-Oil Substitution by Acetone Pretreatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Acetone Pretreatment
2.3. Preparation of Bio-Oil/Epoxy Resins
2.4. Properties of Bonding Strength
2.5. ATR-FTIR Analysis
2.6. Properties of Soluble Resistance
2.7. DSC, TGA and UV-Vis Analysis
3. Results and Discussion
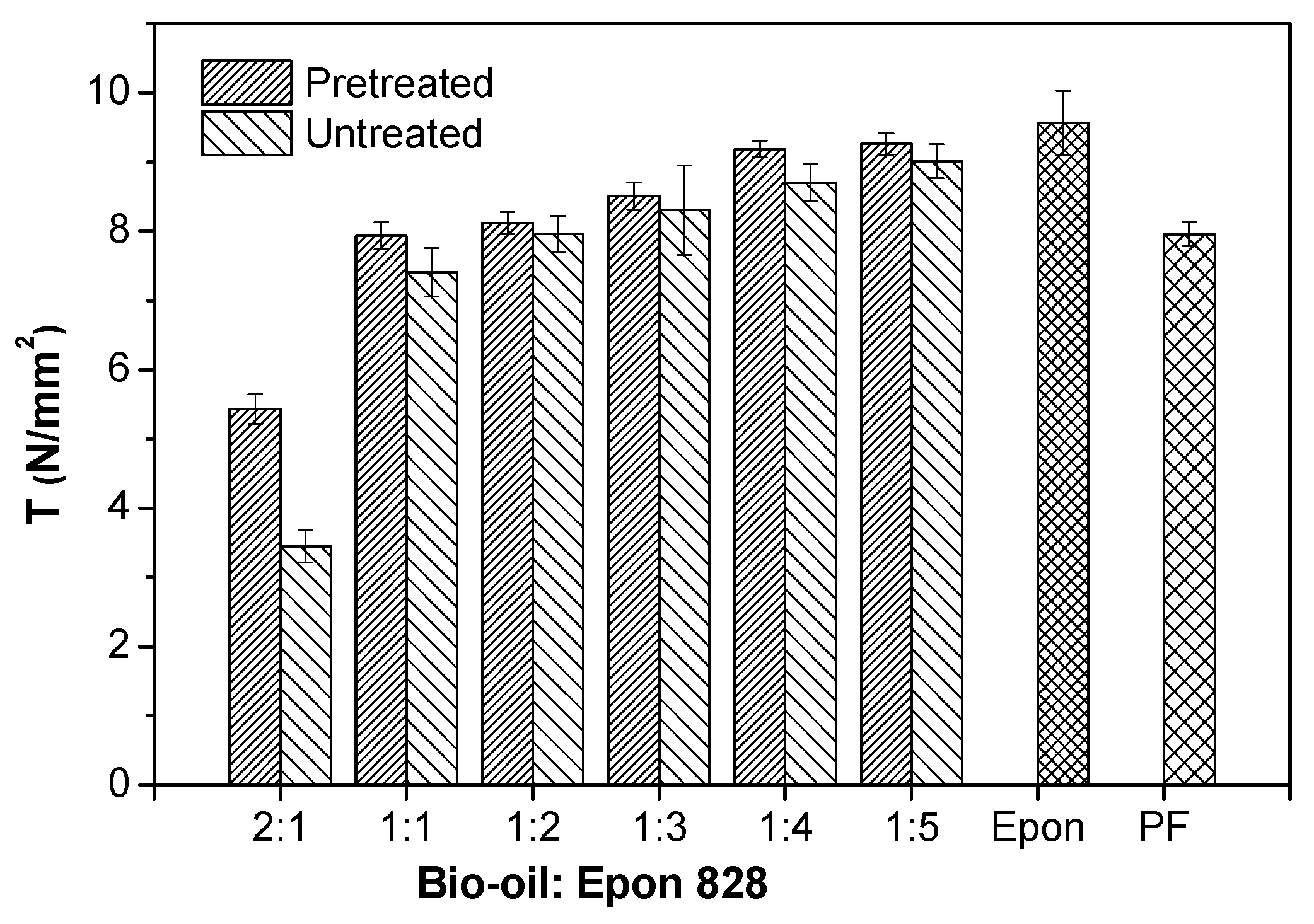
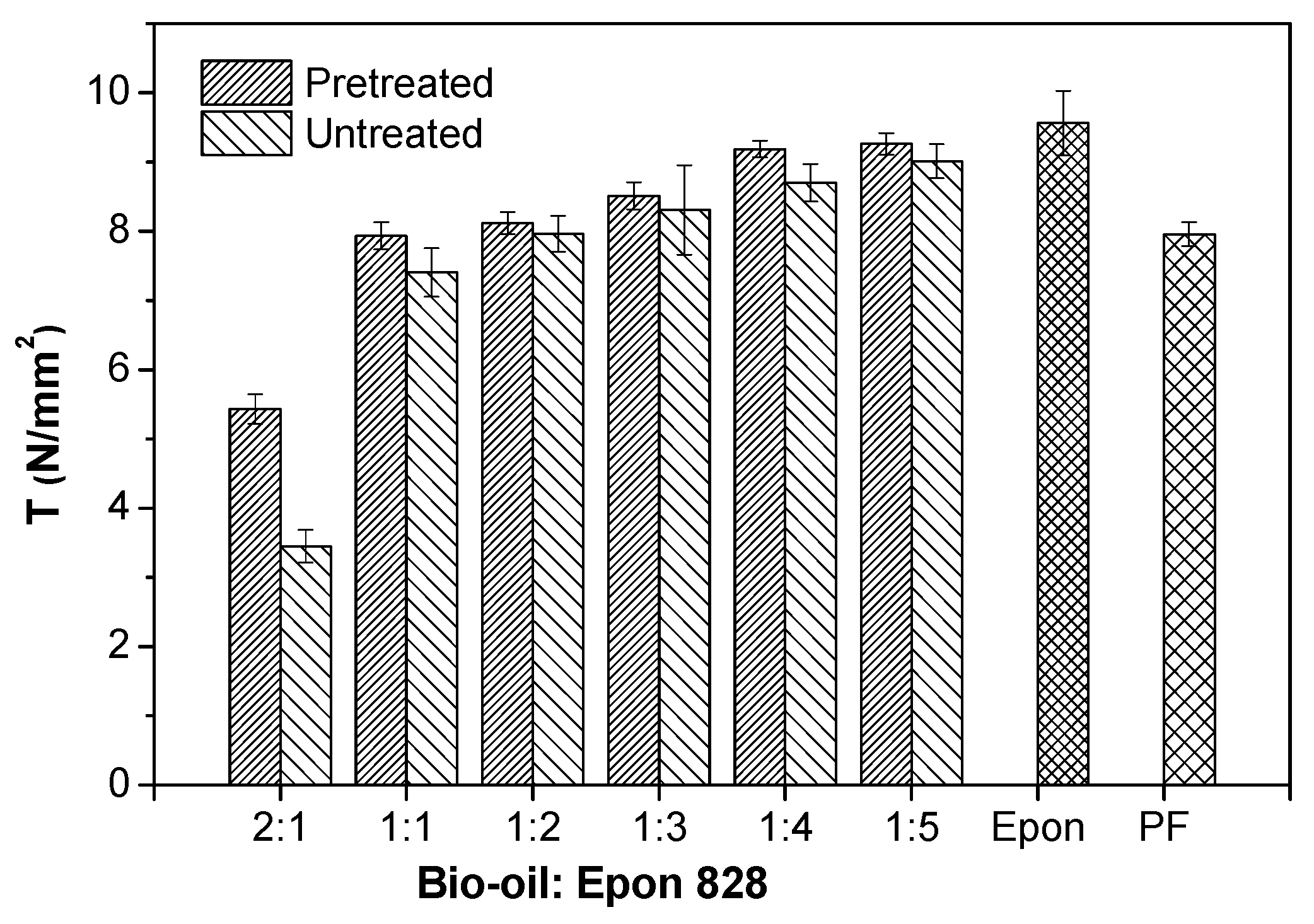
3.1. Performance of Wood Bonded Shear Strength
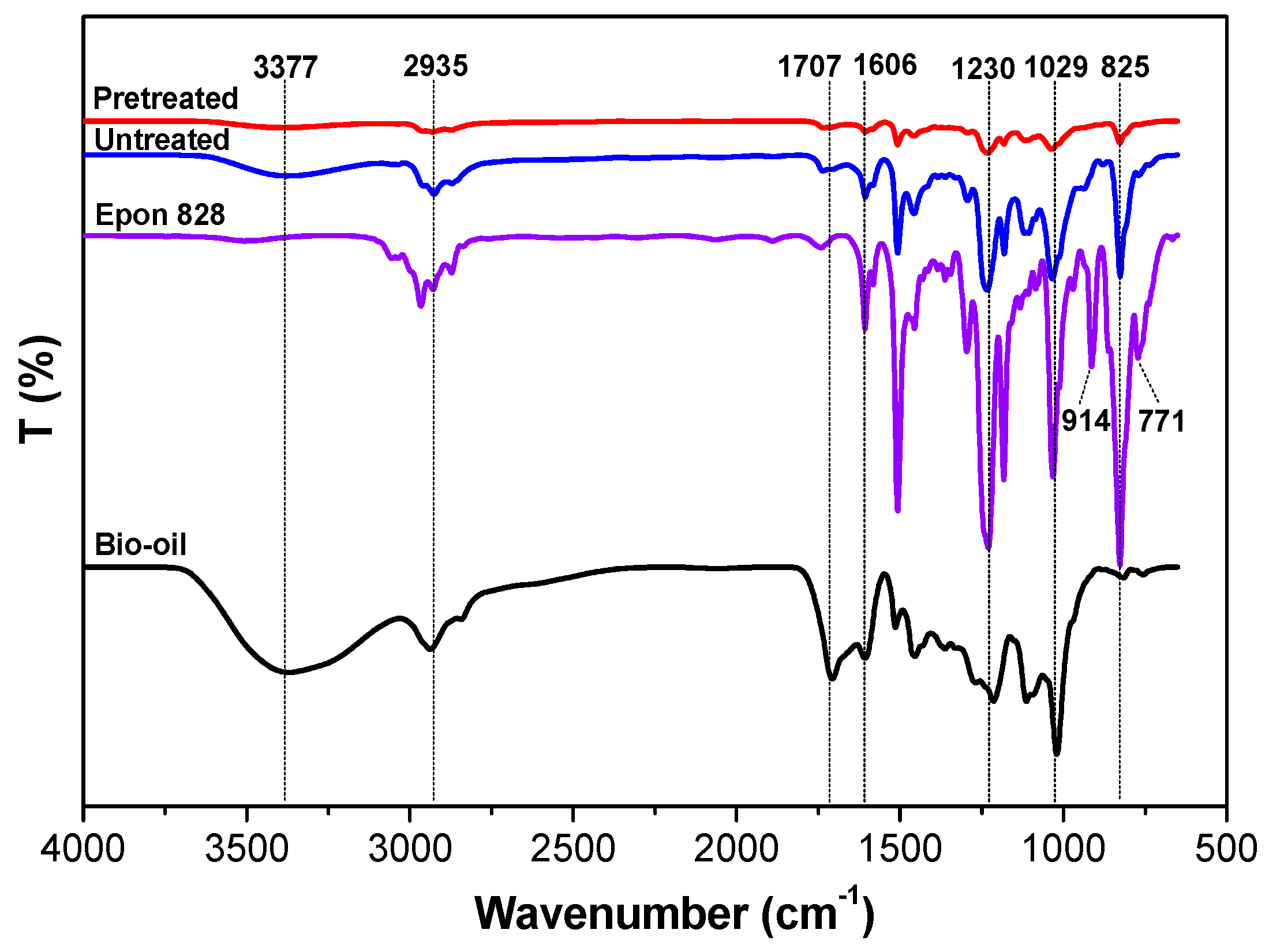
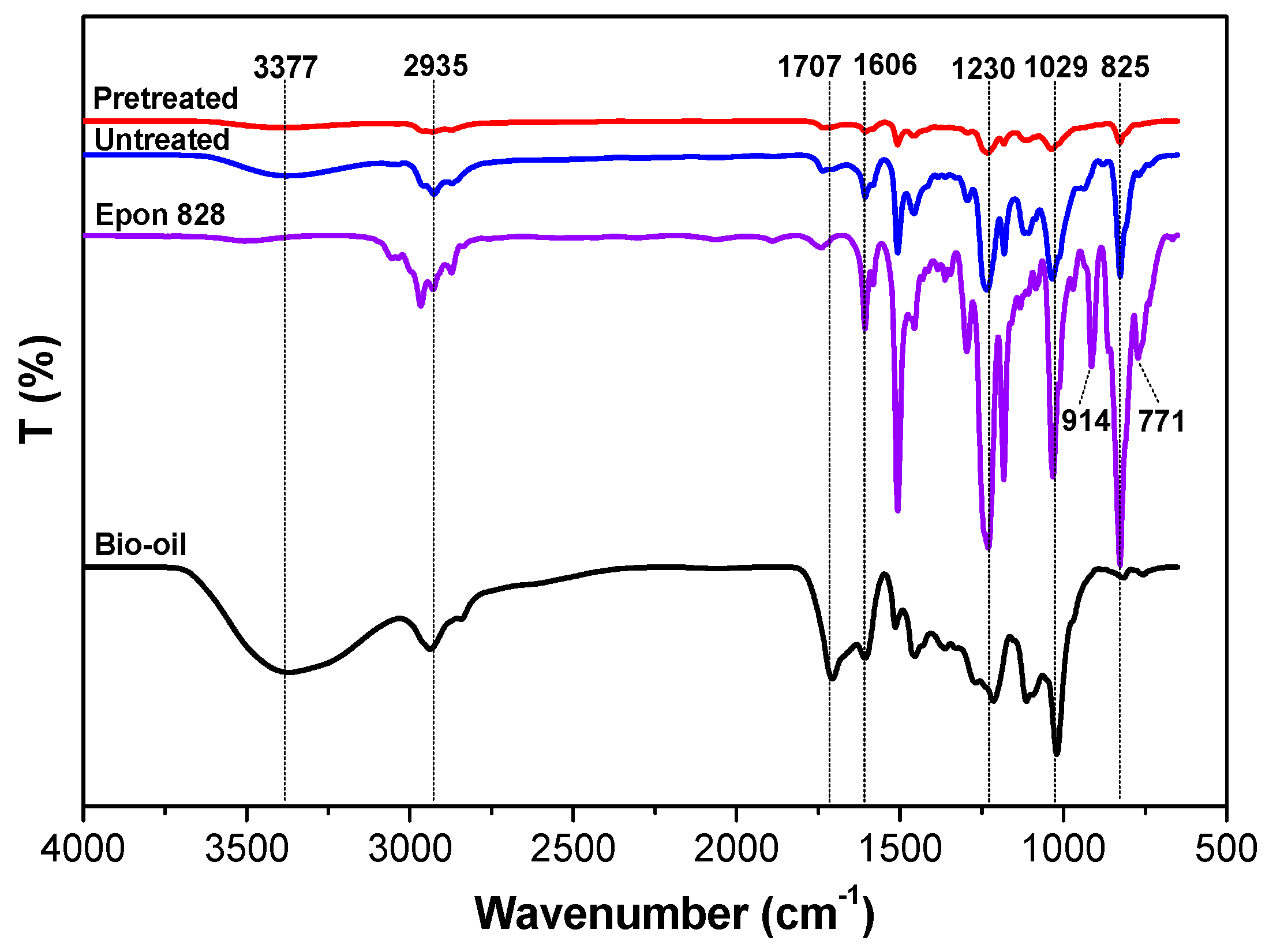
3.2. ATR-FTIR Characterization
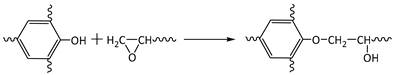
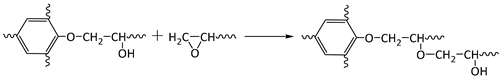
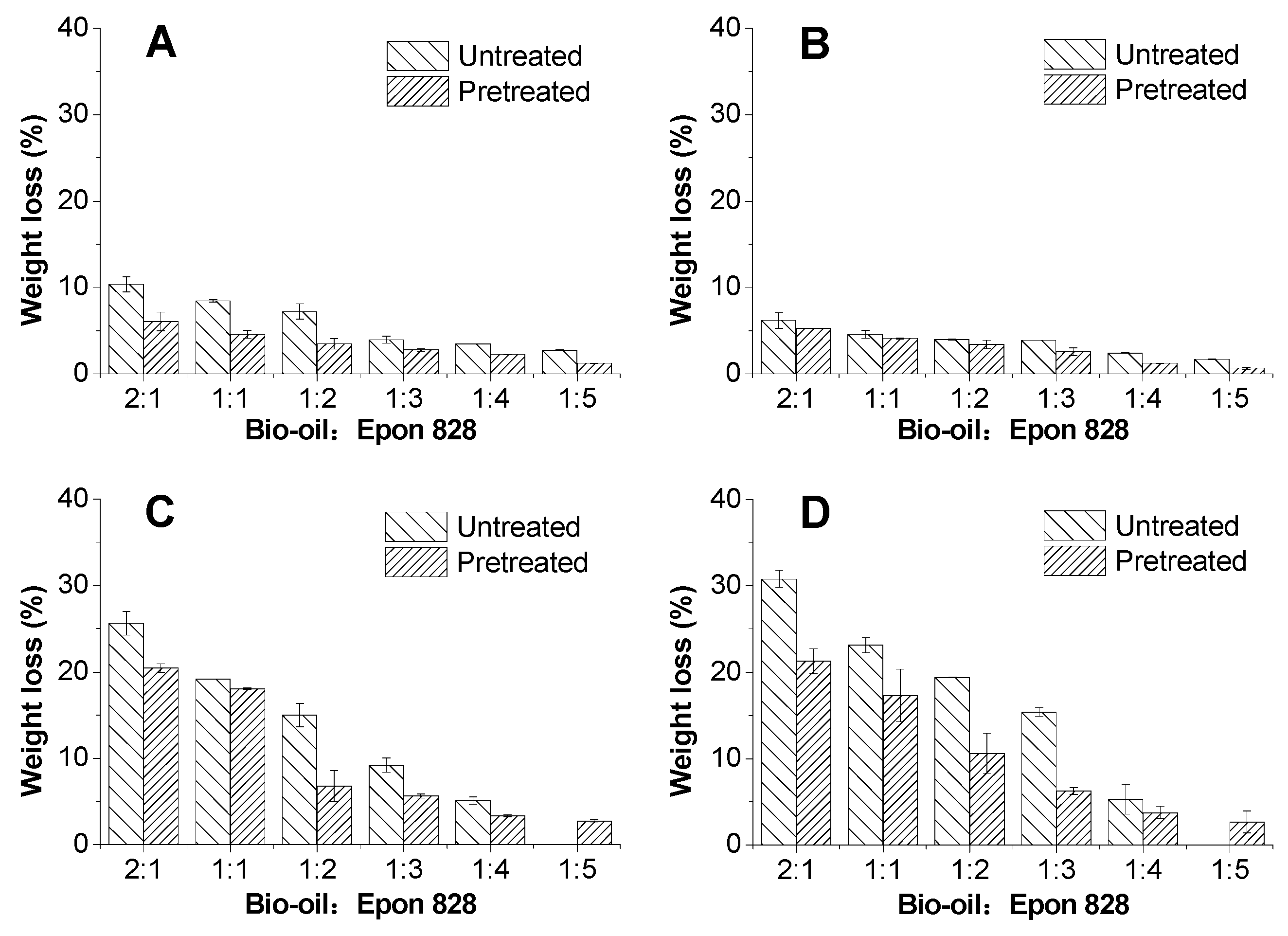
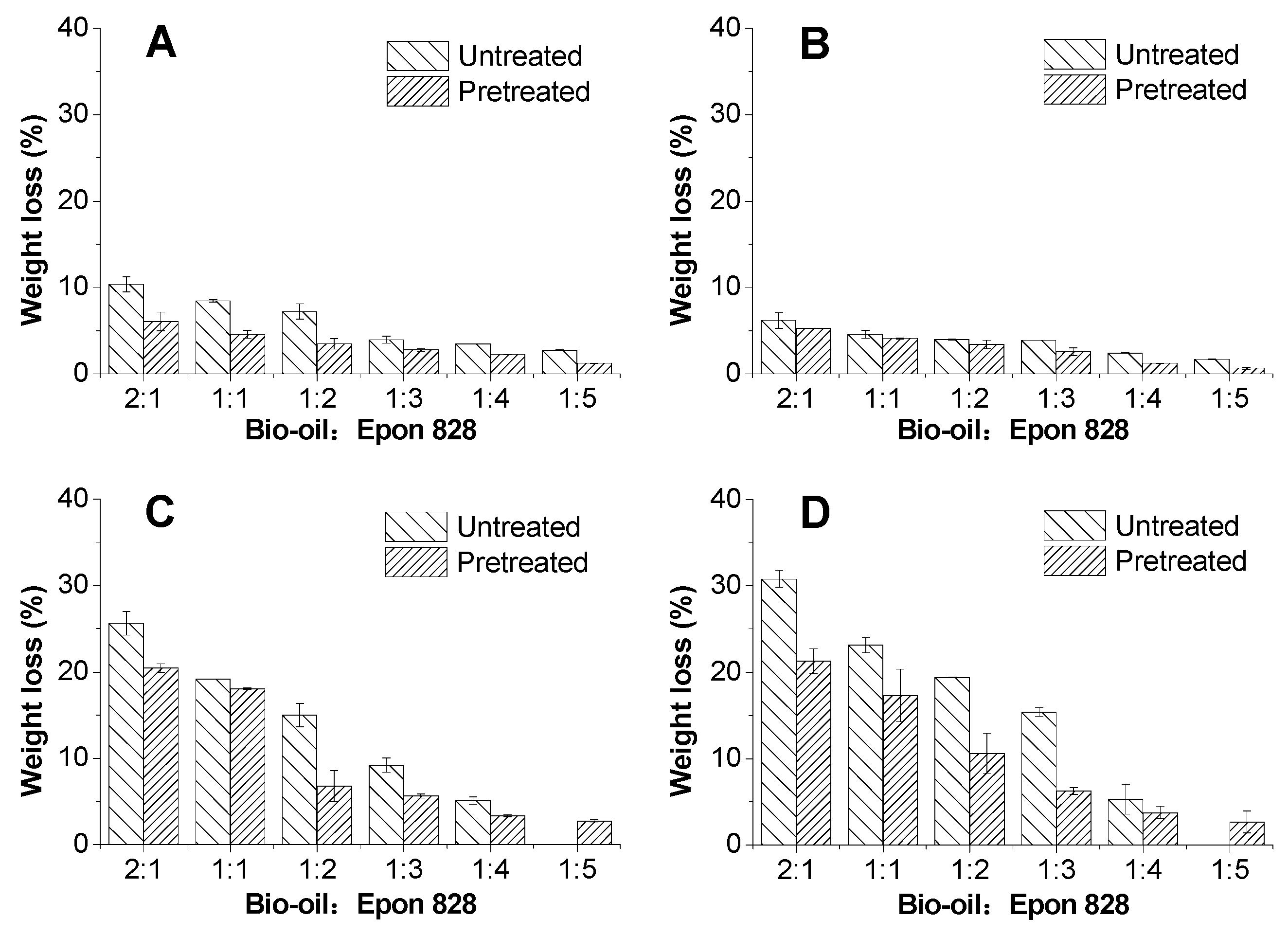
3.3. Solvent Resistance
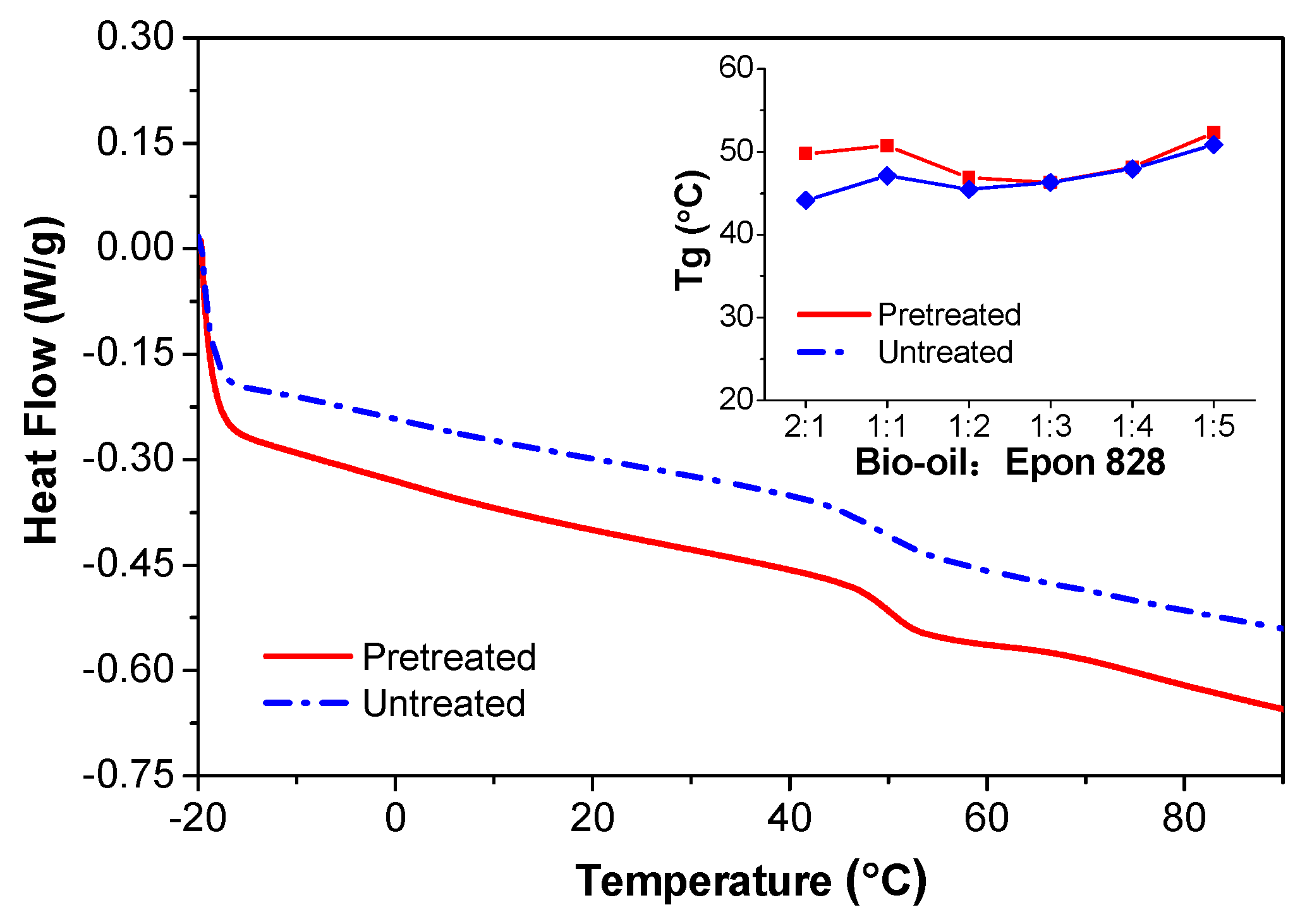
3.4. DSC Analysis
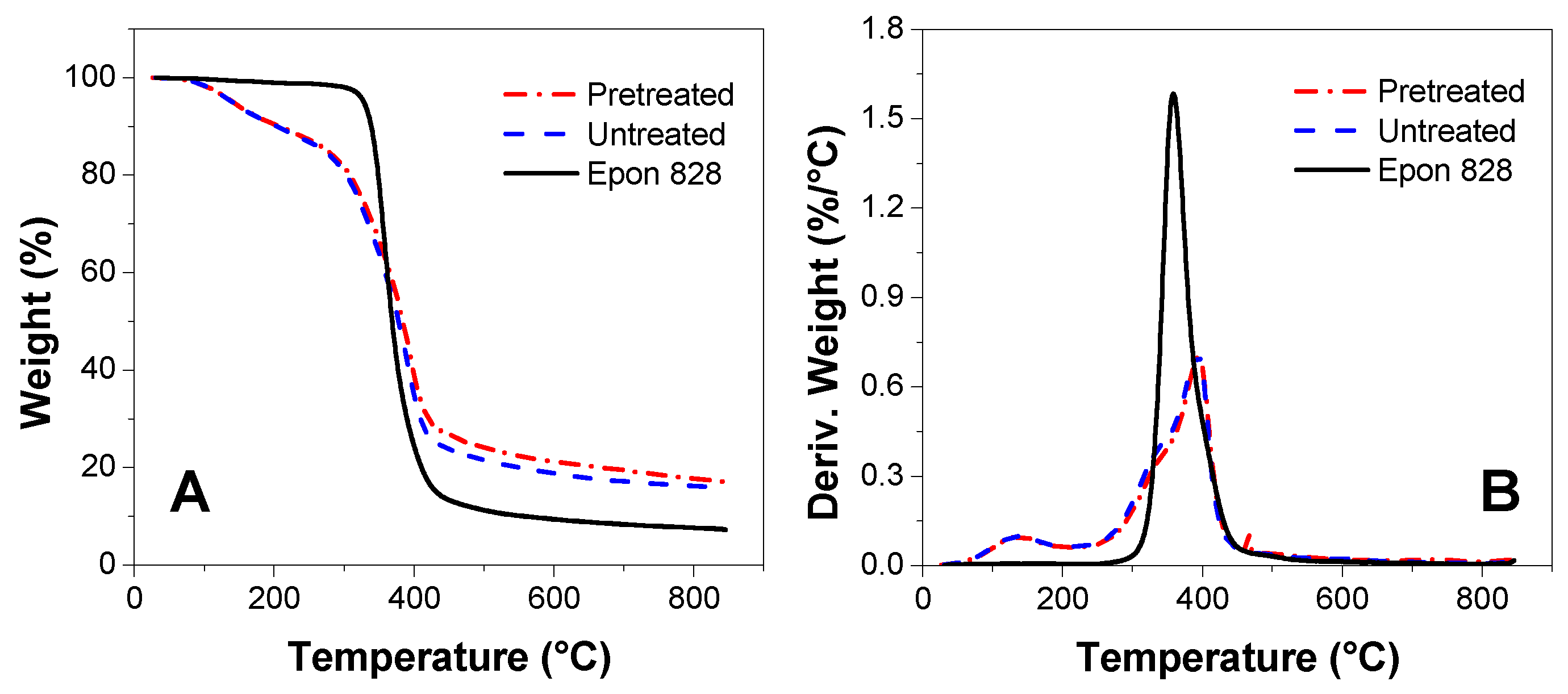
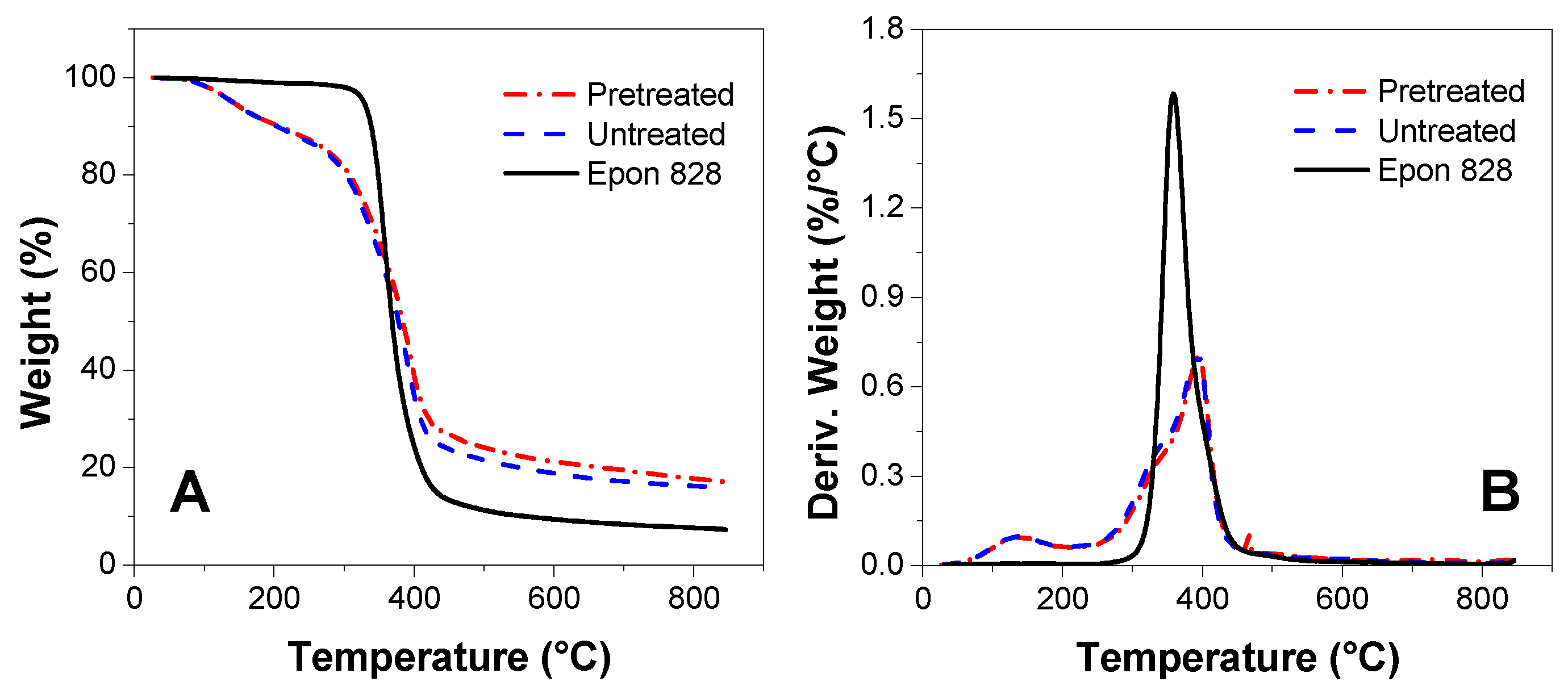
3.5. TGA Measurement
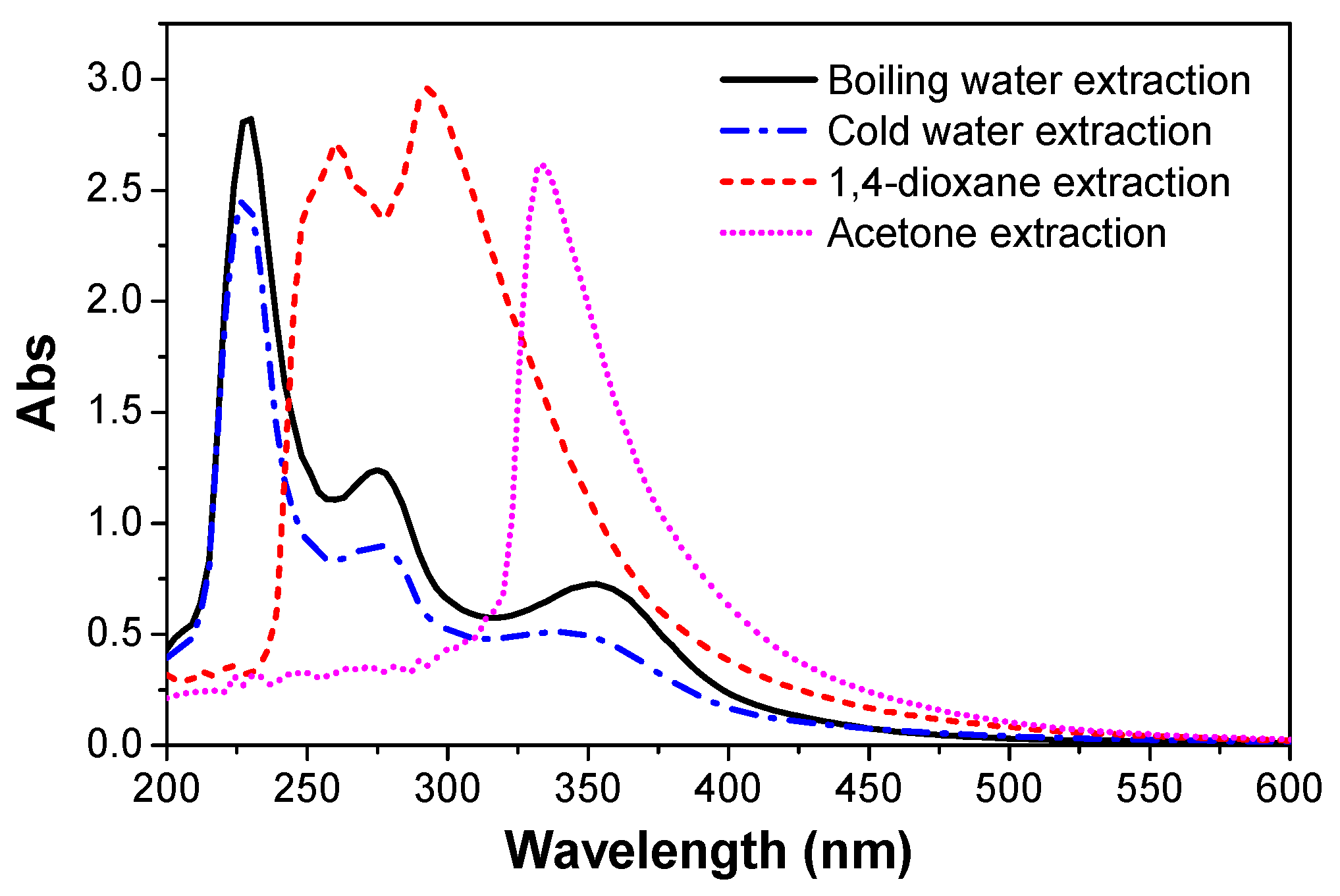
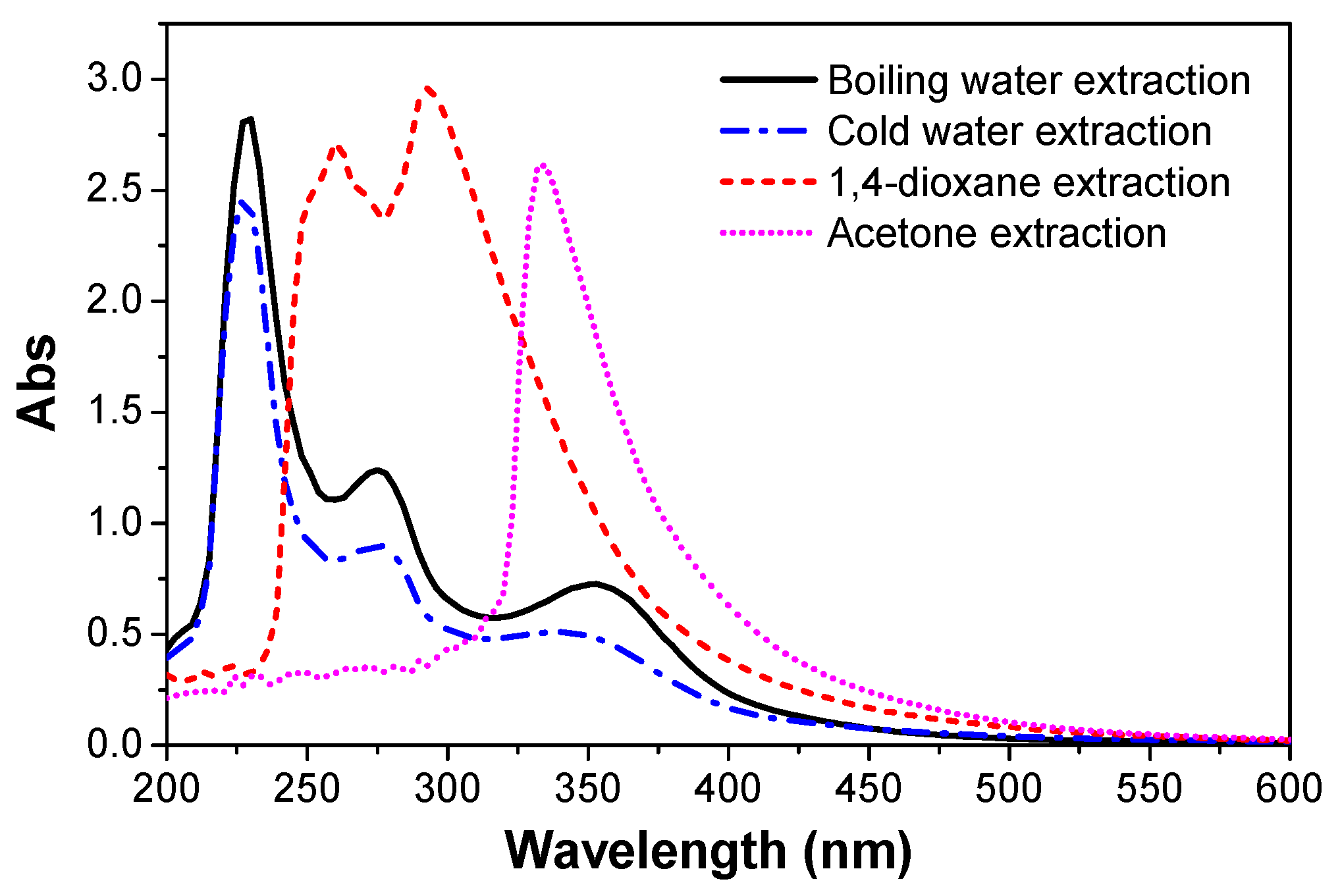
3.6. UV-Vis Spectra
4. Conclusions
- (1)
- The bond strengths and solvent resistance of the bio-oil/epoxy resins were improved after pretreatment for all bio-oil: epoxy ratios. However, as the replacement rate increased, so did the positive promotion effect of acetone on mechanical and thermal properties of the resin.
- (2)
- The shear strength of the bio-oil/epoxy resins decreased as the bio-oil content increased. Increased bio-oil concentration resulted in decreased cross-linking density. However, after pretreatment of the bio-oil/epoxy mixture with acetone and then polymerization while curing, up to a 50% replacement was possible while still meeting usage requirements.
- (3)
- The ATR-FTIR confirmed more –OH and –CH(O)CH– groups consumption in pretreated samples and a higher cross-linked structure after pretreatment of the bio-oil/epoxy mixture with acetone. DSC and TGA analysis demonstrated an improvement in thermal stability with acetone pretreatment. UV-Vis analysis revealed some potentially unreacted compounds within the bio-oil and this may have contributed to the weight loss observed when the cured polymer was exposed to various solvents.
- (4)
- The pretreatment process coupled with precise tuning of the bio-oil to epoxy ratio was an effective method to control cross-linking and consequent bonding of the resin to the wood substrate.
- (5)
- This study is industrially novel and relevant because the low variation in mechanical properties and polymer stability across a wide range of substitution rates will allow the manufacturer to adjust day-to-day changes in substitution rates without risk of a dramatic loss in material performance.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mohan, D.; Pittman, C.U., Jr.; Steele, P.H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Celikbag, Y.; Via, B.K.; Adhikari, S.; Wu, Y. Effect of liquefaction temperature on hydroxyl groups of bio-oil from loblolly pine (Pinus taeda). Bioresour. Technol. 2014, 169, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Via, B.K.; Wang, Y.; McDonald, T.; Auad, M.L. Liquefaction and substitution of switchgrass (Panicum virgatum) based bio-oil into epoxy resins. Ind. Crops Prod. 2014, 57, 116–123. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Amen-Chen, C.; Riedl, B.; Wang, X.-M.; Roy, C. Softwood bark pyrolysis oil-PF resols. Part 1. Resin synthesis and OSB mechanical properties. Holzforschung 2002, 56, 167–175. [Google Scholar] [CrossRef]
- Ferdosian, F.; Pan, Z.; Gao, G.; Zhao, B. Bio-based adhesives and evaluation for wood composites application. Polymers 2017, 9, 70–99. [Google Scholar] [CrossRef]
- Ren, X.; Gou, J.; Wang, W.; Li, Q.; Chang, J.; Li, B. Optimization of bark fast pyrolysis for the production of phenol-rich bio-oil. BioResources 2013, 8, 6481–6492. [Google Scholar] [CrossRef]
- Czernik, S.; Bridgwater, A.V. Overview of applications of biomass fast pyrolysis oil. Energy Fuels 2004, 18, 590–598. [Google Scholar] [CrossRef]
- Effendi, A.; Gerhauser, H.; Bridgwater, A.V. Production of renewable phenolic resins by thermochemical conversion of biomass: A review. Renew. Sustain. Energy Rev. 2008, 12, 2092–2116. [Google Scholar] [CrossRef]
- Chan, F.; Riedl, B.; Wang, X.-M.; Lu, X.; Amen-Chen, C.; Roy, C. Performance of pyrolysis oil-based wood adhesives in OSB (composites and manufactured products) (oriented strand board) (statistical data included). For. Prod. J. 2002, 52, 31–38. [Google Scholar]
- Roy, C.; Calve, L.; Lu, X.; Pakdel, H.; Amen-Chen, C. Wood composite adhesives from softwood bark derived vacuum pyrolysis oils. In Biomass, a Growth Opportunity in Green Energy and Value-Added Products, Proceedings of the 4th Biomass Conference of the Americas, Oakland, CA, USA, 29 August–2 September 1999; Overend, R.P., Chornet, E., Eds.; Elsevier Science: Pergamon, Turkey, 1999; pp. 521–526. [Google Scholar]
- Amen-Chen, C.; Riedl, B.; Roy, C. Softwood bark vacuum pyrolysis oil-PF resols for bonding OSB Panels. Part II. Thermal analysis by DSC and TG. Holzforschung 2002, 56, 273–280. [Google Scholar] [CrossRef]
- Nakos, P.; Tsiantzi, S.; Athanassiadou, E. Wood adhesives made with pyrolysis oils. In Proceedings of the 3rd European Wood-Based Panel Symposium, Hanover, Germany, 12–14 September 2001; European Panel Federation and Wilhelm Klauditz Institute: Hanover, German, 2001. [Google Scholar]
- Zheng, K. Research on Pyrolysis Oil of Larch Bark Modified Phenolic Resin Adhesives; Beijing Forestry University: Beijing, China, 2007; p. 18. [Google Scholar]
- Gagnon, M.; Roy, C.; Rodrigue, D.; Riedl, B. Calorimetric and rheological study of isocyanate-pyrolysis oil blends. J. Appl. Polym. Sci. 2003, 89, 1362–1370. [Google Scholar] [CrossRef]
- Gagnon, M.; Roy, C.; Riedl, B. Adhesives made from isocyanate and pyrolysis oils for wood composites. Holzforschung 2004, 58, 400–407. [Google Scholar] [CrossRef]
- Mao, A.; Shi, S.Q.; Philip, S. Flakeboard bonded with polymeric diphenylmethane diisocyanate/bio-oil adhesive systems. For. Prod. J. 2011, 61, 240–245. [Google Scholar] [CrossRef]
- Pizzi, A. Advanced Wood Adhesives Technology; CRC Press: New York, NY, USA, 1994. [Google Scholar]
- Pan, Y.F.; Pan, Y.; Cheng, Q.Z.; Liu, Y.; Essien, C.; Via, B.; Wang, X.Y.; Sun, R.C.; Taylor, S. Characterization of epoxy composites reinforced with wax encapsulated microcrystalline cellulose. Polymers 2016, 12, 415–427. [Google Scholar] [CrossRef]
- Vick, C.B.; Richter, K.; River, B.H.; Fried, A.R. Hydroxymethylated resorcinol coupling agent for enhanced durability of bisphenol-A epoxy bonds to sitka spruce. Wood Fiber Sci. 1995, 27, 2–12. [Google Scholar]
- Robinson, T.J.; Via, B.K.; Fasina, O.; Adhikari, S.; Carter, E. Impregnation of bio-oil from small diameter pine into wood for moisture resistance. BioResources 2011, 6, 4747–4761. [Google Scholar]
- Ugovšek, A.; Škapin, S.A.; Humar, M.; Sernek, M. Microscopic analysis of the wood bond line using liquefied wood as adhesive. J. Adhes. Sci. Technol. 2013, 27, 1247–1258. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, J.; Guo, H.; Pan, Y.; Zhou, C.; Cheng, Q.; Via, B.K. Interfacial properties of loblolly pine bonded with epoxy/wood pyrolysis bio-oil blended system. BioResources 2015, 10, 638–646. [Google Scholar] [CrossRef]
- Kishi, H.; Fujita, A.; Miyazaki, H.; Matsuda, S.; Murakami, A. Synthesis of wood-based epoxy resins and their mechanical and adhesive properties. J. Appl. Polym. Sci. 2006, 102, 2285–2292. [Google Scholar] [CrossRef]
- Kishi, H.; Akamatsu, Y.; Noguchi, M.; Fujita, A.; Matsuda, S.; Nishida, H. Synthesis of epoxy resins from alcohol-liquefied wood and the mechanical properties of the cured resins. J. Appl. Polym. Sci. 2011, 120, 745–751. [Google Scholar] [CrossRef]
- Wu, C.-C.; Lee, W.-J. Curing behavior and adhesion properties of epoxy resin blended with polyhydric alcohol-liquefied Cryptomeria japonica wood. Wood Sci. Technol. 2010, 45, 559–571. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hatano, Y.; Tomita, B. Viscoelastic properties of liquefied wood/epoxy resin and its bond strength. Holzforschung 2001, 55, 667–671. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tukamoto, K.; Tomita, B. Application of liquefied wood to a new resin system-Synthesis and properties of liquefied wood/epoxy resins. Holzforschung 2000, 54, 93–97. [Google Scholar] [CrossRef]
- Thangalazhy-Gopakumar, S.; Adhikari, S.; Ravindran, H.; Gupta, R.B.; Fasin, O.; Tu, M.; Fernando, S.D. Physiochemical properties of bio-oil produced at various temperatures from pine wood using an auger reactor. Bioresour. Technol. 2010, 101, 8389–8395. [Google Scholar] [CrossRef] [PubMed]
- Ringer, M.; Putsche, V.; Scahill, J. Large-Scale Pyrolysis Oil Production: A Technology Assessment and Economic Analysis; National Renewable Energy Laboratory: Golden, CO, USA, 2006. [Google Scholar]
- Steele, P.; Puettmann, M.E.; Penmetsa, V.K.; Cooper, J.E. Life-cycle assessment of pyrolysis bio-oil production. For. Prod. J. 2012, 62, 326–334. [Google Scholar] [CrossRef]
- Hilten, R.N.; Das, K.C. Comparison of three accelerated aging procedures to assess bio-oil stability. Fuel 2010, 89, 2741–2749. [Google Scholar] [CrossRef]
- Atta-Obeng, E.; Via, B.K.; Fasina, O.; Auad, M.L.; Jiang, W. Cellulose reinforcement of phenol formaldehyde: Characterization and chemometric elucidation. Int. J. Compos. Mater. 2013, 3, 61–68. [Google Scholar]
- Ugovšeka, A.; Sernek, M. Effect of pressing parameters on the shear strength of beech specimens bonded with low solvent liquefied wood. J. Adhes. Sci. Technol. 2012, 27, 182–195. [Google Scholar] [CrossRef]
- Auad, M.L.; Nutt, S.R.; Stefani, P.M.; Aranguren, M.I. Rheological study of the curing kinetics of epoxy-phenol novolac resin. J. Appl. Polym. Sci. 2006, 102, 4430–4439. [Google Scholar] [CrossRef]
- Mao, A.; Shi, S.Q. Dynamic mechanical properties of polymeric diphenylmethane diisocyanate/bio-oil adhesive system. For. Prod. J. 2012, 62, 201–206. [Google Scholar] [CrossRef]
- Gao, Z.H.; Li, D. Chemical modification of poplar wood with foaming polyurethane resins. J. Appl. Polym. Sci. 2007, 104, 2980–2985. [Google Scholar]
- Via, B.K.; Adhikari, S.; Taylor, S. Modeling for proximate analysis and heating value of torrefied biomass with vibration spectroscopy. Bioresour. Technol. 2013, 133, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Devi, R.R.; Maji, T.K. Chemical modification of simul wood with styrene-Acrylonitrile copolymer and organically modified nanoclay. Wood Sci. Technol. 2012, 46, 299–315. [Google Scholar] [CrossRef]
- Sasakia, C.; Wanakaa, M.; Takagib, H.; Tamurac, S.; Asadaa, C.; Nakamura, Y. Evaluation of epoxy resins synthesized from steam-exploded bamboo lignin. Ind. Crops Prod. 2013, 43, 757–761. [Google Scholar] [CrossRef]
- Chen, H.; Lang, Q.; Zhang, H.; Wu, G.; Zheng, X.; Pu, J. Study of Chemical Modification by impregnation of fresh poplar log and by hot-press drying process. BioResources 2013, 8, 3924–3933. [Google Scholar] [CrossRef]
- Khalil, H.P.S.A.; Marliana, M.M.; Alshammari, T. Material properties of epoxy-reinforced biocomposites with lignin from empty fruit bunch as curing agent. BioResources 2011, 6, 5206–5223. [Google Scholar]
- Wu, G.; Sun, E.; Huang, H.; Chang, Z.; Xu, Y. Preparation and properties of biodegradable planting containers made with straw and starch adhesive. BioResources 2013, 8, 5358–5368. [Google Scholar] [CrossRef]
- Zhang, H.; Dinga, F.; Luoa, C.; Xiong, L.; Chen, X. Liquefaction and characterization of acid hydrolysis residue of corncob in polyhydric alcohols. Ind. Crops Prod. 2012, 39, 47–51. [Google Scholar] [CrossRef]
- Rowell, R.M. Handbook of Wood Chemistry and Wood Composites; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Wang, W.; Zhu, Y.; Cao, J. Morphological, thermal and dynamic mechanical properties of Cathay poplar/organoclay composites prepared by in situ process. Mater. Des. 2014, 59, 233–240. [Google Scholar] [CrossRef]
- Ren, X.; Du, H.; Wang, W.; Gou, J.; Chang, J. Analysis of pyrolysis process and gas evolution rule of larch wood by TG-FTIR. Spectrosc. Spectr. Anal. 2012, 32, 944–948. [Google Scholar]
- Liu, Q.; Wang, S.; Zheng, Y.; Luo, Z.; Cen, K. Mechanism study of wood lignin pyrolysis by using TG-FTIR analysis. J. Anal. Appl. Pyrolysis 2008, 82, 170–177. [Google Scholar] [CrossRef]
- Ke, Y.; Dong, H. Analytical Chemistry Handbook, 2nd ed.; Book III—Spectral Analysis; Chemical Industry Press: Beijing, China, 1998. [Google Scholar]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Via, B.K.; Pan, Y.; Cheng, Q.; Guo, H.; Auad, M.L.; Taylor, S. Preparation and Characterization of Epoxy Resin Cross-Linked with High Wood Pyrolysis Bio-Oil Substitution by Acetone Pretreatment. Polymers 2017, 9, 106. https://doi.org/10.3390/polym9030106
Liu Y, Via BK, Pan Y, Cheng Q, Guo H, Auad ML, Taylor S. Preparation and Characterization of Epoxy Resin Cross-Linked with High Wood Pyrolysis Bio-Oil Substitution by Acetone Pretreatment. Polymers. 2017; 9(3):106. https://doi.org/10.3390/polym9030106
Chicago/Turabian StyleLiu, Yi, Brian K. Via, Yuanfeng Pan, Qingzheng Cheng, Hongwu Guo, Maria L. Auad, and Steven Taylor. 2017. "Preparation and Characterization of Epoxy Resin Cross-Linked with High Wood Pyrolysis Bio-Oil Substitution by Acetone Pretreatment" Polymers 9, no. 3: 106. https://doi.org/10.3390/polym9030106
APA StyleLiu, Y., Via, B. K., Pan, Y., Cheng, Q., Guo, H., Auad, M. L., & Taylor, S. (2017). Preparation and Characterization of Epoxy Resin Cross-Linked with High Wood Pyrolysis Bio-Oil Substitution by Acetone Pretreatment. Polymers, 9(3), 106. https://doi.org/10.3390/polym9030106