3.1. Effect of the Grafted Polymer Chain Length
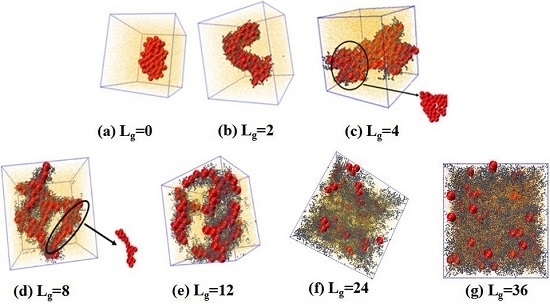
We firstly examined the self-assembly structures, potentially formed by NPs uniformly grafted with the same chains as the matrix polymer chains. Note that we fix the grafting density equal to 0.18, given by:
where
denotes the number of grafting chains grafted on each nanoparticle, and
denotes the surface area of each nanoparticle, while the grafted chain length is varied as follows:
(bare particles),
,
,
,
,
and
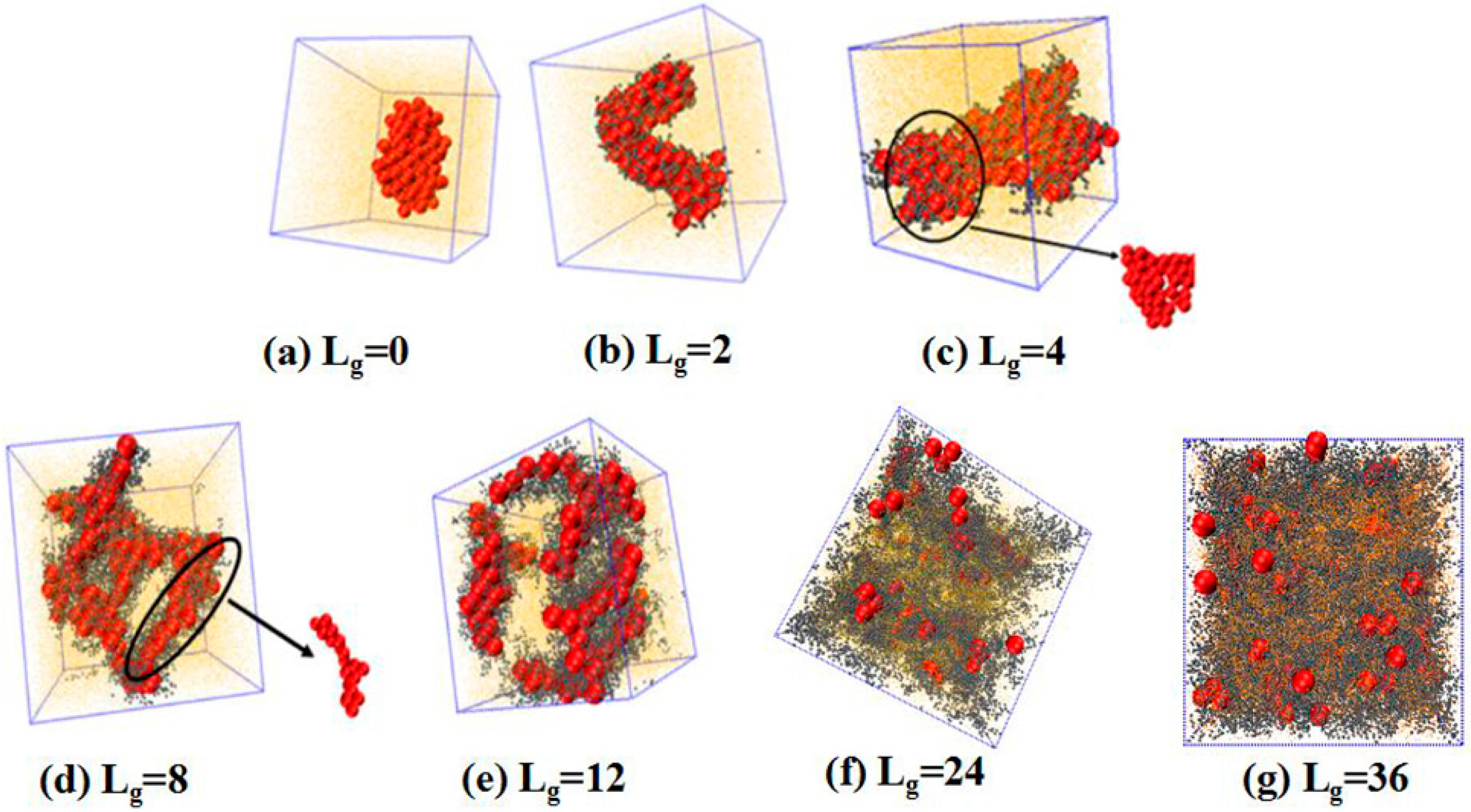
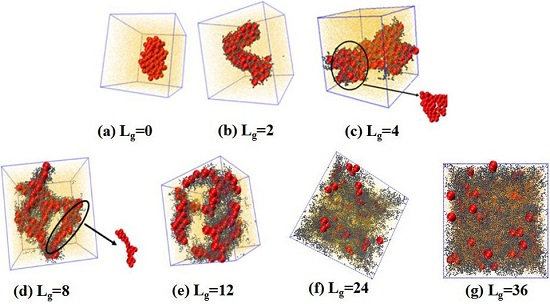
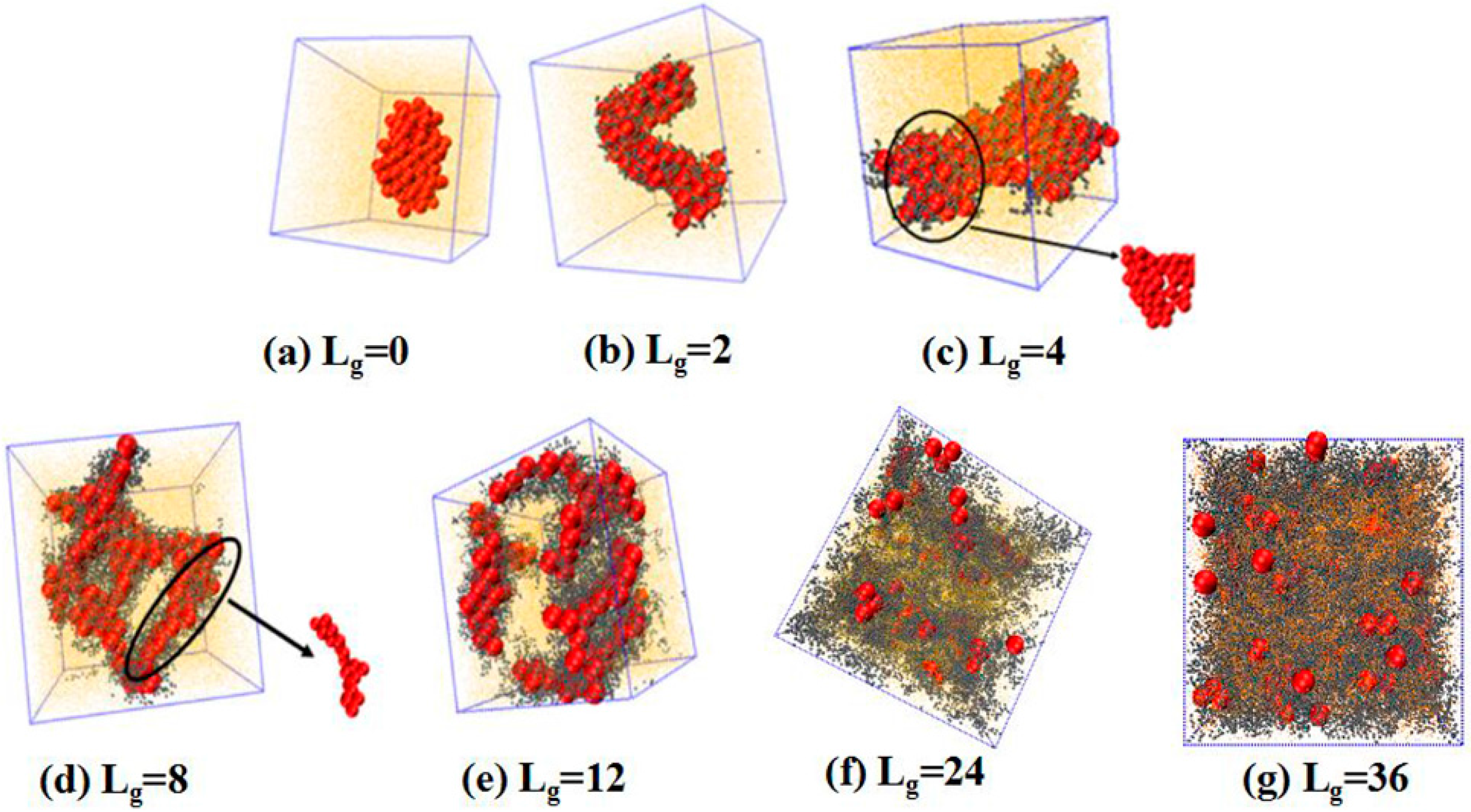
. We place 100 NPs in the simulation box to observe the self-assembled structures. The simulated results are shown in
Figure 2. Obviously, without any grafting, the bare NPs tend to aggregate into an isotropic cluster, because of the incompatibility between the NPs and the matrix polymer chains. In other cases, by gradually increasing the grafted chain length, a variety of structures are formed. For instance, when the grafted chains contain two beads (
), the NPs self-assemble to form a cylinder structure, which is changed to sheets with the length of the grafted chains
. Furthermore, a connected string structure and a bunch of short strings occur in the case of
and
, respectively. These observations are in qualitative agreement with the work carried out by Akcora et al. [
9]. When the grafted chain length increases to
, most NPs become separated with each other and dispersed, and when the grafted chain length is increased to
, all NPs become well dispersed in the polymer matrix. This observation is consistent with our previous work that NPs tend to become dispersed in the polymer matrix when they are densely grafted or grafted with long polymer chains [
20]. The underlying mechanism results from the competition between the entropy contributed by the conformation of the grafted polymer chains, and enthalpy between the NPs. When the grafted chain length becomes long enough, the long grafted polymer chains repel the NPs apart, and meanwhile a strong interlocking occurs between the long grafted polymer chains and the matrix polymer chains. The formed structure represents the maximum contact between NPs via the un-shielded surfaces. Mahynski et al. [
15] pointed out that different chain lengths lead to different configurations around NPs, that is, longer grafted chains lead to a larger area of coverage onto the surface of NPs, in analogy to the “patchy particle” offering the rest of the exposed surfaces to contact with each other. Thus, it is our speculation that when the NPs are grafted by relatively short chains such as
, the grafted chains can provide little coverage for the surface of the NPs, since the surface area of the NPs is 16 times greater than that of each polymer bead, offering opportunities for the NPs to attract with each other. However, in the cases of
and
, a string structure is formed, whereas, for
, the sheet structure is more likely to form when the interaction energy between NPs is strong enough to overcome the entropy penalty of the grafted chains.
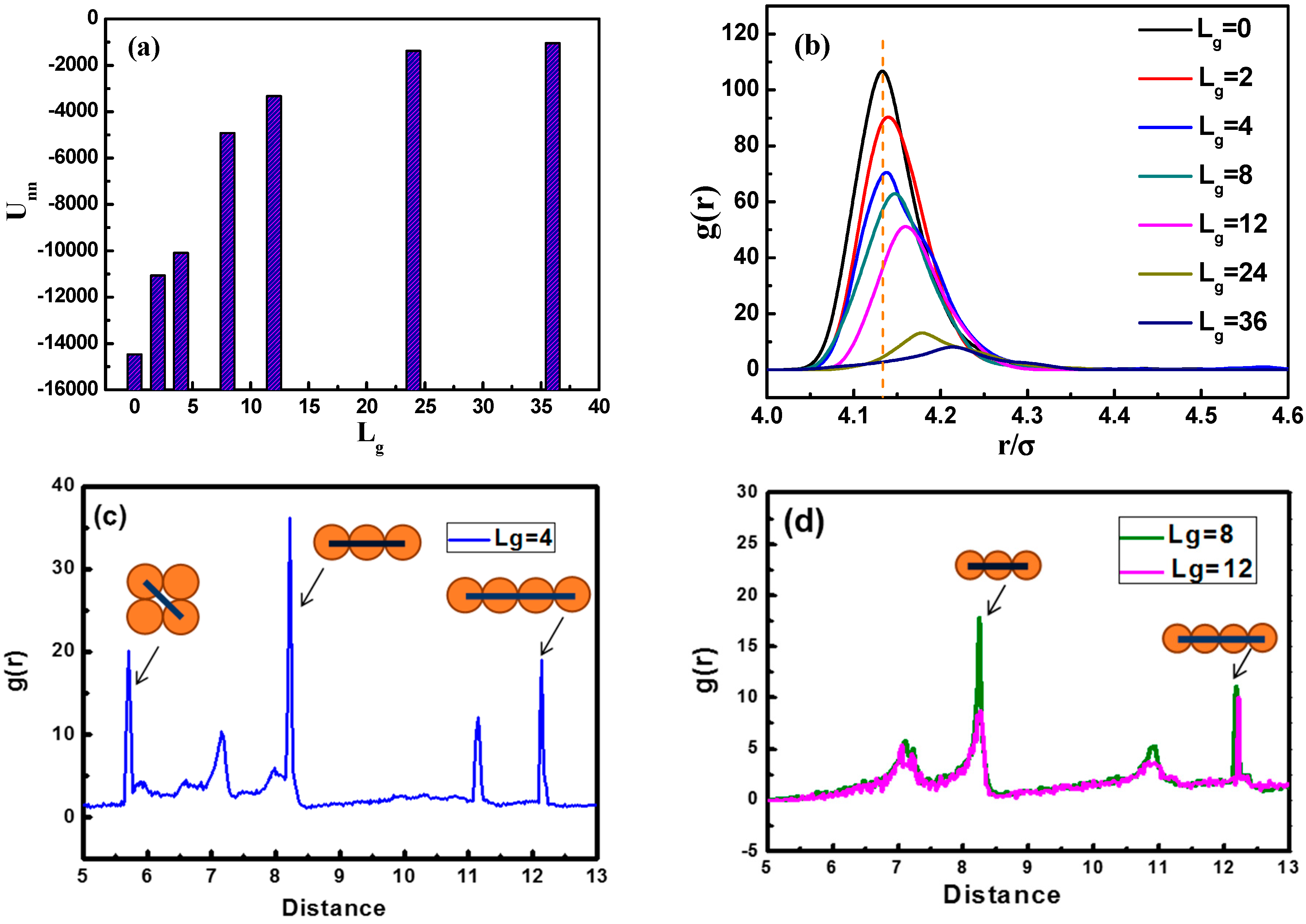
In
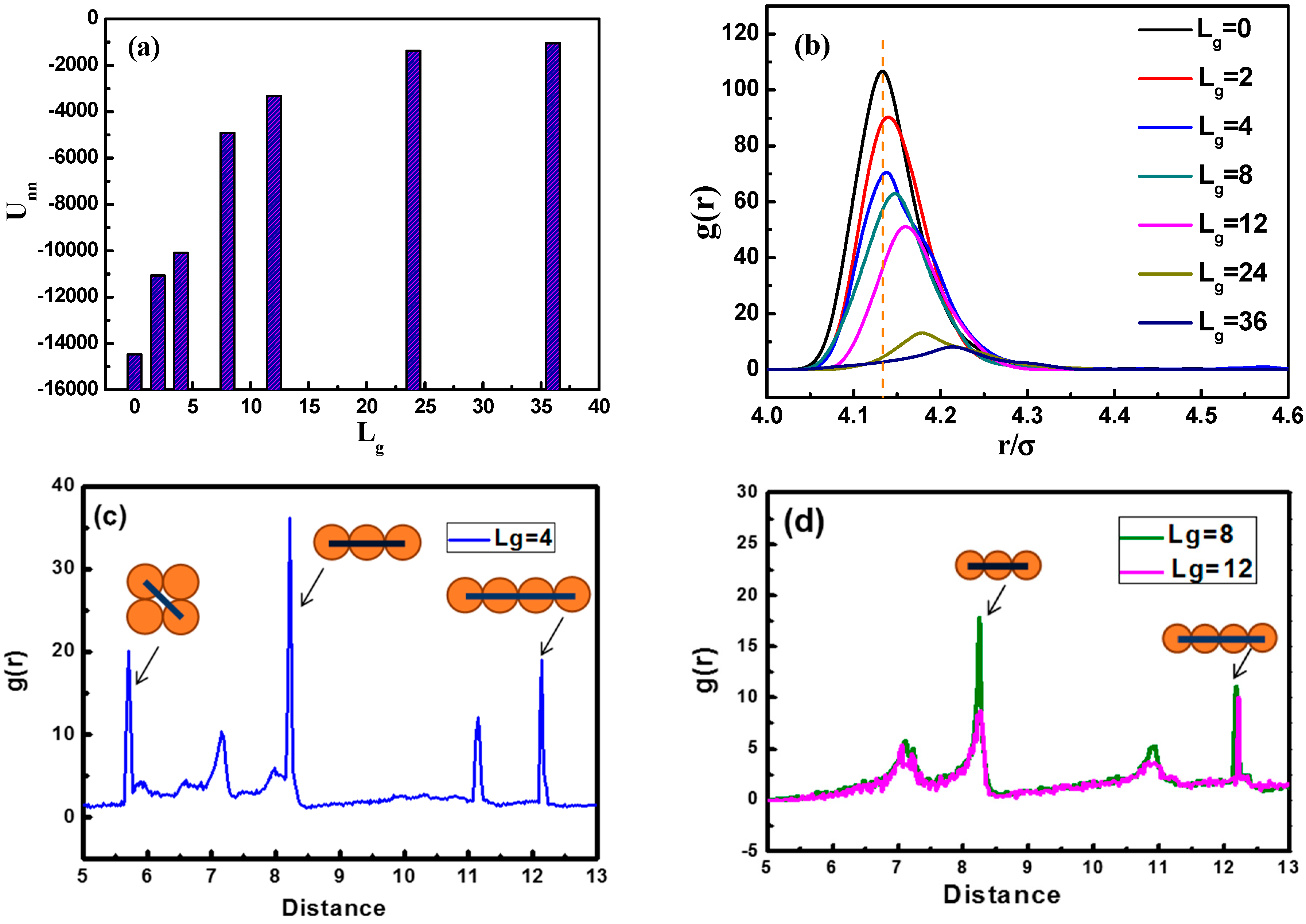
Figure 3a, we calculate the total interaction energy between NPs as a function of the grafted chain length to reflect the aggregation state. It is clearly shown that the total interaction energy between NPs for
is much larger than that of
and
. This may serve as evidence that when
is equal to fourpolymer beads, the NPs self-assemble into sheets instead of strings. It is evident that as the grafting chain length becomes longer, the total interaction energy between NPs becomes weaker. A weaker total interaction energy indicates a relatively good dispersion of NPs, as supported by the snapshots in
Figure 2 As the grafted polymer chains become longer, the matrix polymer chains gradually become inter-locked with the grafted chains attributed to the attractive interaction between each other, leading to a better dispersion of NPs.
To better characterize the self-assembled structures, we use the radial distribution function (RDF), as shown in
Figure 3b. It can be seen that the height of the first peak declines gradually with the increase of the grafted chain length. Note that the first peak appears at the distance of approximately
, corresponding to the direct contact equilibrium distance between NPs. When the grafted chain length
is equal to 36, the first peak almost disappears, implying that the NPs become well dispersed. From
Figure 3c,d, we demonstrate the RDF plots of NPs with grafted chains of
,
, and
, at a distance from
to
.
Figure 3c displays that three main peaks are located at a distance of
,
, and
, separately. The diameter of each NP is
, and if four NPs are arranged into a square-like structure, the length of the diagonal line is equal to
, so the peak at the distance of around
makes sense as NPs pack into a square-like form. This gives an evidence of the occurrence of the sheet network.
Figure 3d displays situations concerning
and
, where two main peaks located at a distance of
and
can be observed, and peaks at a distance of
imply the fact that two NPs are separated by the number of n NPs, forming a chain-like structure, exhibiting the string morphology.
We then investigate the tensile process of various self-assembled structures, expecting to find the most optimal structure that exhibits the best mechanical performance. We simulate all the systems by stretching the simulation box along the y direction while keeping the box volume constant.
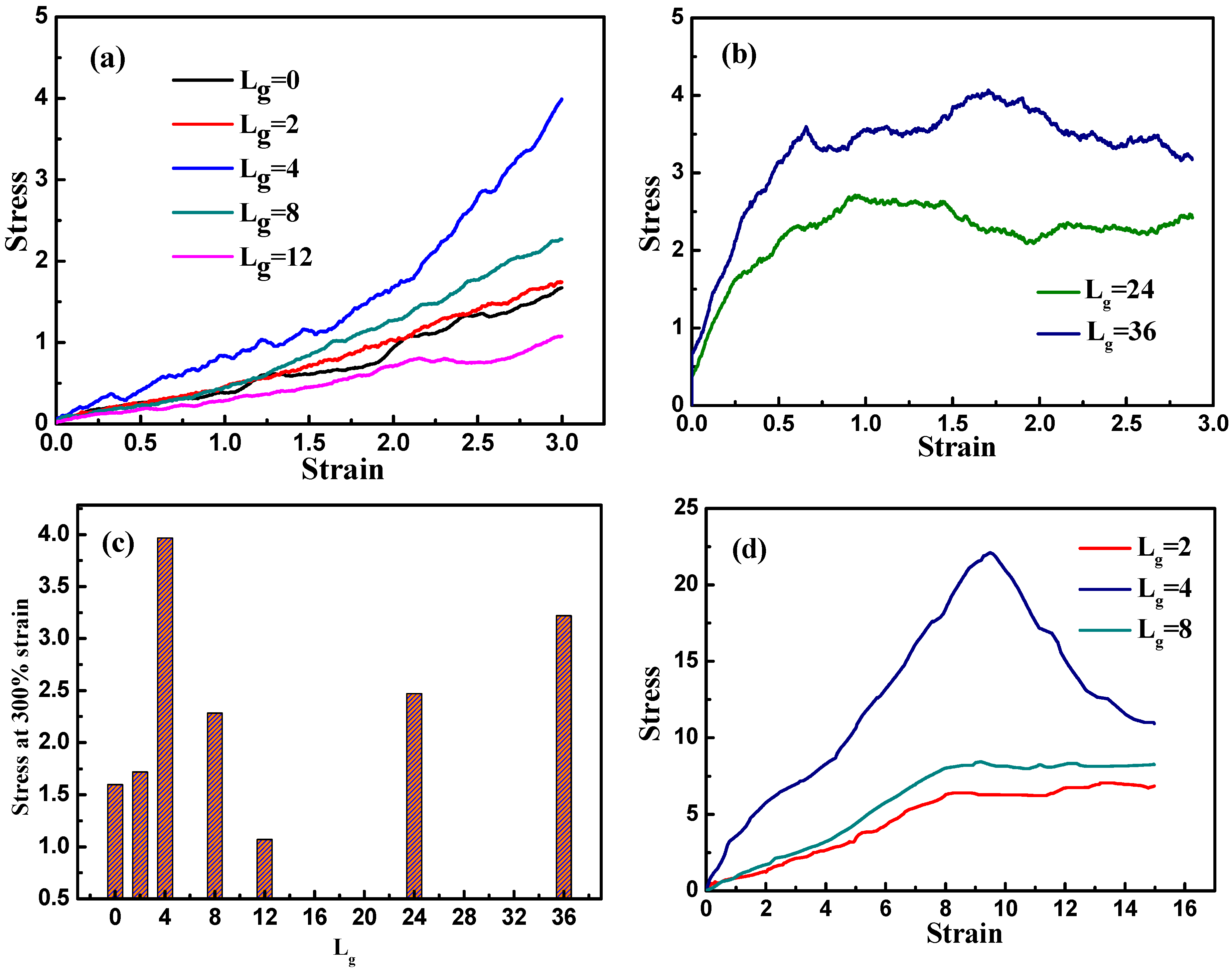
Figure 4a displays the stress–strain curves corresponding to the various grafted chain length. It is evident that as the self-assembly structure transforms from agglomeration and cylinder to sheet, the tensile stress is enhanced greatly, while the slope of the stress–strain curve declines gradually as the anisotropic structure turns into string. This clearly shows that among these self-assembly structures, the sheet structure displays the best mechanical property. When the length of the grafted chains is increased to
and
, the stress–strain curve shows a stress plateau at a rather small strain (approximately 75%). This stress plateau is generated because of the blocking effect of well-dispersed nanoparticles, and thus instead of stretching the self-assembled structures formed by nanoparticles, the matrix undertakes most of the stress. As the interactions among matrix chains are relatively small, they are able to slide over and deform quite easily; consequently, the stress barely changes with the increase of strain. For better illustration, we examine the mechanical reinforcement by comparing the tensile stress at the strain equal to 300%, as is shown in
Figure 4c. Again, it is further confirmed that the best tensile property occurs when the self-assembly structure of the grafted NPs form sheets, compared to other morphologies.
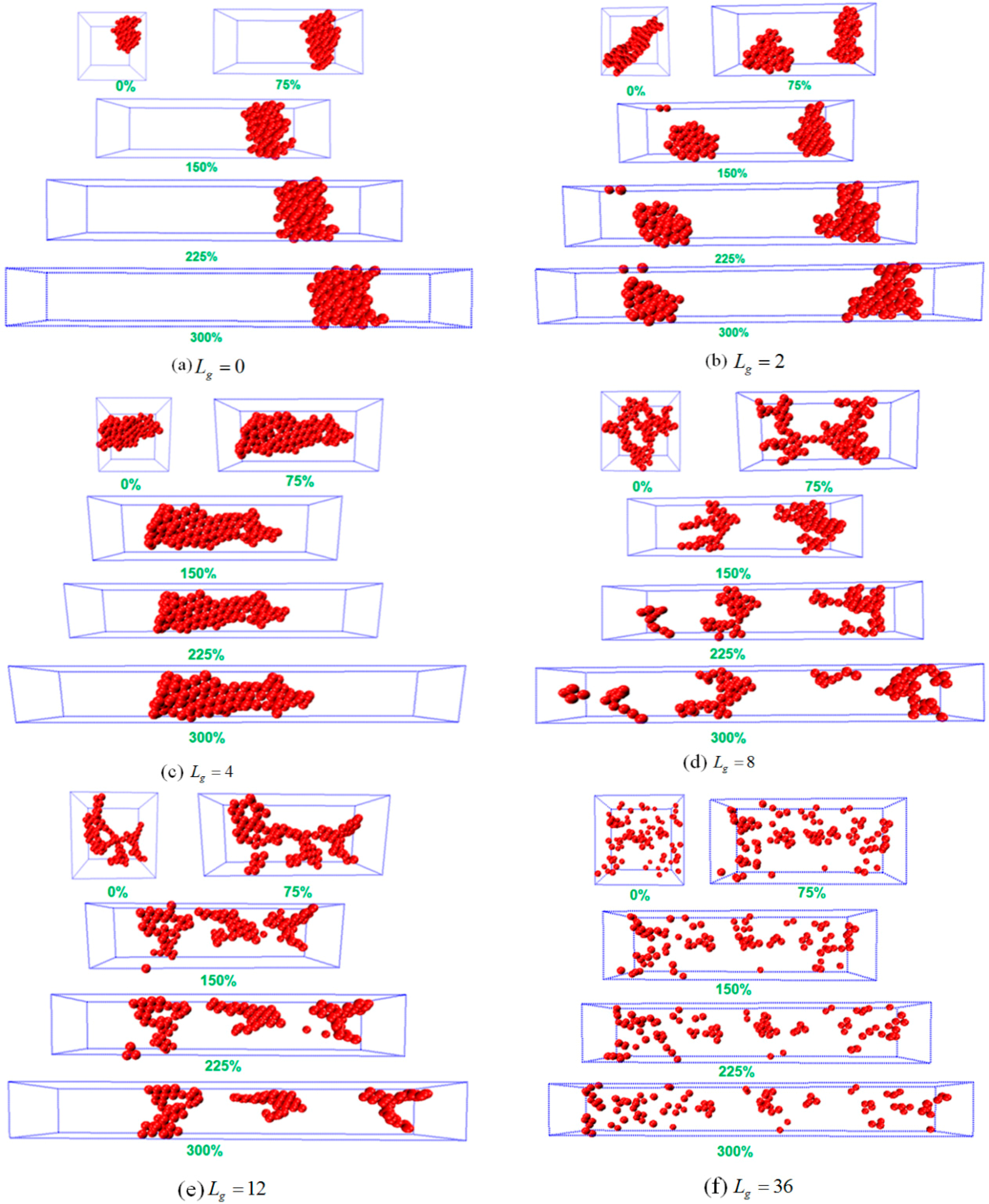
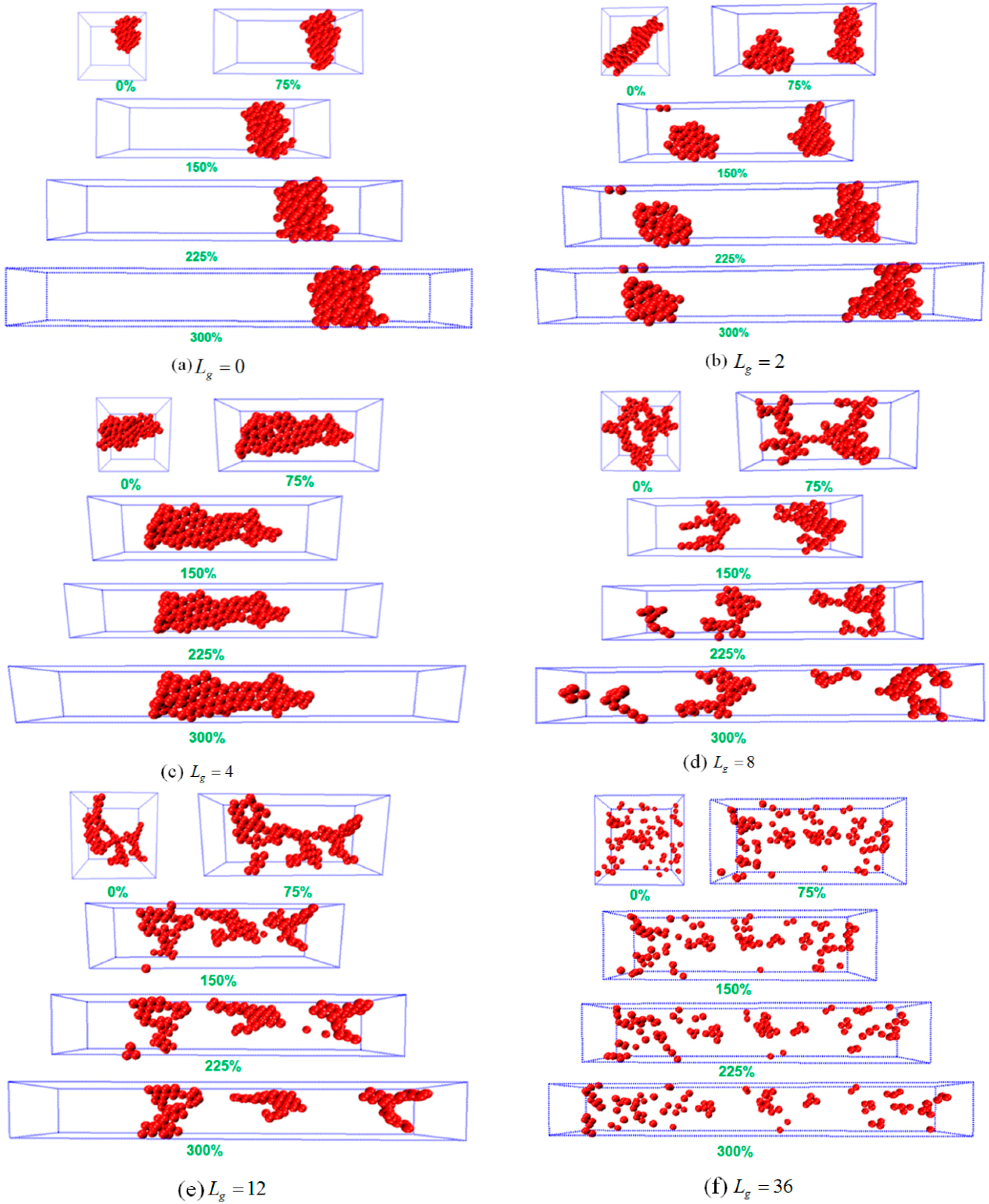
To analyze the possible reason of why the sheet structure endows the best mechanical performance, we probe the snapshots during the tensile process as shown in
Figure 5. For
, the agglomerate formed by the bare NPs remains intact during the tensile deformation, indicating no breakage because of the strong attractive interaction between NPs. Similar phenomenon is observed for
, in which case the cylinder structure is initially broken into two small clusters, and then the clusters flow and align along the tensile direction. For
and
, it can be seen that both systems exhibit an almost linear stress–strain curve, which explains the fact that the polymer matrix plays a dominant role during the tensile process. Furthermore, when the self-assembly structure transforms into sheets, and this structure gradually breaks and stretches into a rectangle with a longer side on the y direction. This observation implies that the percolating network itself undertakes a large part of the tensile force to enhance its mechanical property. In addition, when the grafted chain length is increased to
and
, the string structure formed by the grafted NPs initially break, and then coarsen locally into small clusters, followed by the alignment and orientation along the tensile direction. For the sheet and string structures formed by the self-assembled grafted NPs, it is noted that even though the NPs percolating network plays a dominant role in these two cases, the string structure contributes less to the mechanical reinforcement compared to the sheet structure, attributed to much weaker interaction between the grafted NPs for the string structure. While for the dispersed states of
and
, during the tensile deformation the NPs do not show any phase transition, except for a slight agglomeration during the tensile process. However, a strong physical inter-locking occurs between the grafted and matrix polymer chains, in the case of long grafted polymer chains, which could lead to the increase of the mechanical reinforcement.
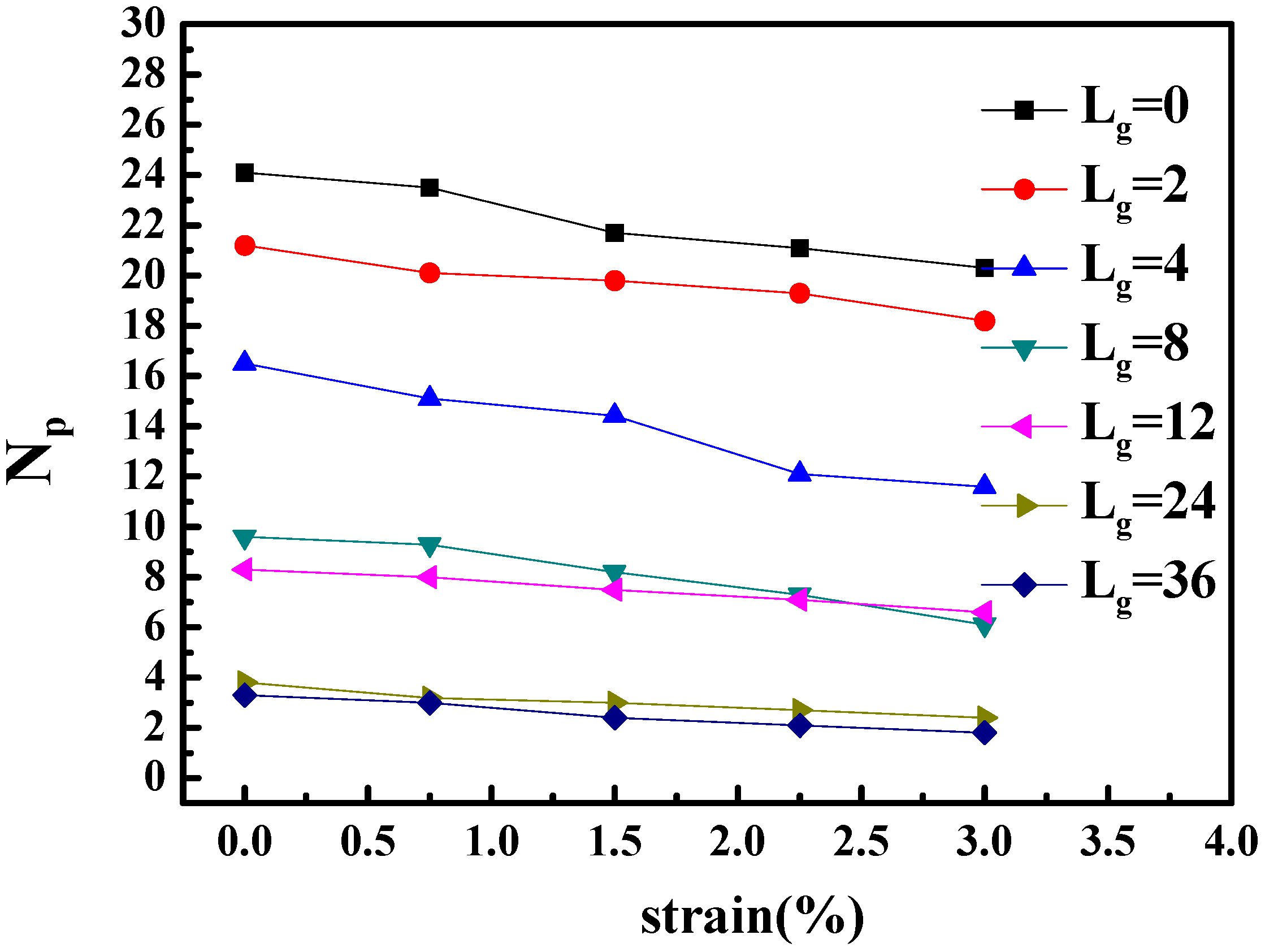
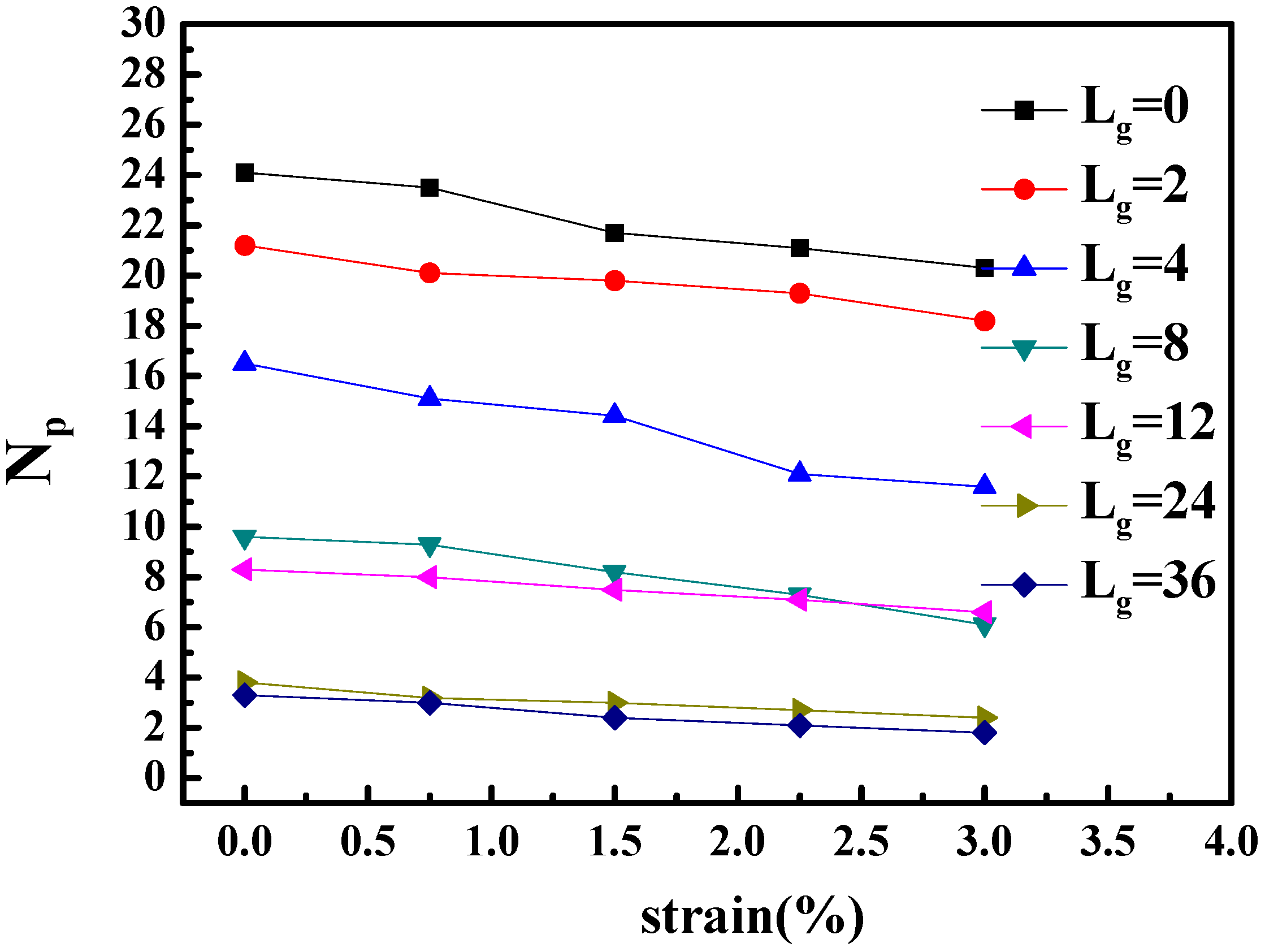
To better identify that the microscopic origin of the stress is from the breakage of clusters, we calculated the shape deformation and cluster size of nanoparticles during the extension. The variation of the number of clusters and the number of nanoparticles per cluster are listed in
Table 2. In addition, the change of the number of neighbor nanoparticles within the distance of
(radius excluded) with regard of time evolution is illustrated in
Figure 6. It can be seen that with the increase of strain, the cluster breaks into smaller pieces, and within the same cluster, the distance between particles become significantly smaller. This means that the clusters, i.e., the nanoparticle structures undertake the major part of the tensile stress.
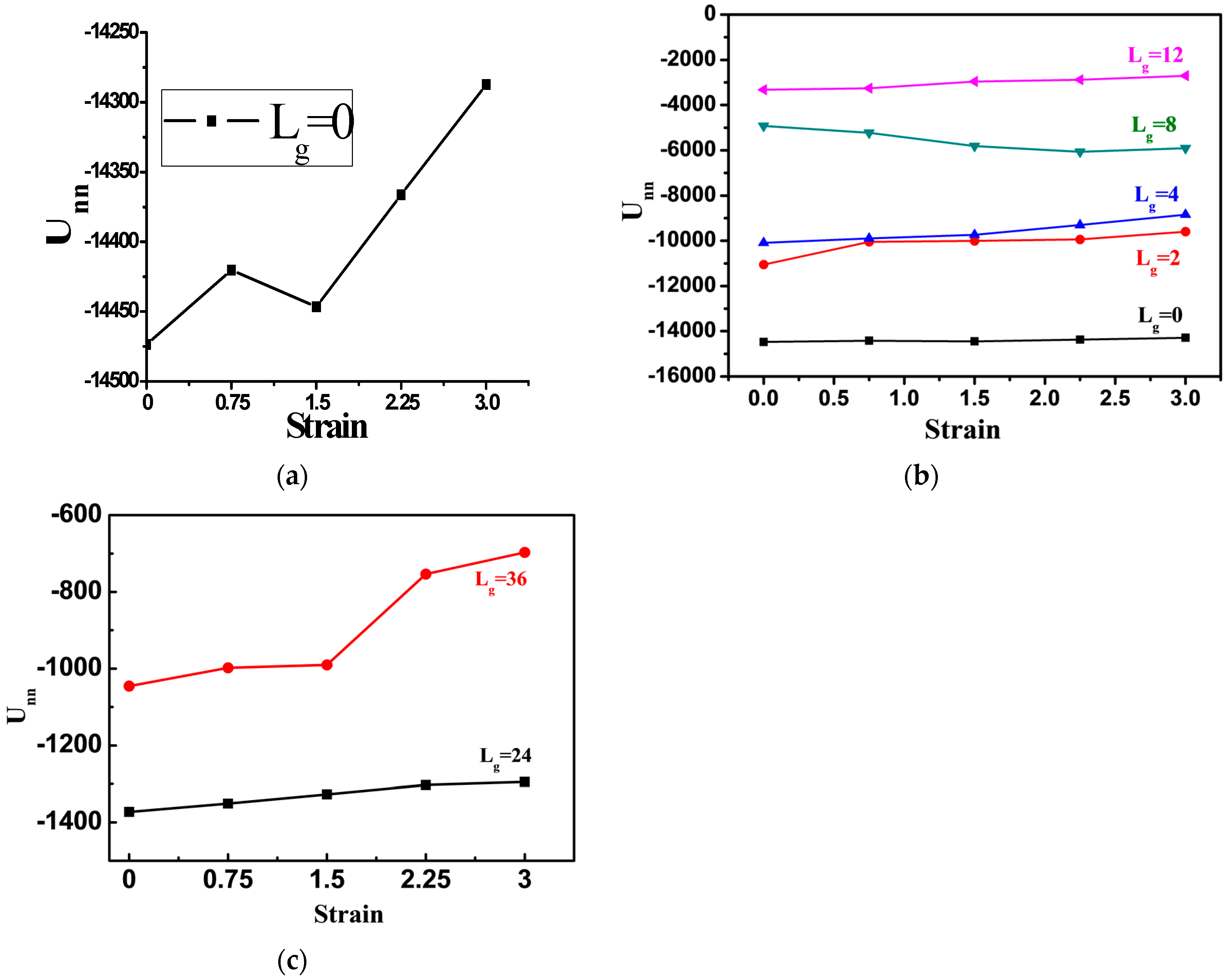
To better quantify the deformation process, we calculate the total interaction energy between NPs during tensile process, as shown in
Figure 7. For the cases of the bare NPs or NPs grafted with chains
, the total interaction energy between NPs is slightly decreased, corresponding to the snapshots in
Figure 5a,b. Namely, during the tensile deformation, the tight structure formed by NPs is broken up only slightly away from each other. When the grafted chain length is extended to
, the variation trend of the total interaction energy is similar to that of the bare NPs. As suggested by the corresponding snapshot, the sheet-like structure formed by the NPs is extended further away from each other, resulting in a monotonic increase of the interaction energy. However, in the case of the grafted chain length equal to
, the total interaction energy is increased during the tensile deformation, implying the break-up of the NPs agglomerates. For the much longer grafted chain length such as
and
, the total interaction energy between the NPs decreases more significantly, as shown in
Figure 7c.
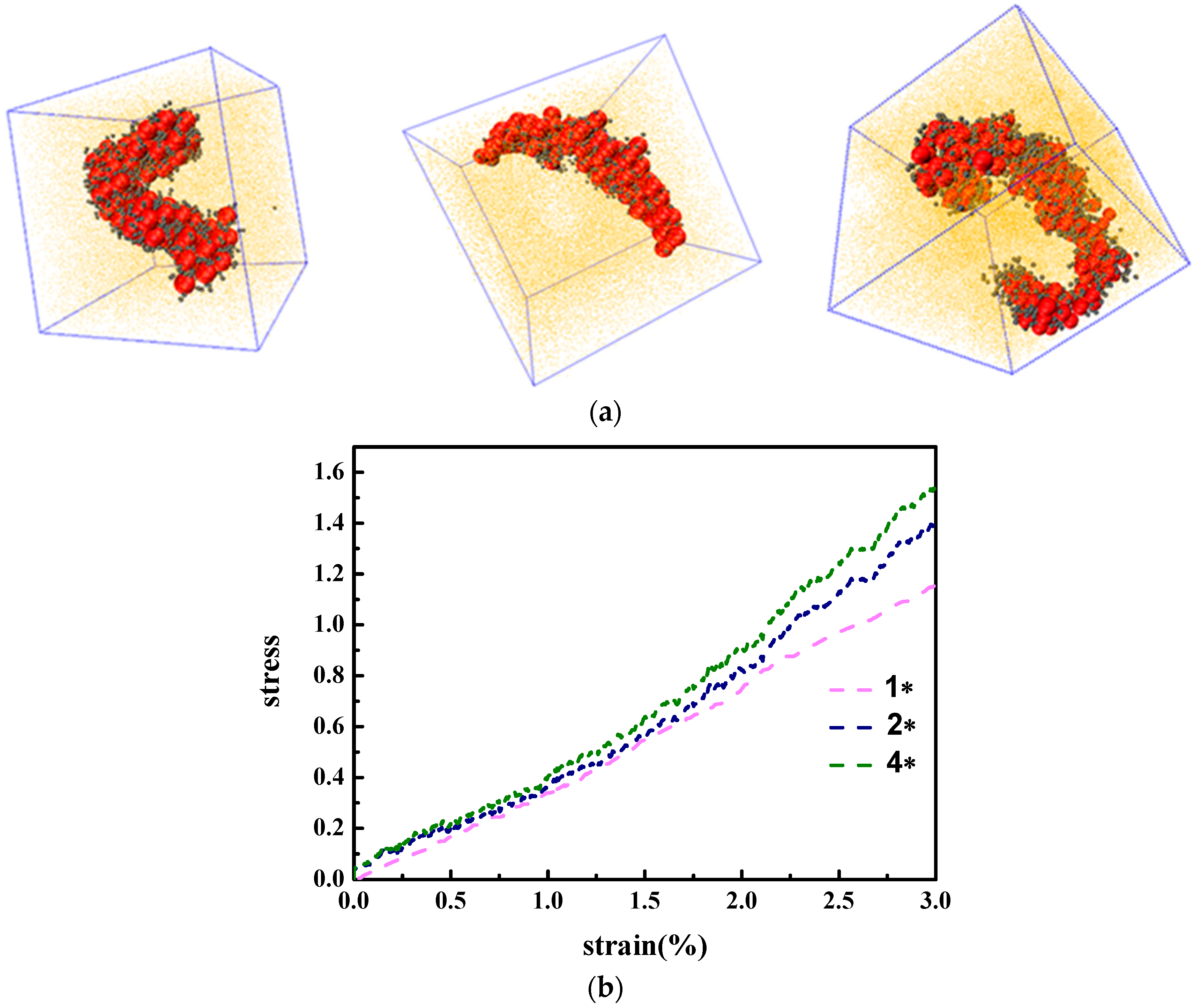
In fact, we have observed that the strongest mechanical reinforcement is obtained when the grafted NPs self-assemble into the sheet structure. Based on the research work carried out by Moll et al. [
13], a maximum stress occurs at the moderate strain, followed by the decrease of the stress as a function of the strain, which is also referred to the “stress overshoot”. The occurrence of the maximum stress is to verify the existence of a solid-like structure, which is often observed in those systems with a percolated structure network rather than a well-dispersed structure. Here we also want to probe this issue, by investigating the following three systems such as
,
and
, and stretch along the y direction to a large strain by keeping the volume unchanged, as displayed in
Figure 4d. Obviously, the stress reaches the maximum value, or an overshoot when the strain is equal to 10 for the grafted chain length
with the sheet structure, while only a stress plateau occurs for the cases of
and
even at the large strain. This also supports the fact that the sheet structure gives the best mechanical reinforcement performance. The decline of the stress could result from the break-up of the percolated network structure of the sheet structure.
Finally, in order to eliminate the effect of the periodic boundary, we demonstrated the finite size effects of the simulation box on the obtained structures and the corresponding mechanical response. We multiplied the whole system by both twice and four times the initial system. After equilibrium of the same conditions, we obtained the self-assembly structure of nanoparticles, as is seen in
Figure 8a. It is clearly illustrated that at both conditions, the nanoparticles self-assemble into a cylinder-shaped structure, indicating that our results regarding the system size is not necessarily modified. As for the mechanical properties, we performed a tensile process on both systems and the results are shown in
Figure 8b. The stress–strain behavior does not show any obvious shift. This means that our boundary conditions and system size choice is reliable.
3.2. Effect of the Flexibility of the Grafted Polymer Chain
In this part, we shift our attention to vary the grafted chain flexibility, which receives little attention about its influence on the relation between the morphology and the mechanical properties. By choosing the grafted chain length
,
,
,
and
, we introduce the angle potential to model the semi-flexible grafted polymer chain. The potential of angles can be described as follows:
in which
is the equilibrium value of the angle, and
is the prefactor demonstrating the stiffness of the bond. Here, we set
and
, and overlap grafted polymer beads by
with each other, leading the persistence length of the grafted chains to be approximately
.
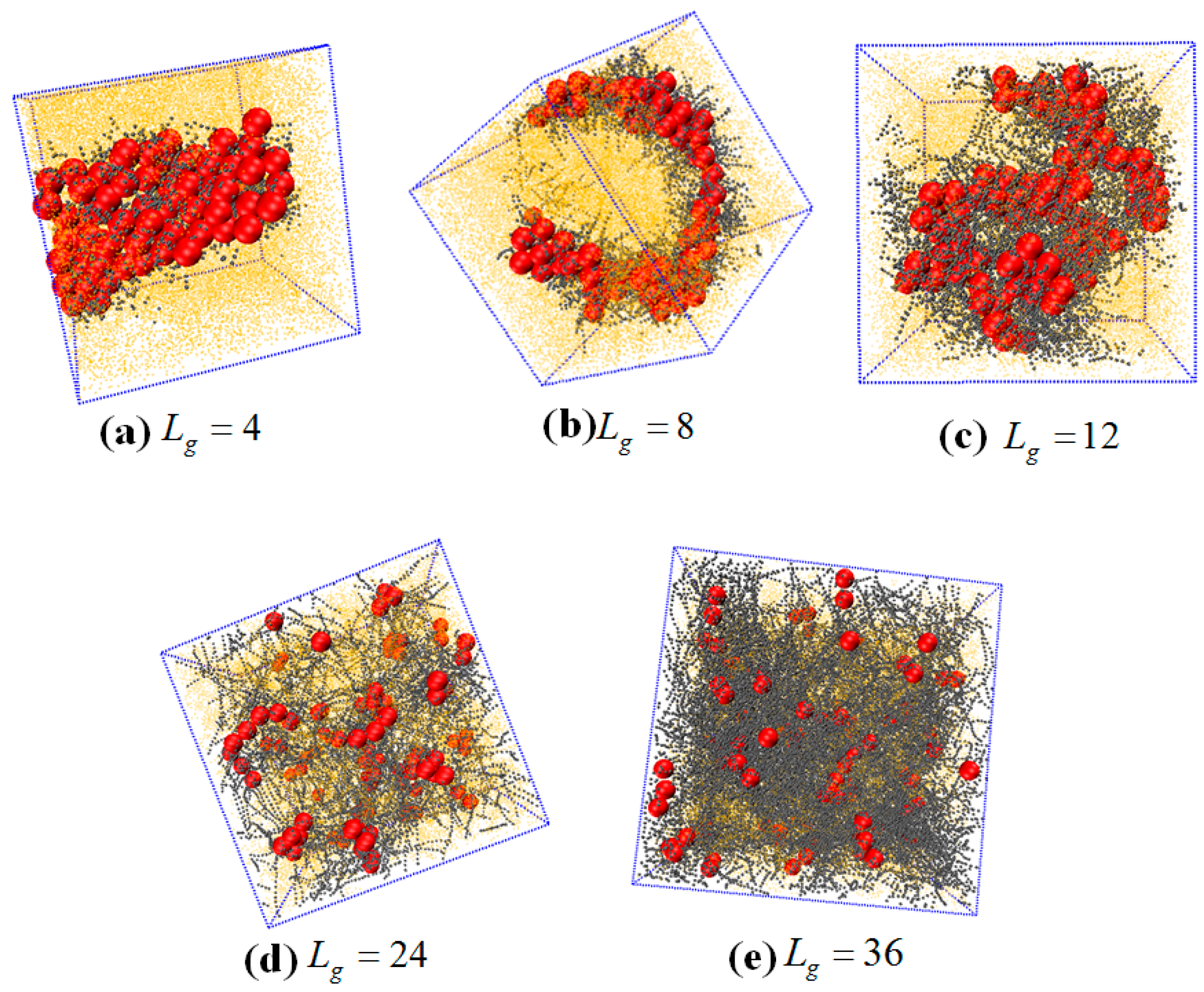
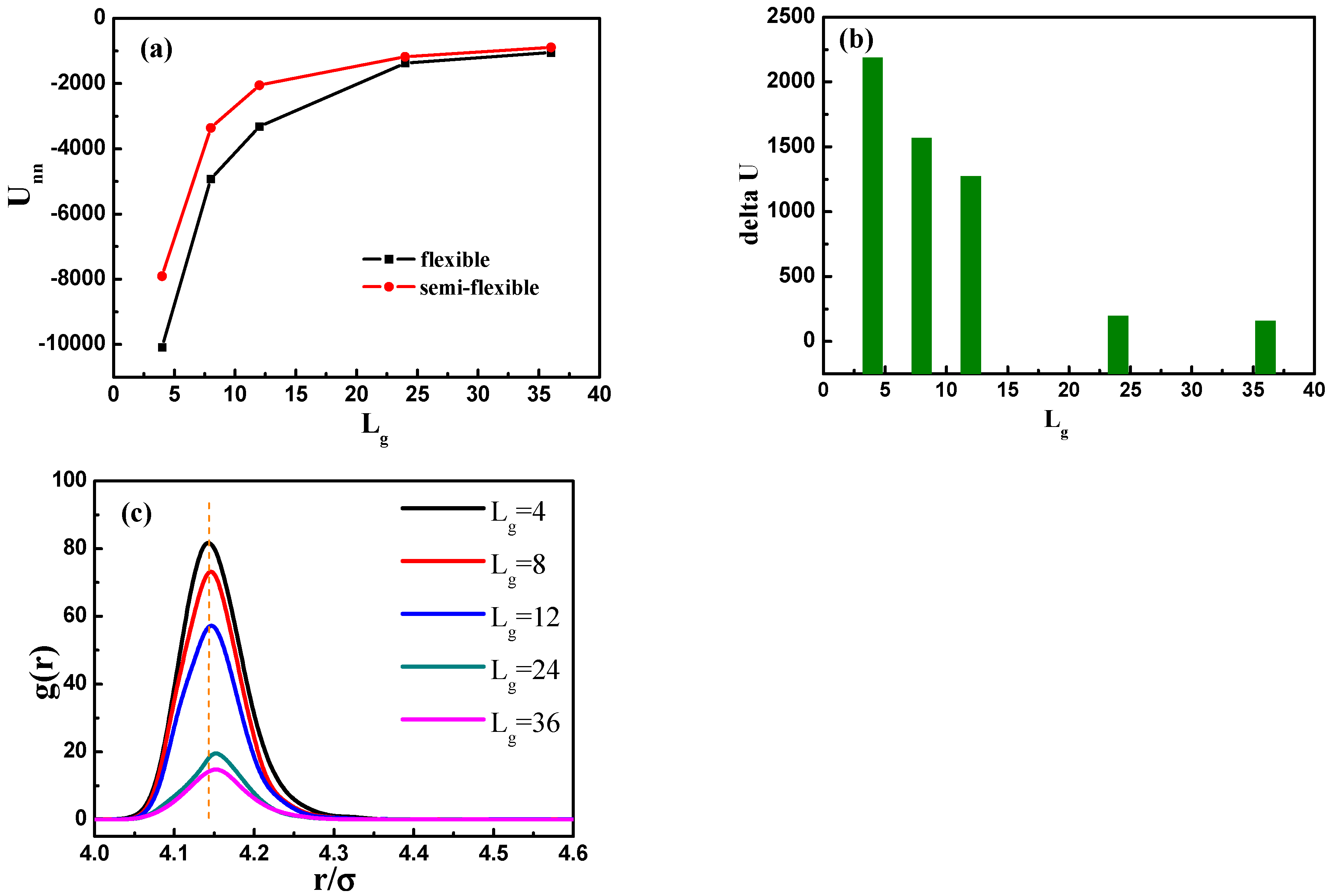
The self-assembled equilibrium structure for various grafted chain length is shown in
Figure 9. In the case of
, a sheet structure is obtained similar to the case of the flexible grafted chain. For
, the NPs self-assemble into a more ordered circle-like string structure. For
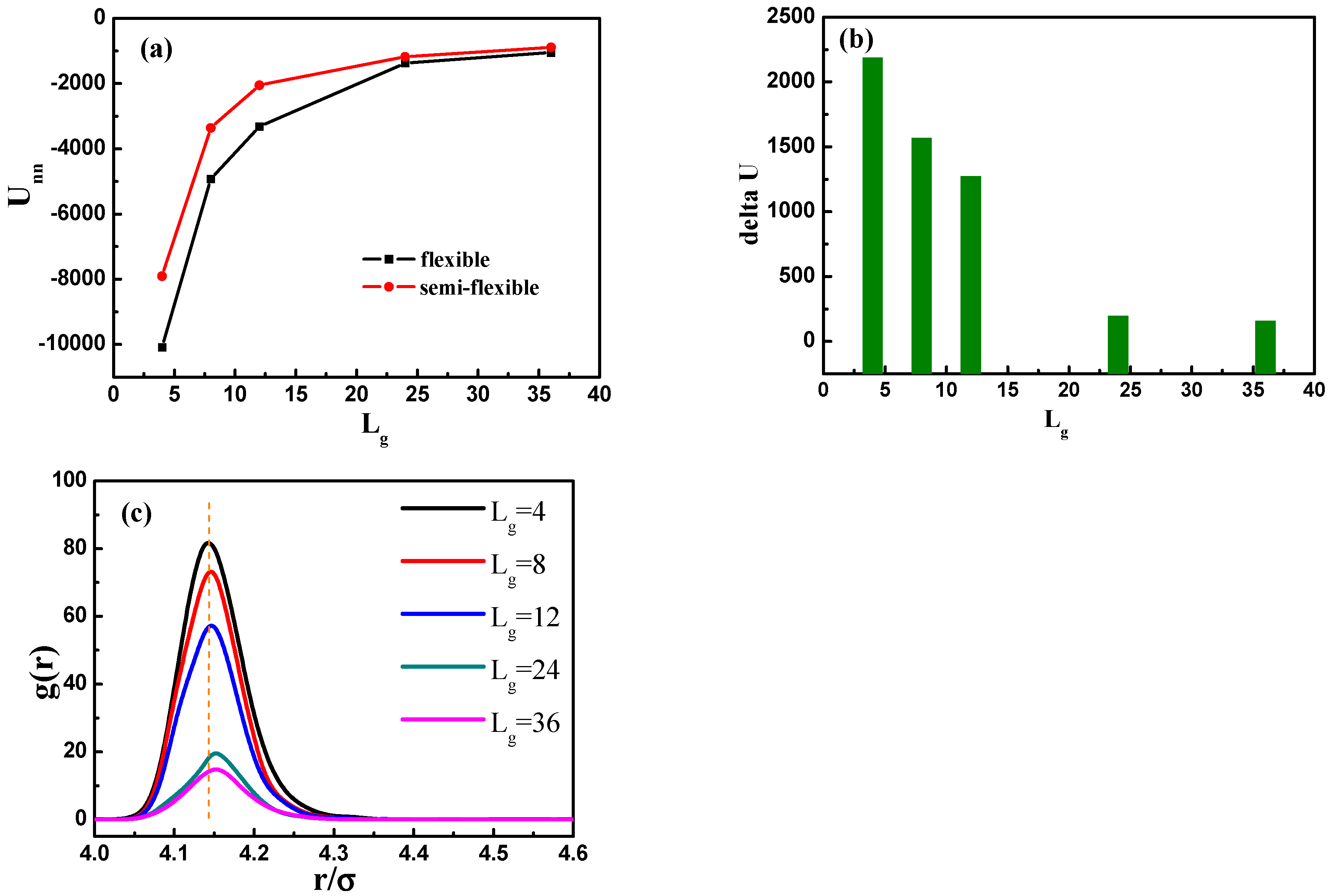
, 24 and 36, the grafted NPs self-assemble to form short string structure, dispersed and well-dispersed states, respectively, which are similar to the cases with flexible grafted chains. We further calculate the total interaction energy between the NPs as a function of the grafted chain length in
Figure 10a. It clearly shows that for the semi-flexible grafted polymer chains, the total interaction between NPs becomes much weaker, compared to the case of the flexible grafted polymer chain at each grafted chain length. The underlying reason is that the semi-flexible grafted polymer chains tend to be extended into a larger corona around each NP, because the semi-flexible polymer chains are less likely to fold to become the coil configuration, making the NPs more difficult to contact with each other and thus resulting in relatively small total interaction energy between NPs. Relevant previous research [
16] has shown with aid of experiments that when flexible grafting chains are shifted to rigid chains, the effect of increasing the length of grafted chains on increasing the dispersion of nanoparticles is reduced. In order to better illustrate this, we calculate the difference of the total interaction energy among NPs between the flexible and semi-flexible grafted polymer chains in
Figure 10b. For the semi-flexible grafted polymer chains, the dispersion state of NPs is improved with
, which, however, is not as obvious as the case of the flexible grafted polymer chains. Moreover, when the grafted polymer chains become long enough, such as
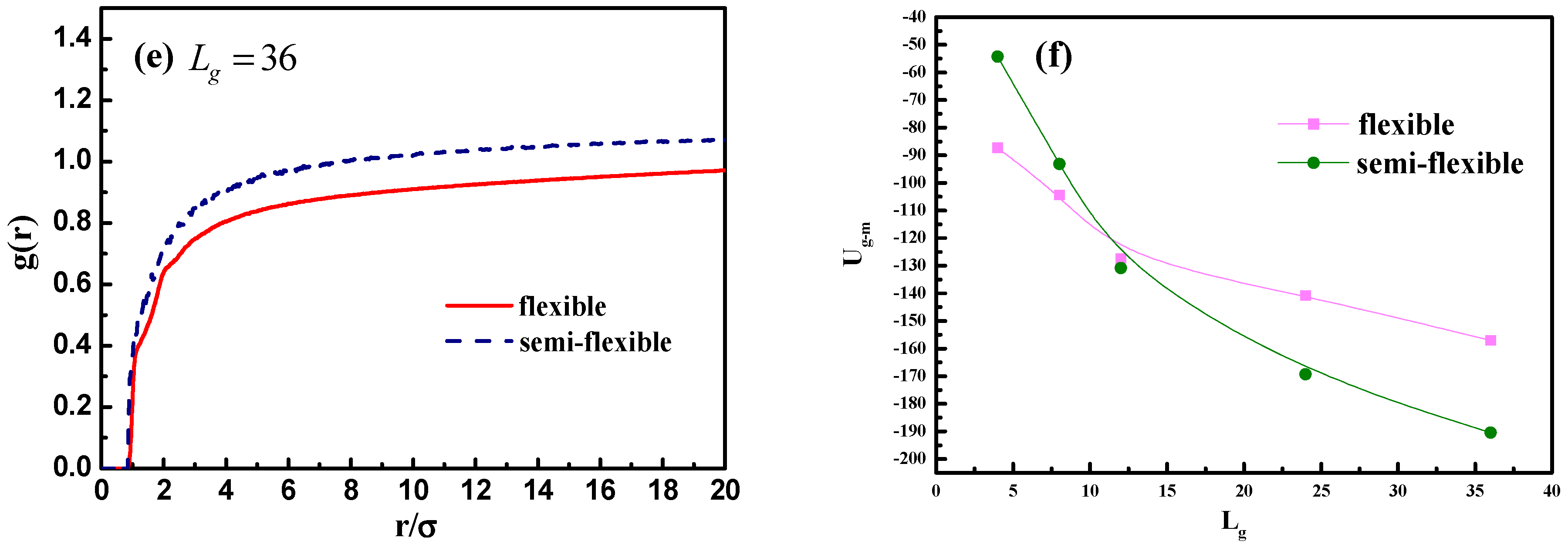
and 36, the difference between the total interaction energy among NPs becomes not so obvious by comparing the flexible and semi-flexible cases, because in this situation the effect of the chain length surpasses the chain configuration.
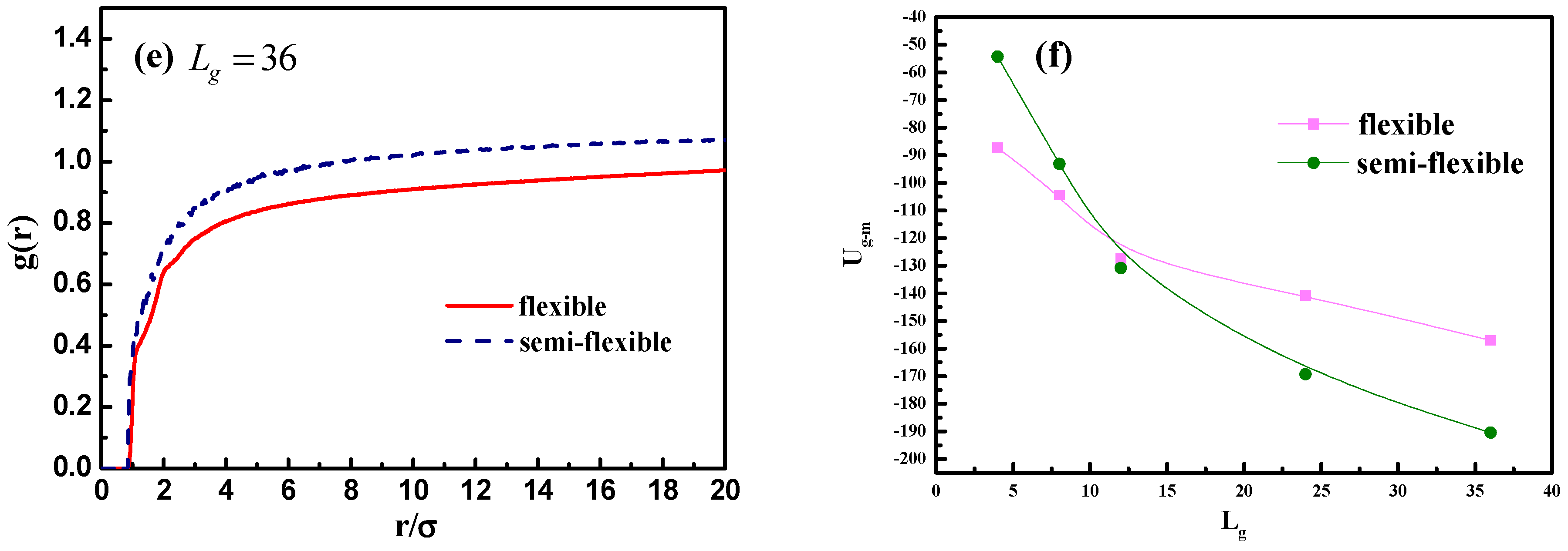
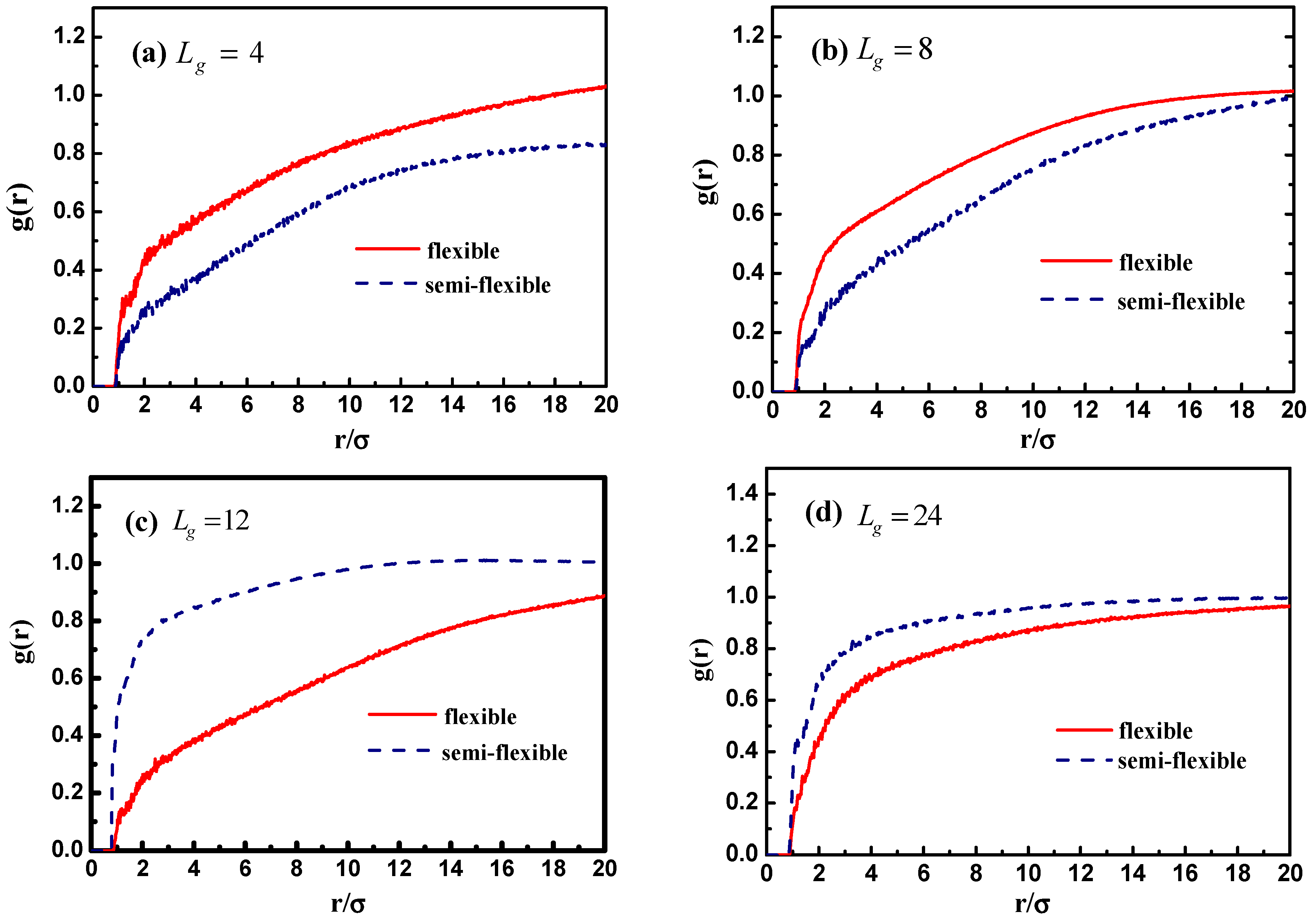
In addition, we calculate the radial distribution function of the NPs in
Figure 10c, and it is rather clear that the density of the other NPs around each NP is largely reduced with the increase of grafted chain length. It is noticeable that the position of each peak is slightly moved to a large inter-particle distance as the grafted chain length becomes longer.
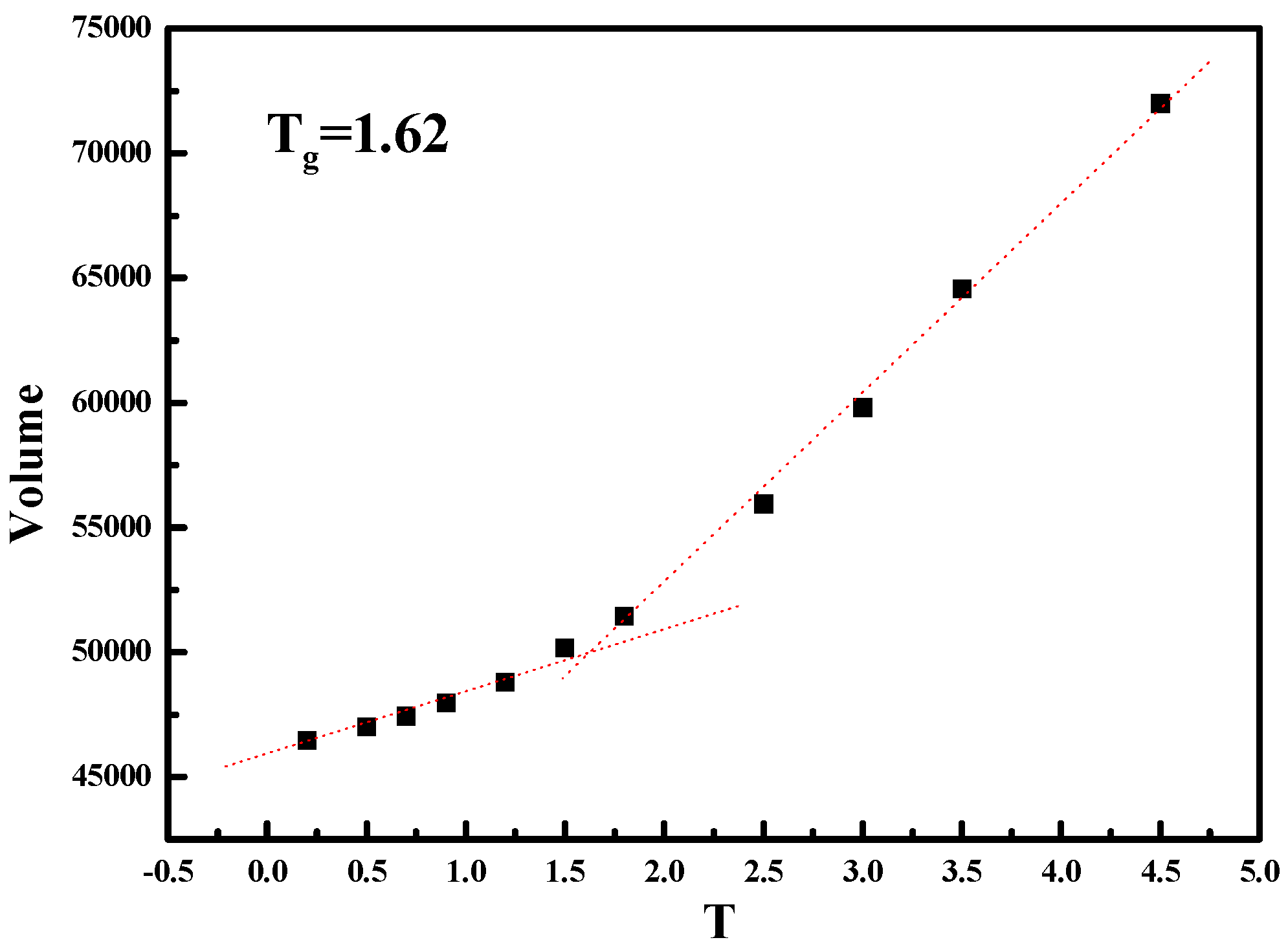
We then characterize the mechanical properties of these five simulated systems with the NPs grafted with semi-flexible polymer chains. It should be noted that our system with semi-flexible grafted chains is still under melting state with temperature
, and thus the stress–strain behavior is absolutely observed well above glass-transition point, as shown in
Figure 11.
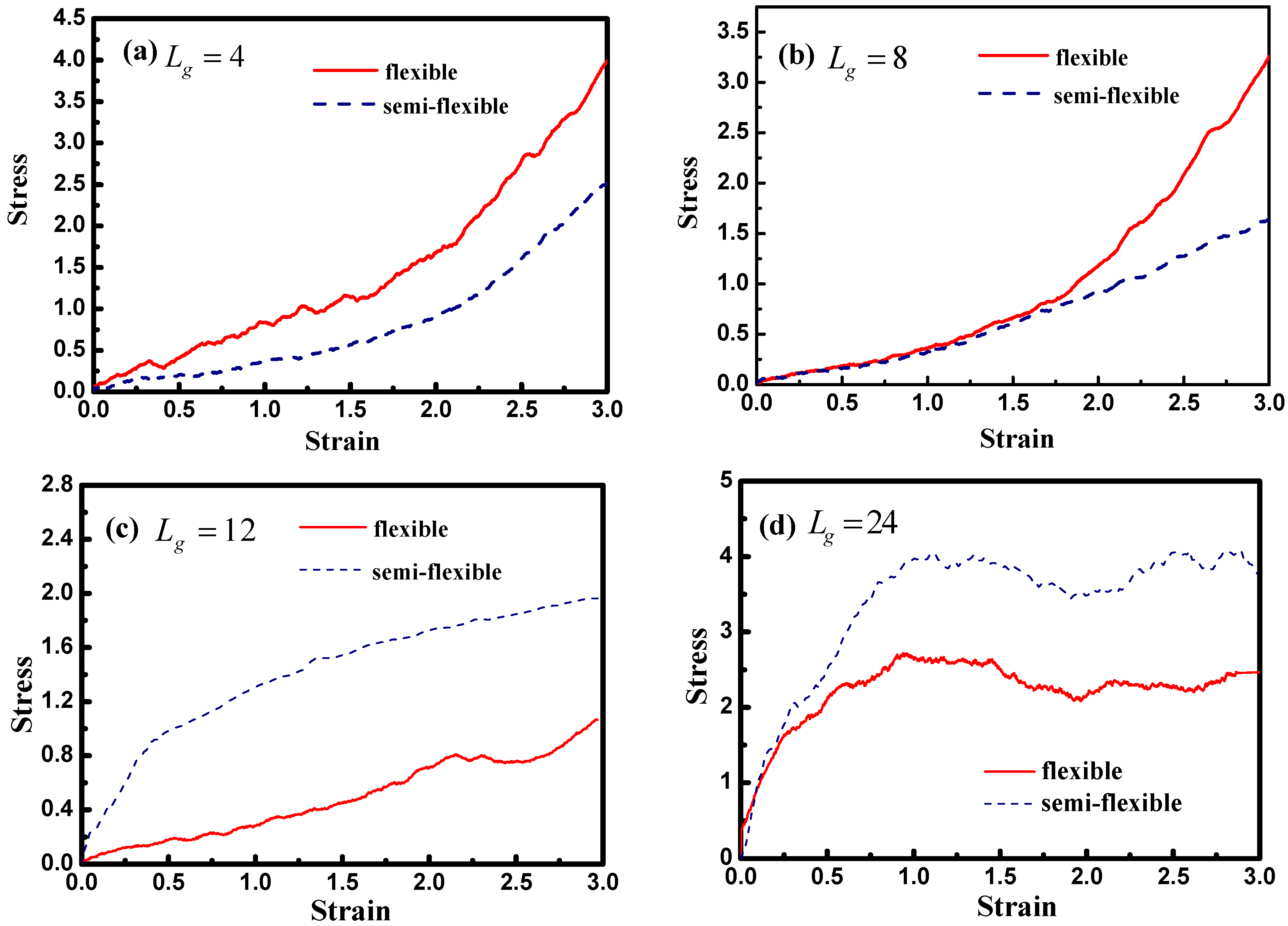
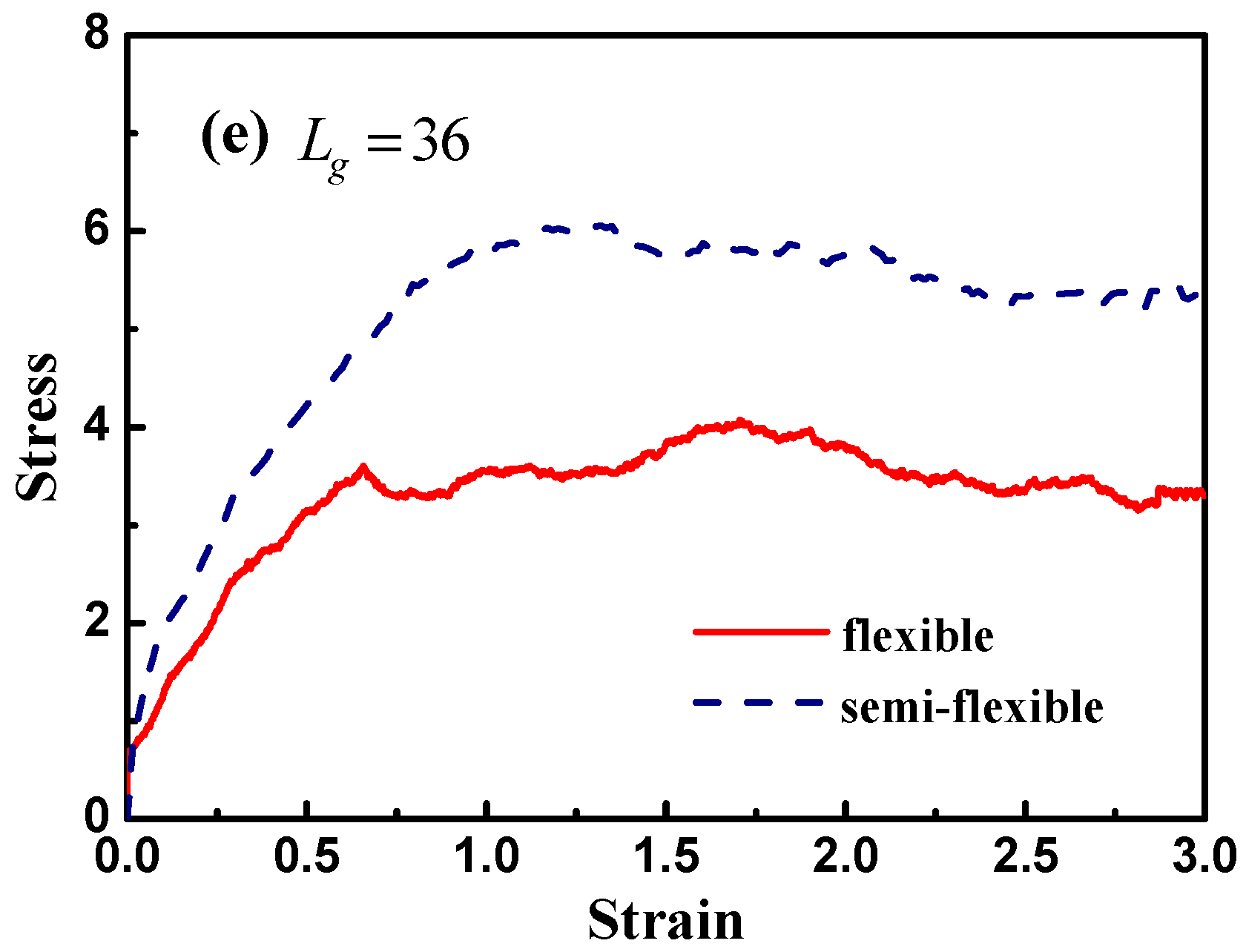
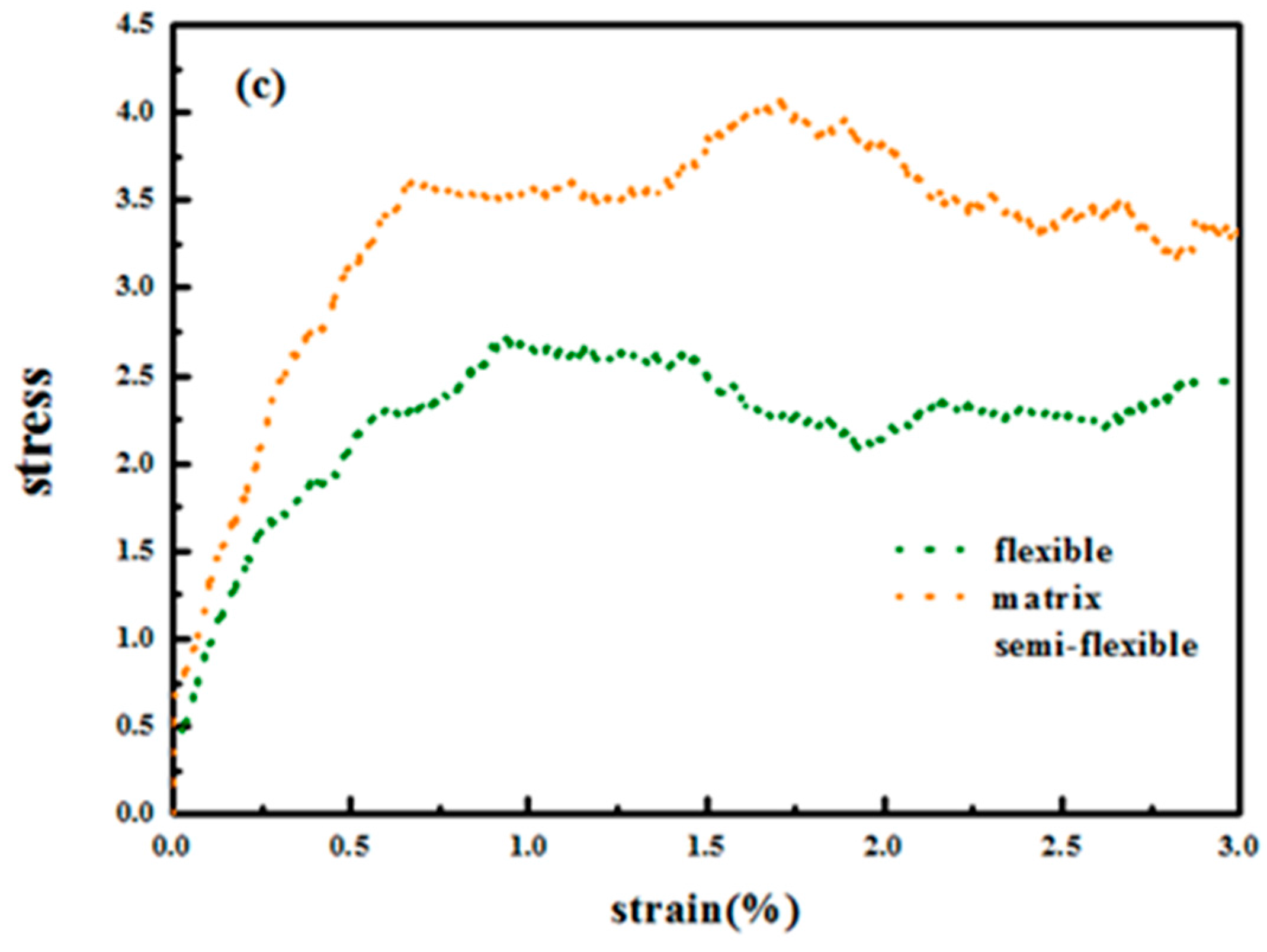
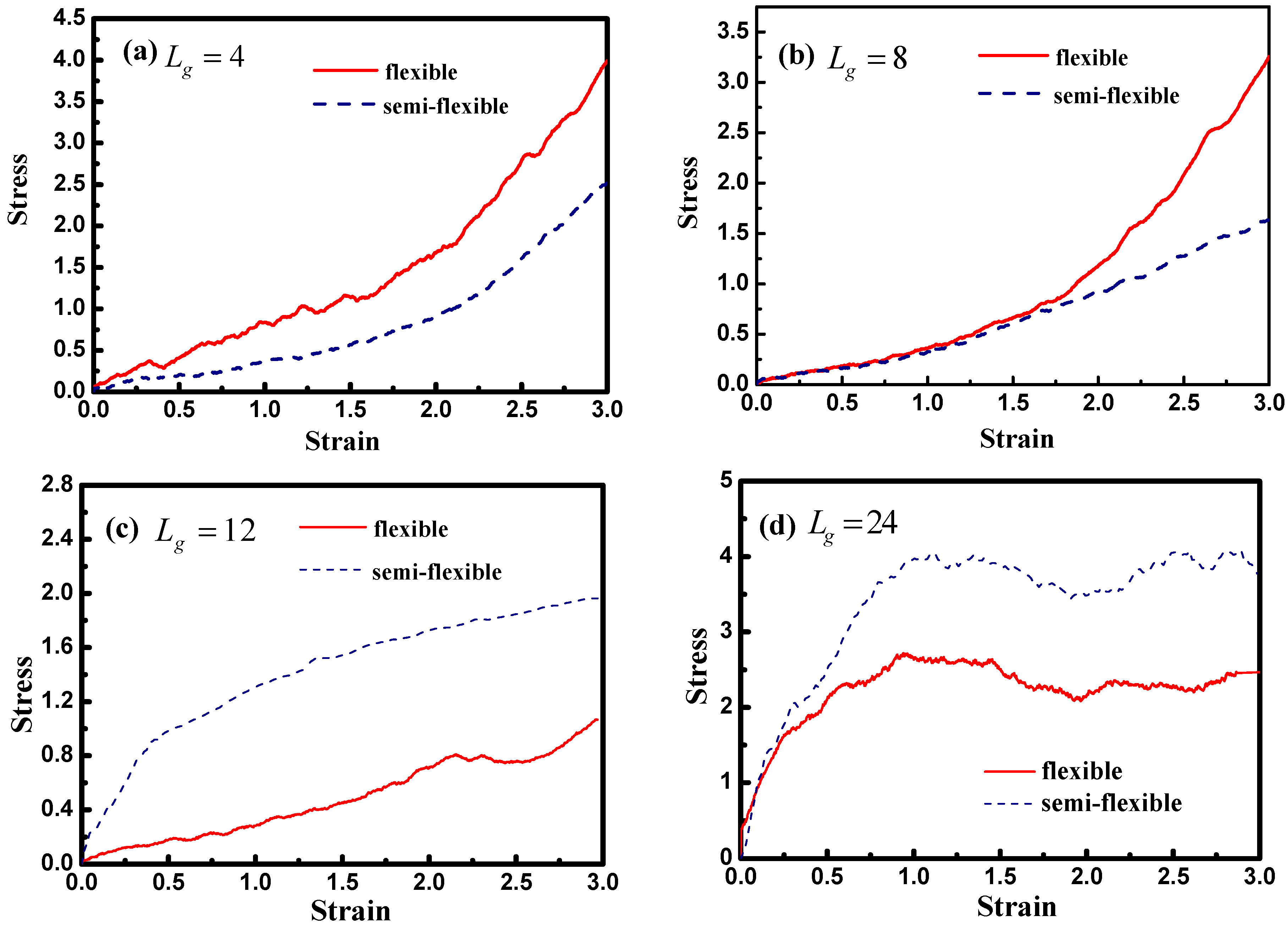
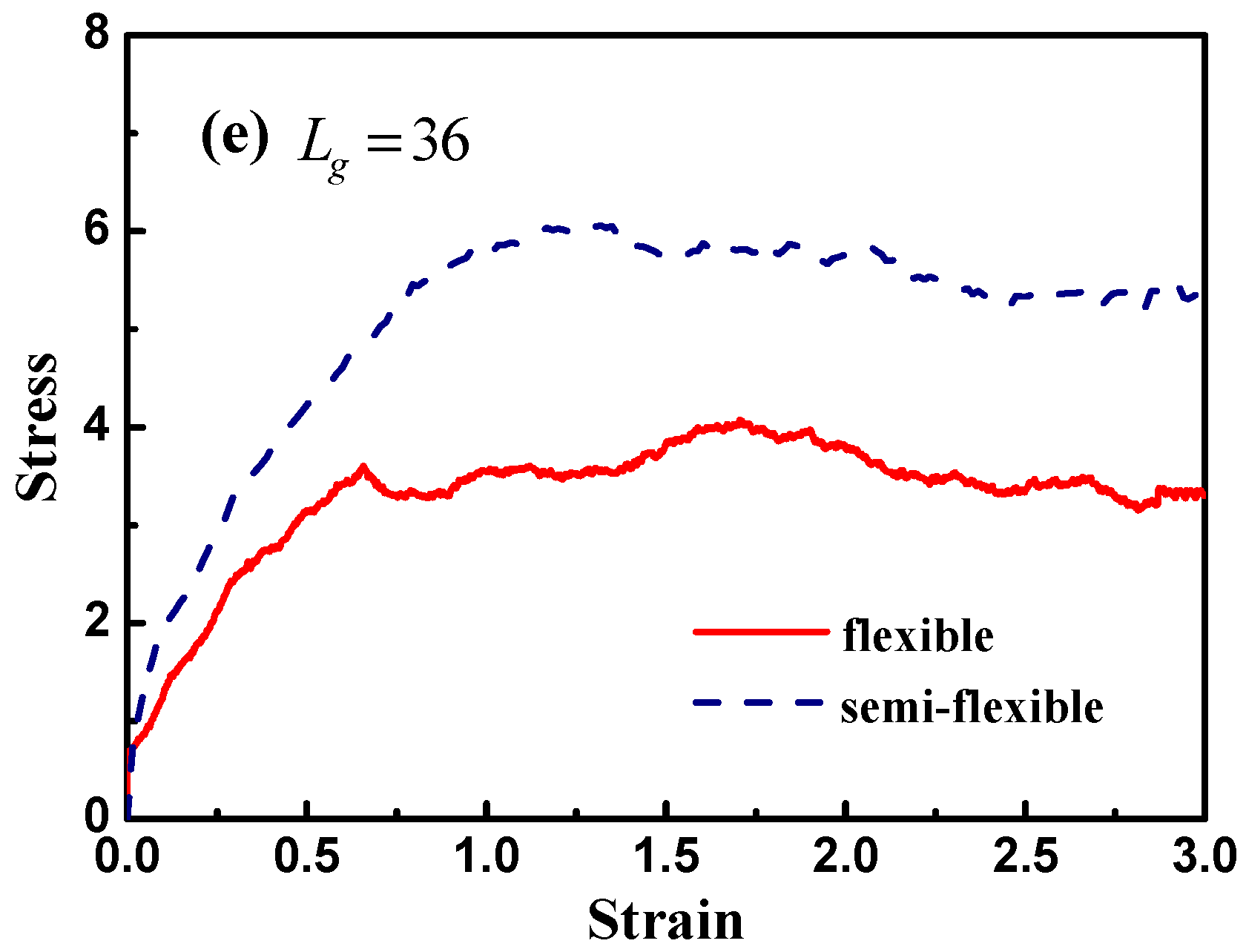
Figure 12 demonstrates the stress–strain curve with different grafted chain length, and we compare the stress–strain curve between semi-flexible grafted chains and flexible grafted chains. It can be seen that for
and
, a much better stress–strain curve occurs for the case of the semi-flexible grafted polymer chains, compared to the case of the flexible grafted polymer chains, at longer grafted chain length such as
, 24 and 36, however, this trend becomes contrary. We infer that at long grafted chain length, the physical inter-locking interaction and wetting between the matrix and grafted polymer chains account for this phenomenon. To better verify our assumption, we calculate the radial distribution function of the polymer matrix beads around each grafted chain polymer bead as shown in
Figure 13. The result is consistent with the observation of the stress–strain behavior; namely, at short grafted chain length the density of the polymer matrix beads around the semi-flexible grafted polymer chains is smaller than that of the flexible grafted polymer chain. While at long grafted polymer chain length, the semi-flexible grafted polymer chains become wetter by the polymer matrix compared to the flexible grafted polymer chains. This transition point happens when the grafted chain length increases from
and
, as indicated by the radial distribution function shown in
Figure 13, which agrees with the transition of the stress–strain behavior versus the grafted chain length. This result just implies that the semi-flexible grafted polymer chain should be long enough to become inter-locking and deeply wet with the surrounding polymer matrix, which is consistent with the experimental observation that the thermo-mechanical properties of PNCs can be critically influenced by polymer–NPs wetting behavior [
23]. In order to quantitatively determine the scale of how much semi-flexible polymer and flexible polymer become compatible with each other regarding wetting, we hereby calculated the interaction parameter between the grafted polymer chains and matrix polymer chains with conditions of both flexible grafted chains and semi-flexible grafted chains. As shown in
Figure 13f, the flexible grafted chains show a better wetting with matrix at shorter lengths, while the semi-flexible grafted chains show a better wetting at longer chain lengths.
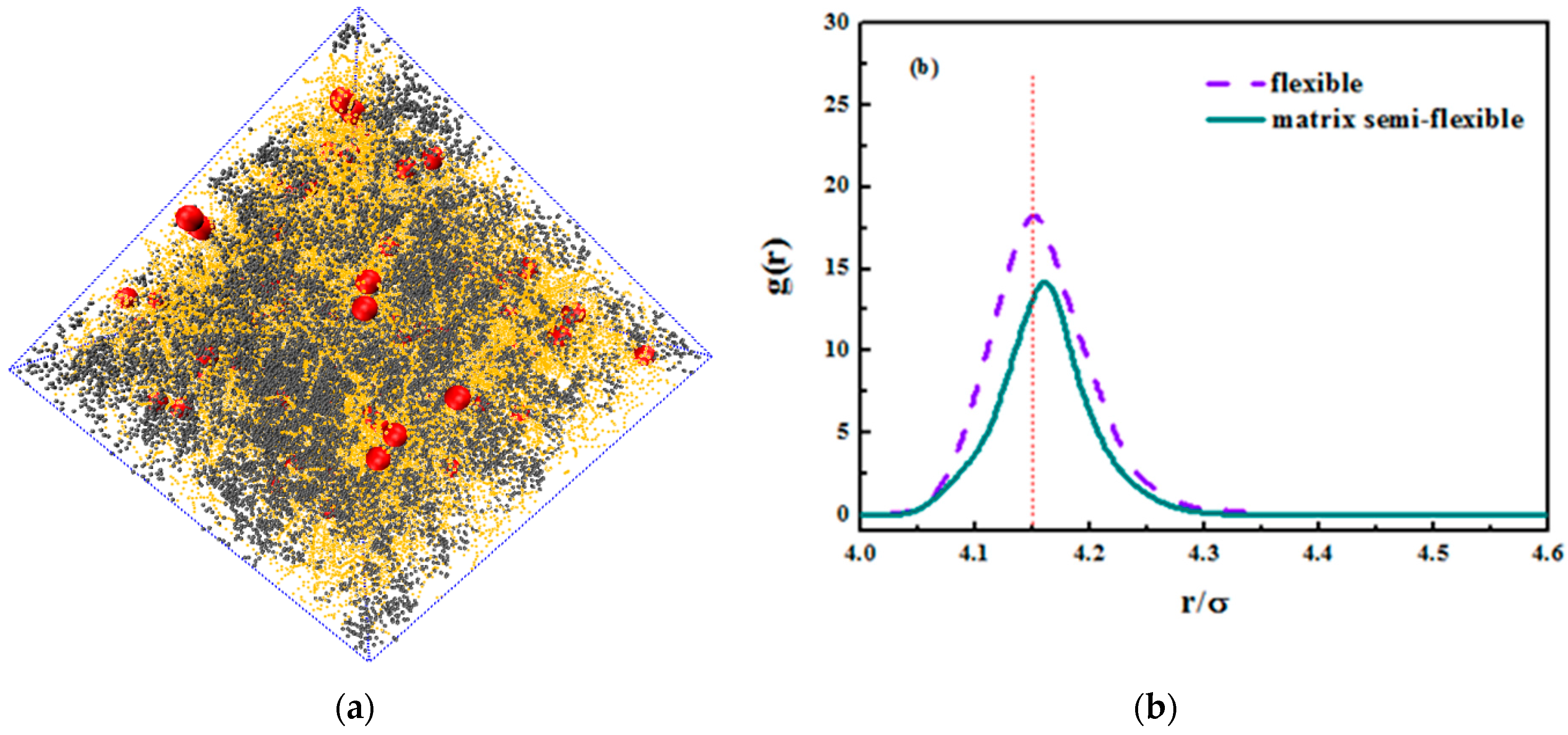
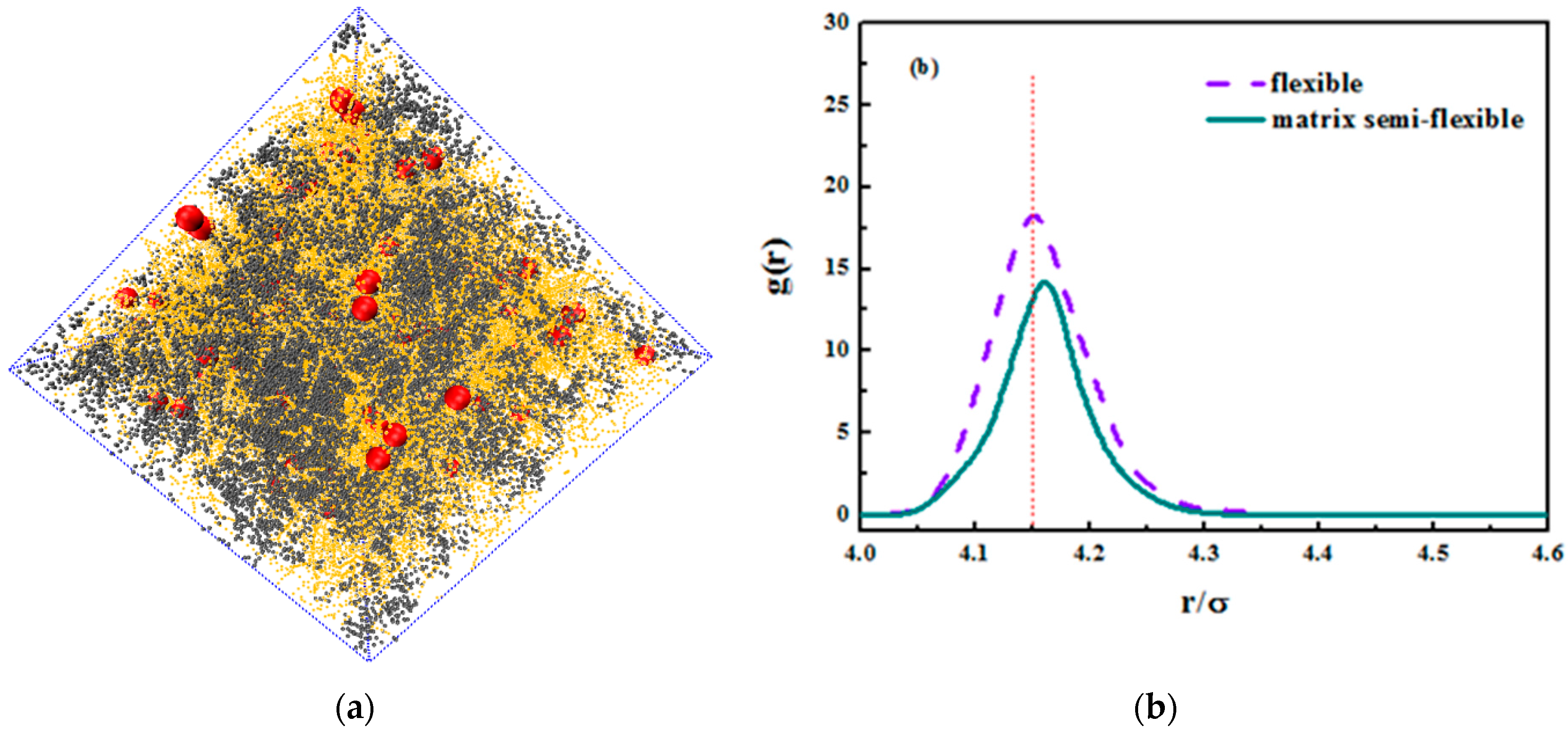
We hereby demonstrate the situation where the matrix polymer chains are semi-flexible and the grafted chains are flexible merely as a reference. We concentrated on the grafted chain length of
, and an equally nice dispersion is seen in the snapshot in
Figure 14a. In addition, we compared the radius distribution function of nanoparticles in both semi-flexible matrix chains and flexible matrix chains. The nanoparticles in semi-flexible matrix chains demonstrate a better dispersion that those in flexible matrix chains, which can validate the former idea that the entanglement between grafted chains and matrix chains promotes the dispersion of nanoparticles. As the matrix chains become semi-flexible, it is less likely that they curl into a coil and the surface area increases, leading to a better interaction with grafted chains. As for its tensile response, the comparison is shown in
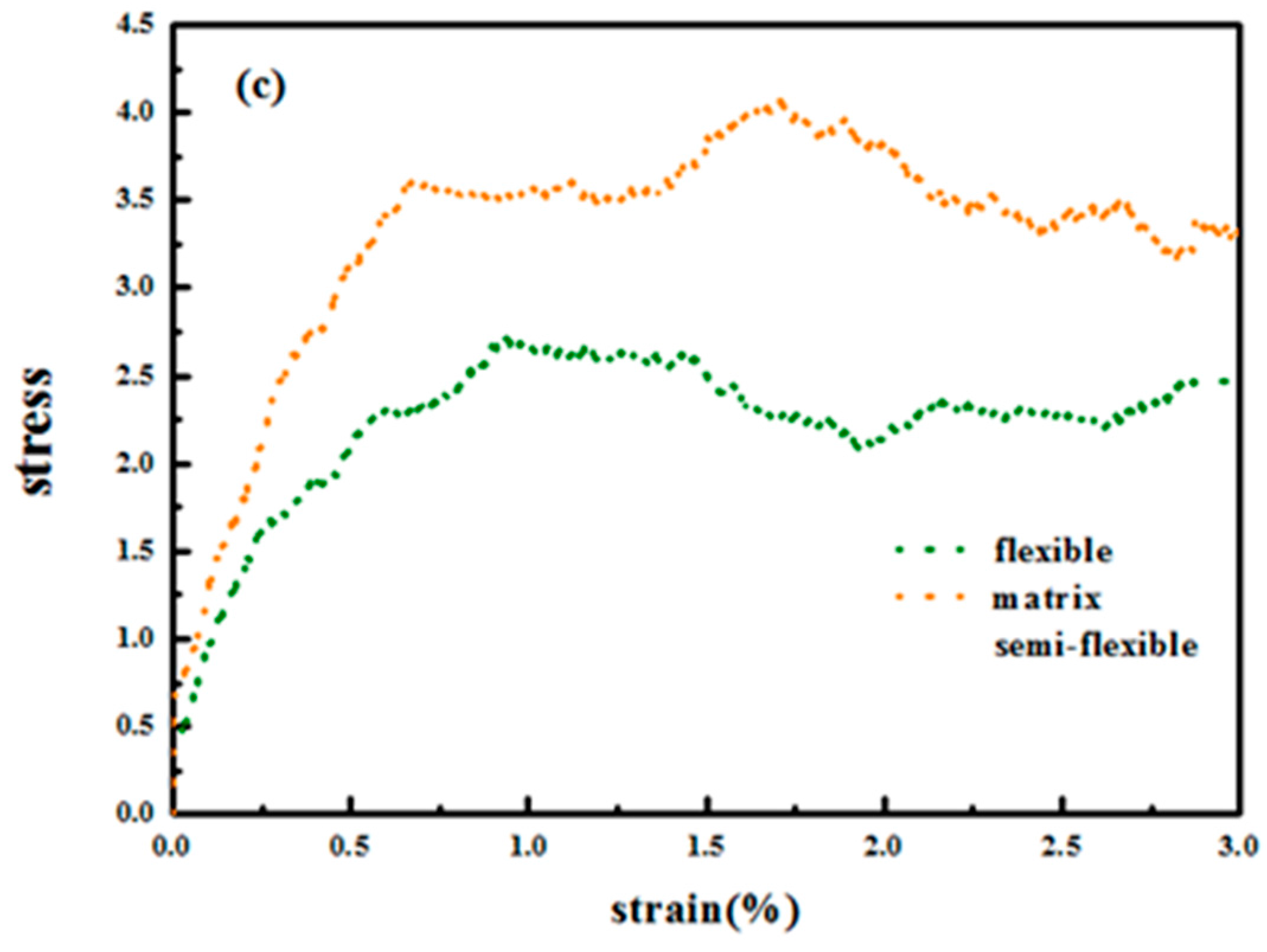
Figure 14c. It is not hard to see that the rigid matrix chains enhance the tensile properties. The matrix chains here undertake most of the stress especially during plateau deformations. As matrix chains become more rigid, they are less likely to slide with each other under tensile and instead they orient with tensile direction and the covalent bonds take up most of the stress. The covalent bonding interaction is larger than the van der Waals’ interaction, so the tensile strength is relatively large for semi-flexible chains.
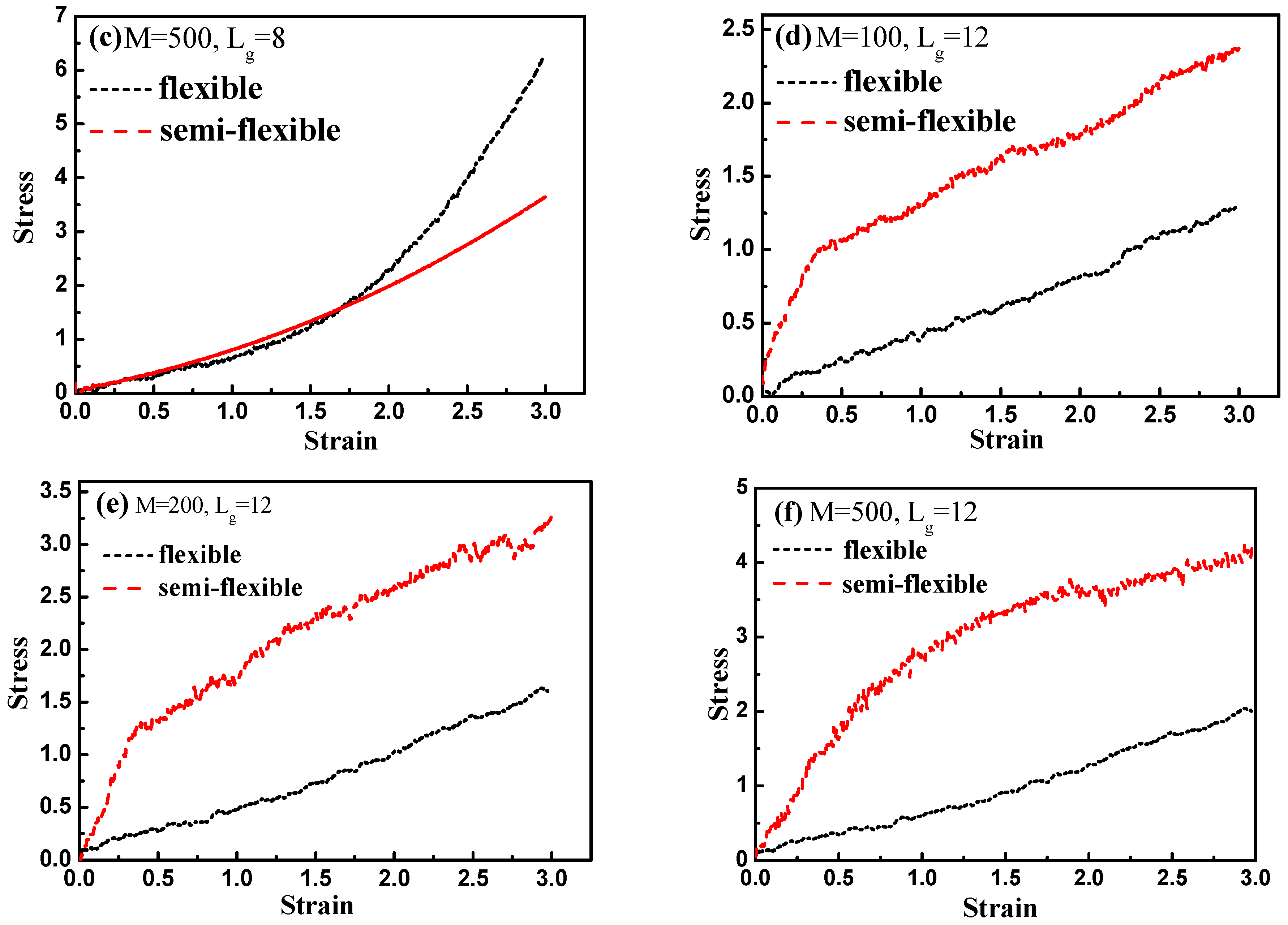
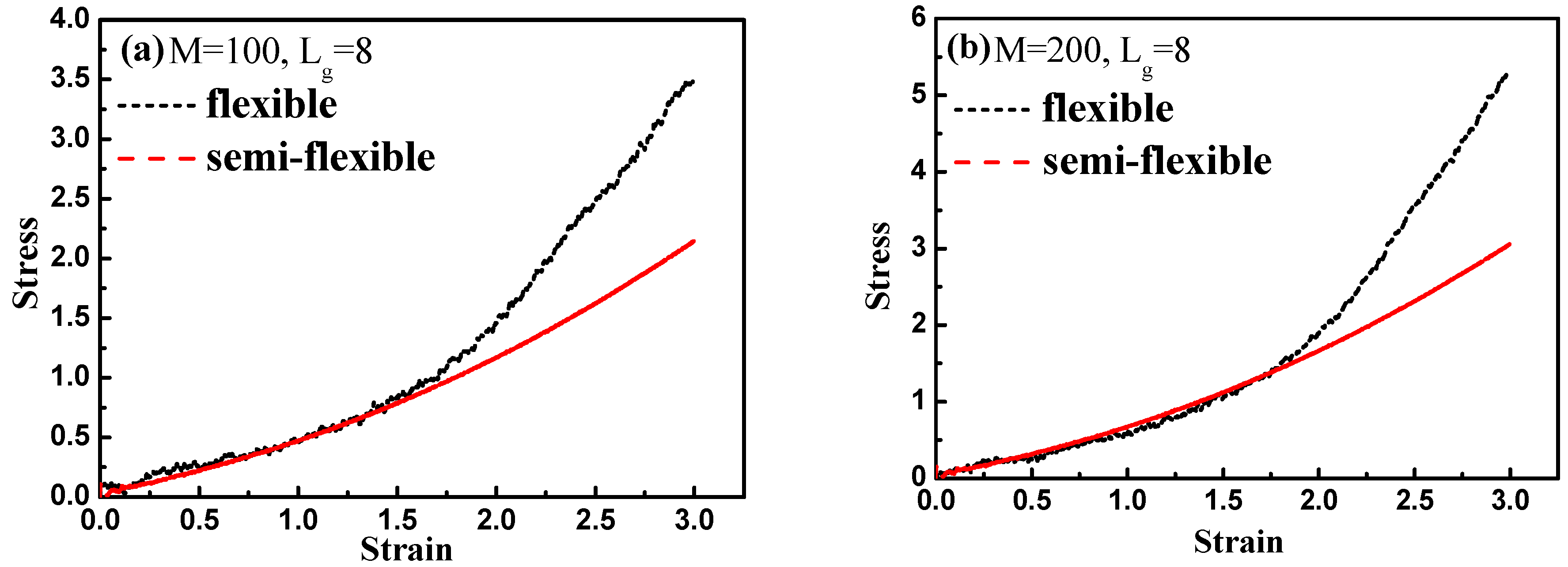
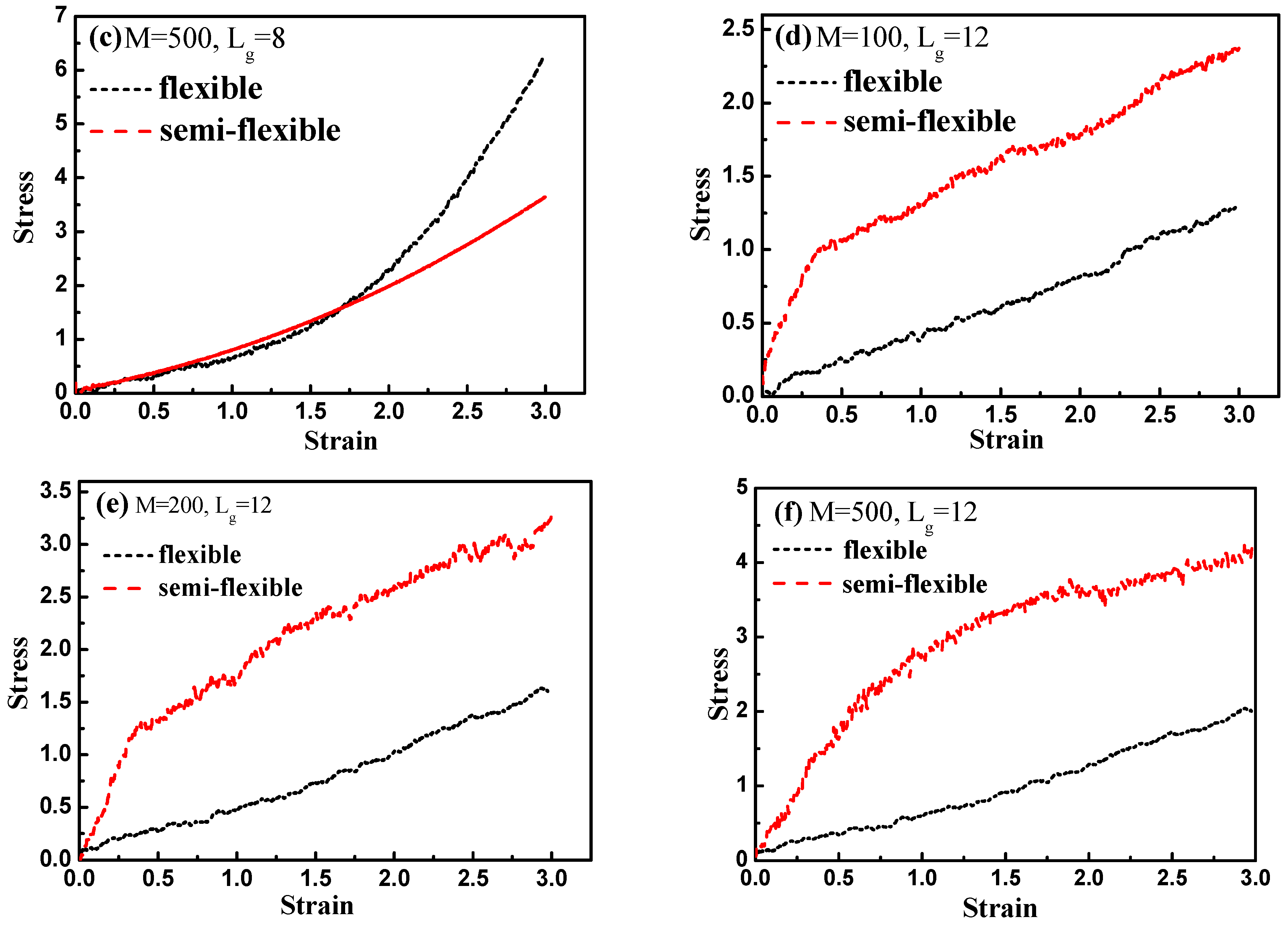
Finally, by introducing interfacial chemical cross-linking between grafted and matrix polymer chains, we compare the stress–strain behavior between the flexible and semi-flexible grafted chains systems like
and
, and we vary the number of the cross-linked bonds
such as 100, 200, and 500. The way we introduce a cross-linking is by generating a chemical bond between one grafted chain bead and one matrix chain bead. We choose the grafted chains and matrix chains randomly, and a maximum of one cross-linking is generated for each chain. In this case, our system is like the system of rubber. Our results are shown in
Figure 15. From
Figure 15a–c, it is clearly shown that with the increase of the interfacial cross-linking density, both flexible and semi-flexible systems exhibit a gradually enhanced stress–strain property. For systems with the grafted polymer chain length
, flexible grafted chains help to enhance the mechanical property better than semi-flexible grafted chains. However, as for systems with the grafted polymer chain length
, an opposite phenomenon is seen. Namely, the semi-flexible grafted polymer chains reinforce more compared to the flexible grafted polymer chains when the length of the grafted chain is long in presence of the interfacial chemical cross-linking. This result is consistent with the observation we find in
Figure 12, in which a “transition point” of the stress–strain curve as well occurs from
to
without any interfacial chemical linking.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}