
A Thixotropic Polyglycerol Sebacate-Based Supramolecular Hydrogel as an Injectable Drug Delivery Matrix


Abstract
:
1. Introduction
2. Methods
2.1. Chemicals and Materials
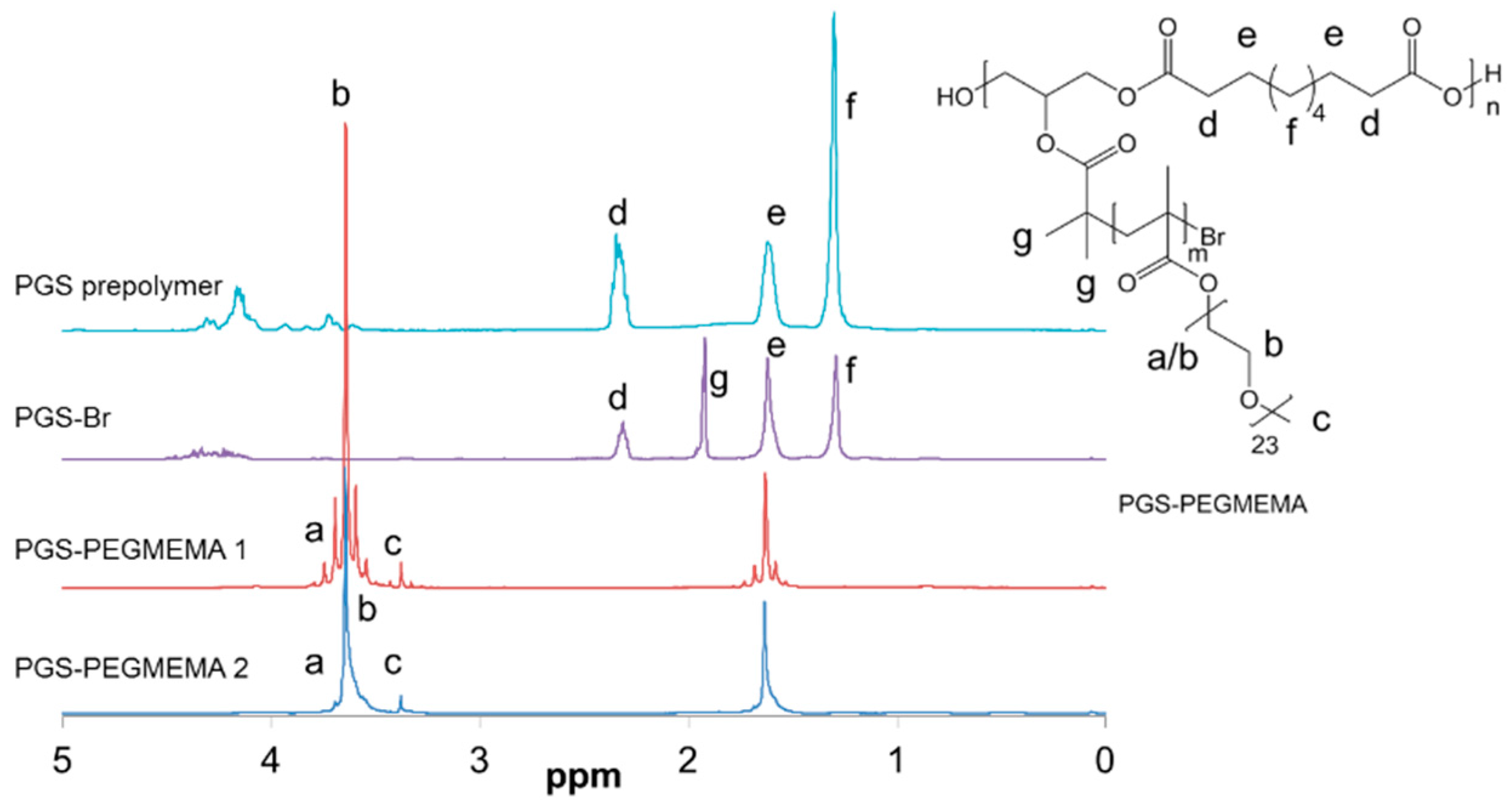
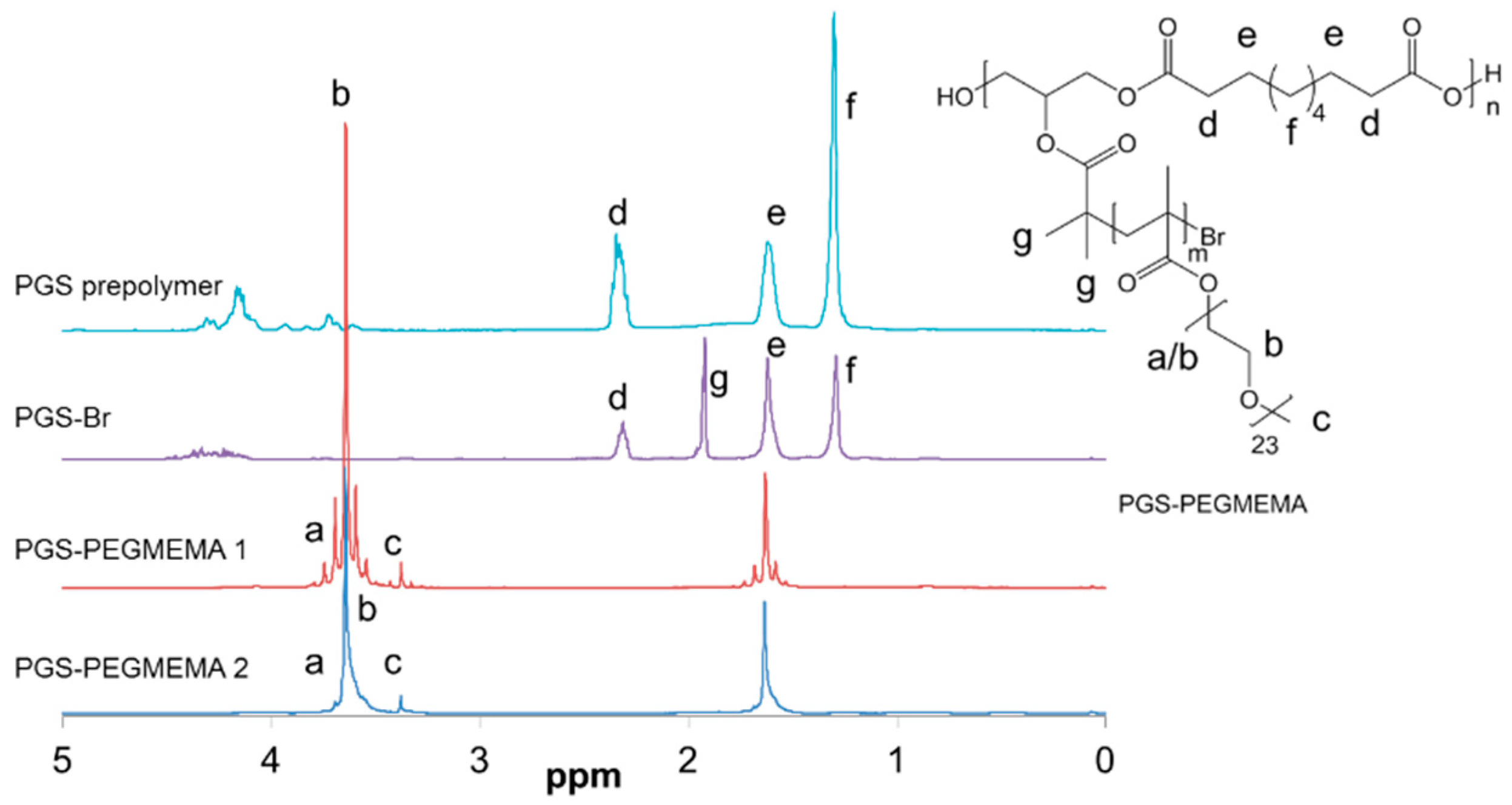
2.2. Synthesis of PGS-PEGMEMA
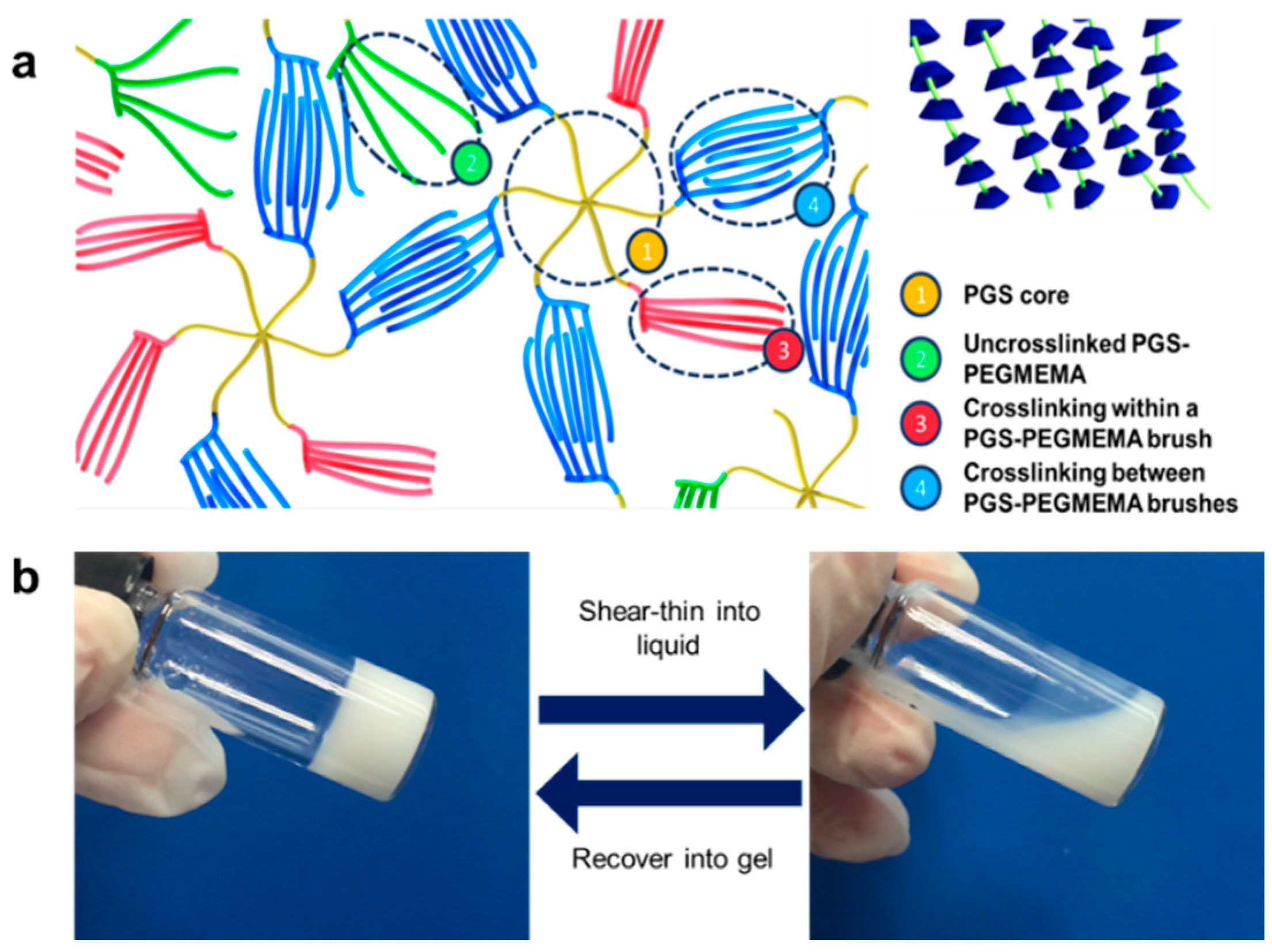
2.3. Preparation of the PGS-PEGMEMA/αCD Hydrogel
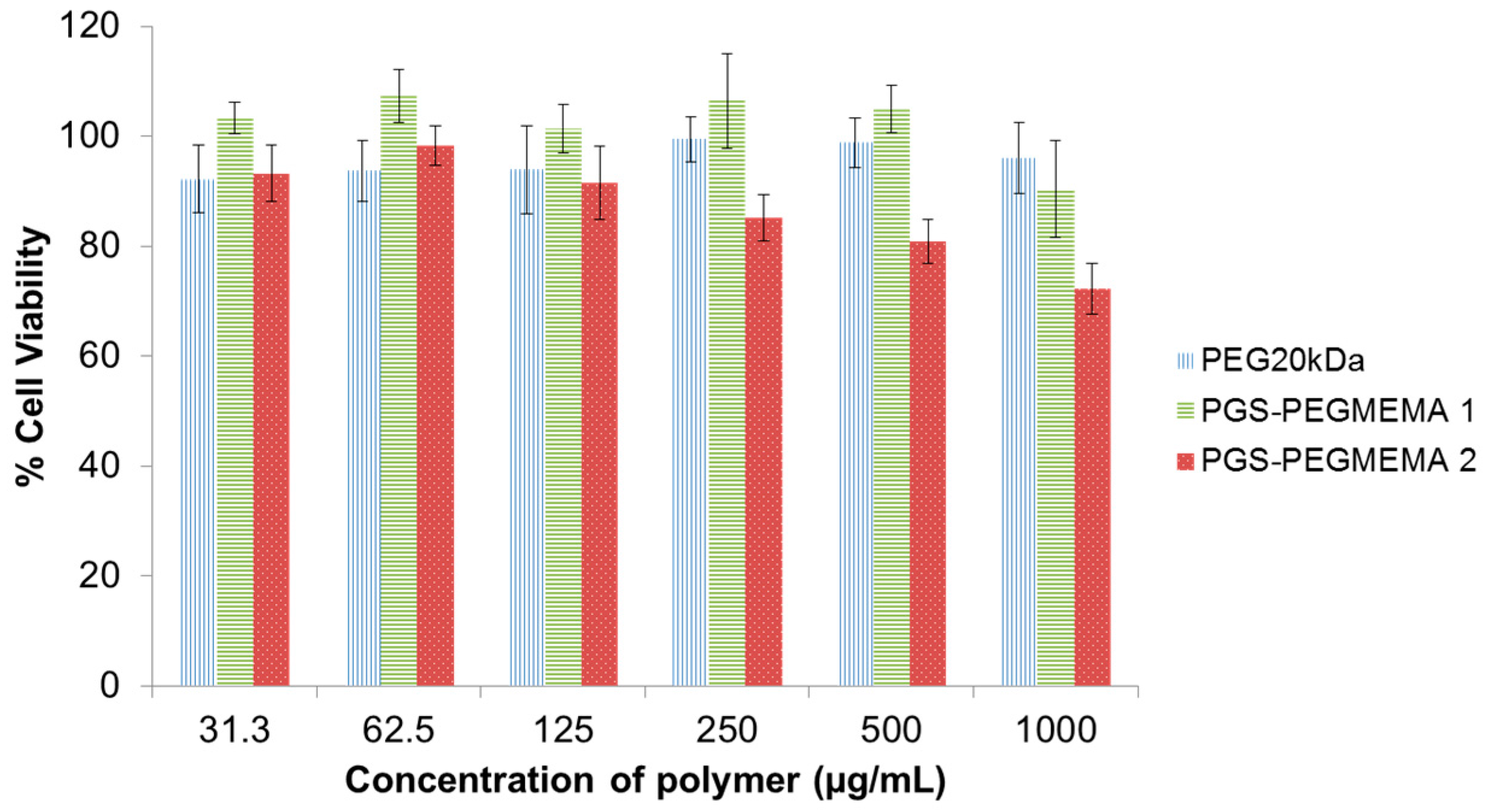
2.4. Biocompatibility of PGS-PEGMEMA
2.5. Biodegradability of PGS-PEGMEMA
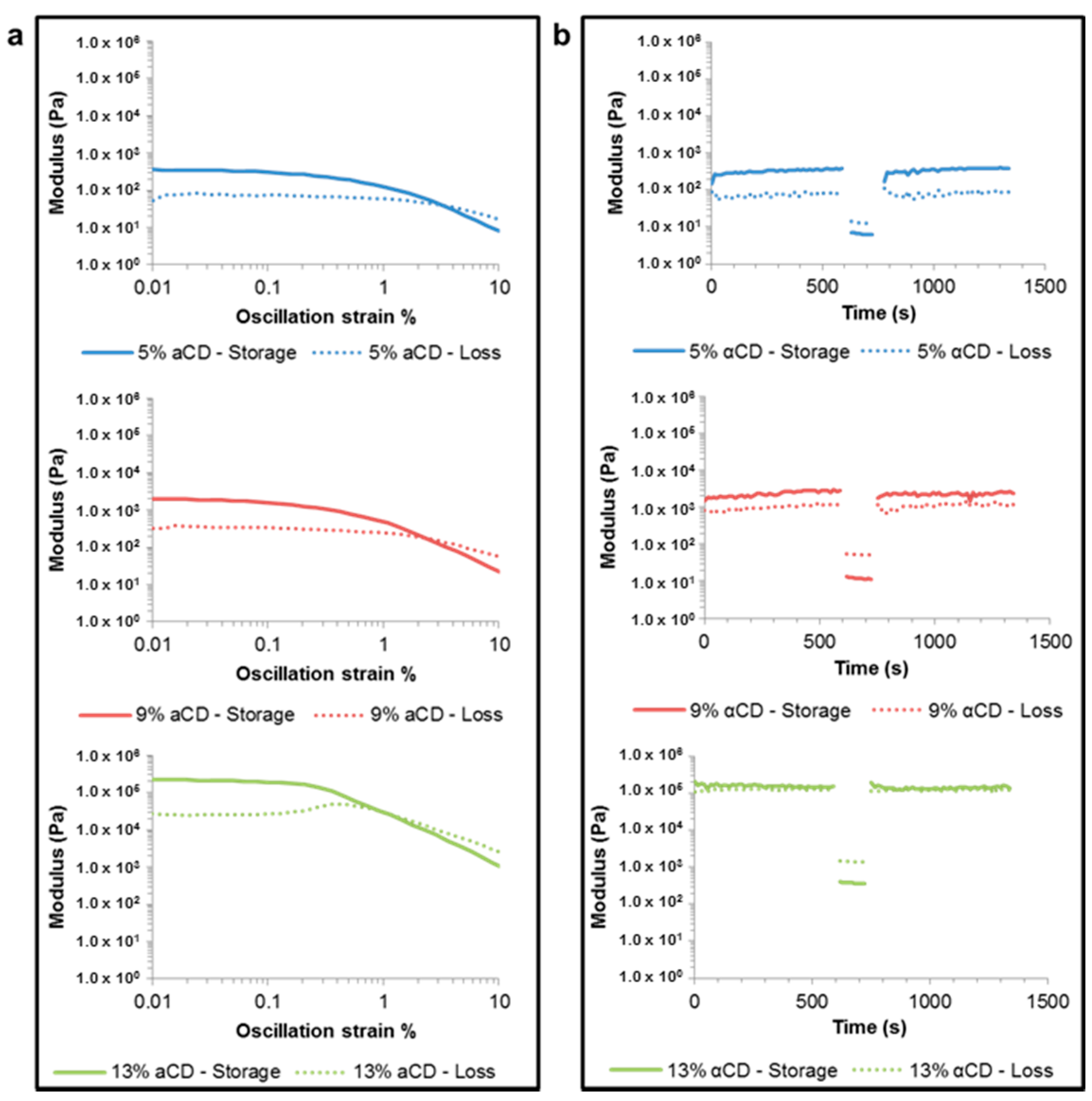
2.6. Rheology of the PGS-PEGMEMA/αCD Hydrogel
2.7. Scanning Electron Microscopy of the PGS-PEGMEMA/αCD Hydrogel
2.8. Hydrogel Erosion of the PGS-PEGMEMA/αCD Hydrogel
2.9. Drug Release from the PGS-PEGMEMA/αCD Hydrogel
3. Results
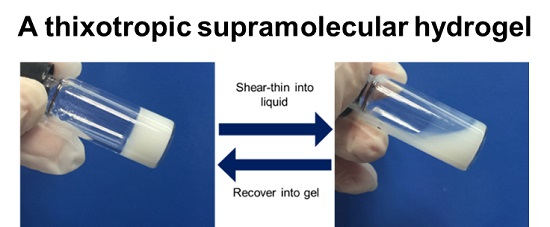
3.1. Synthesis and PGS-PEGMEMA/αCD Hydrogel Preparation
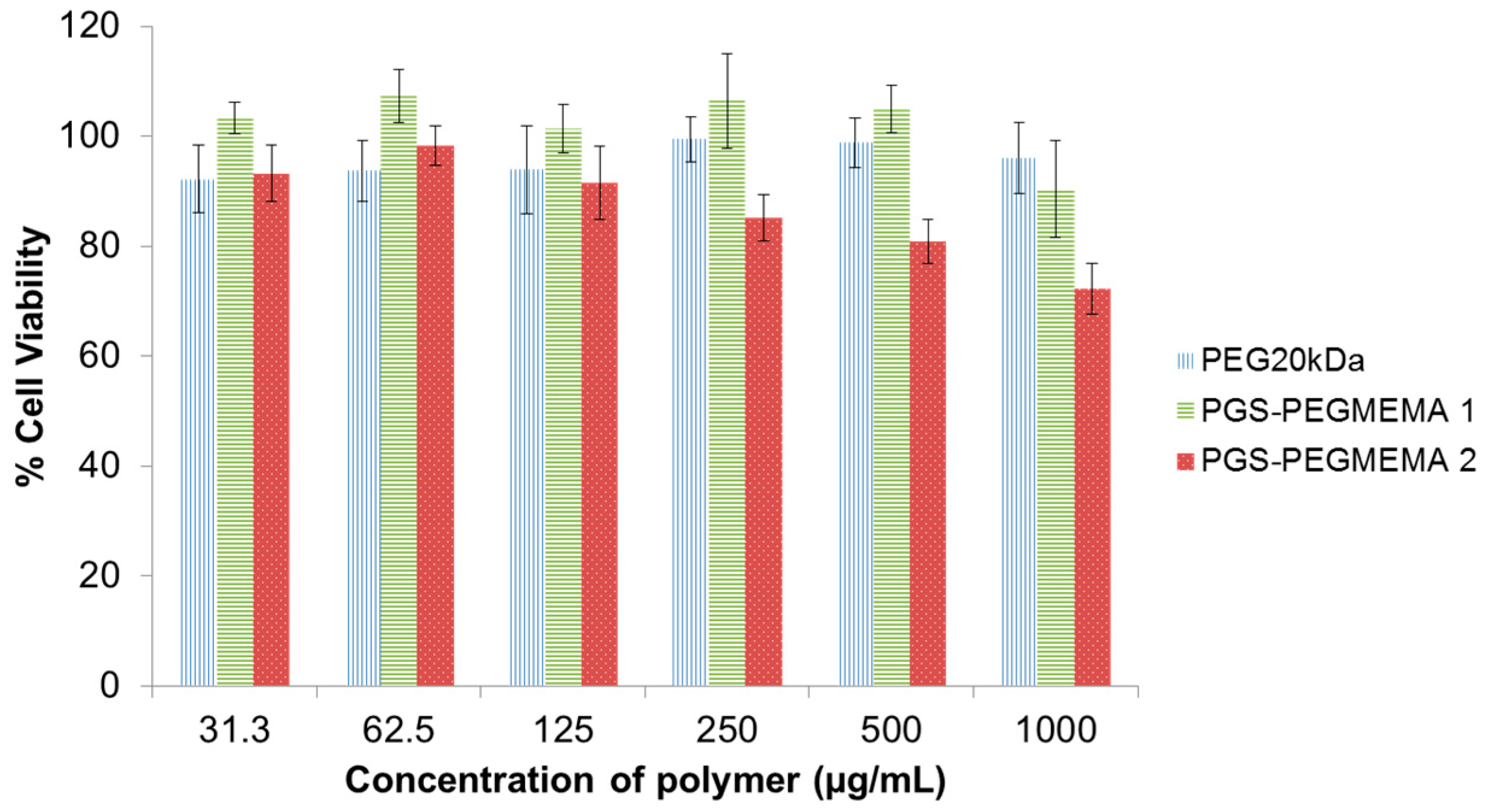
3.2. Biocompatibility of PGS-PEGMEMA
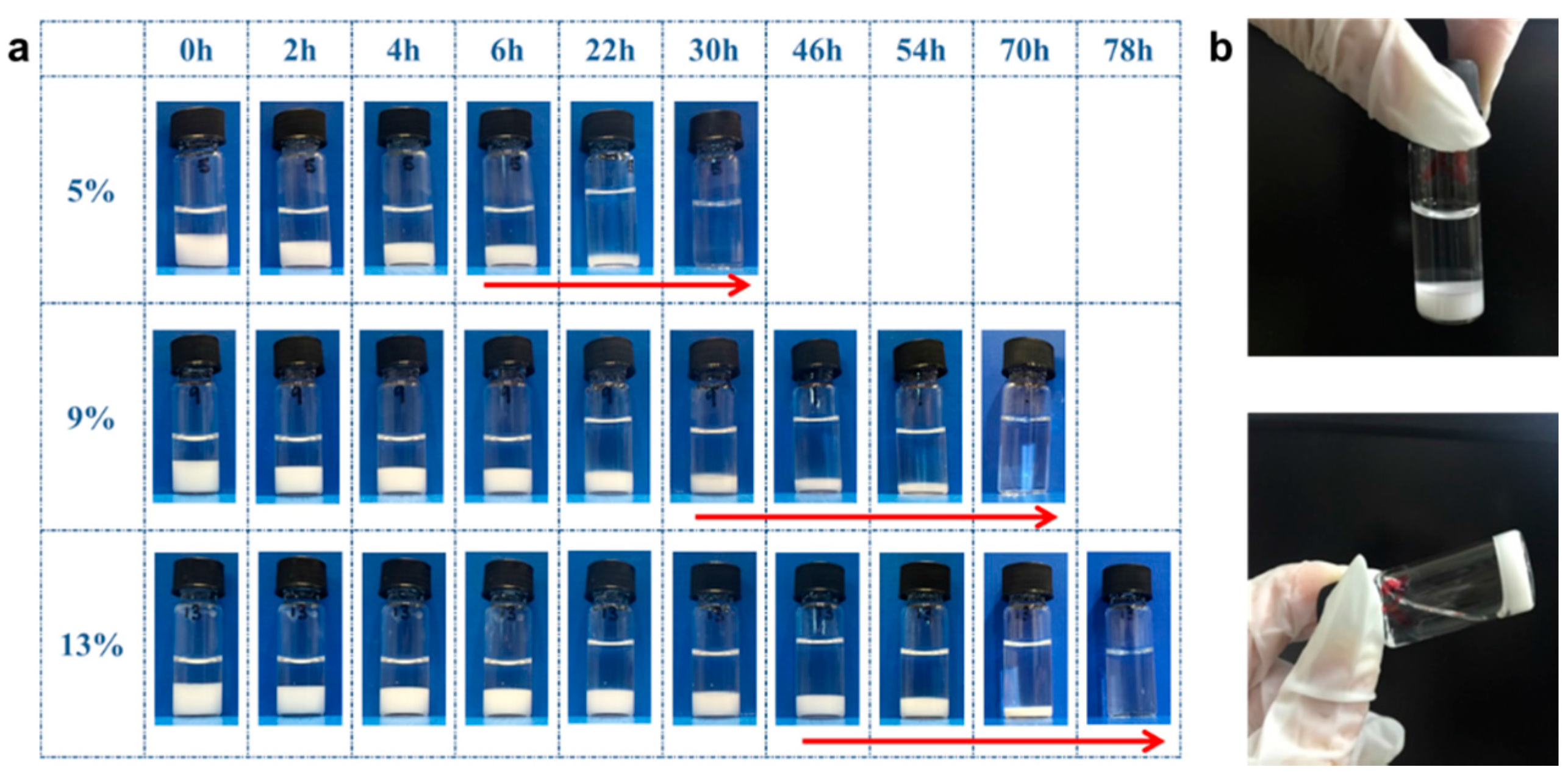
3.3. Biodegradability of PGS-PEGMEMA
3.4. Rheology of the PGS-PEGMEMA/αCD Hydrogel
3.5. Scanning Electron Microscopy of the PGS-PEGMEMA/αCD Hydrogel
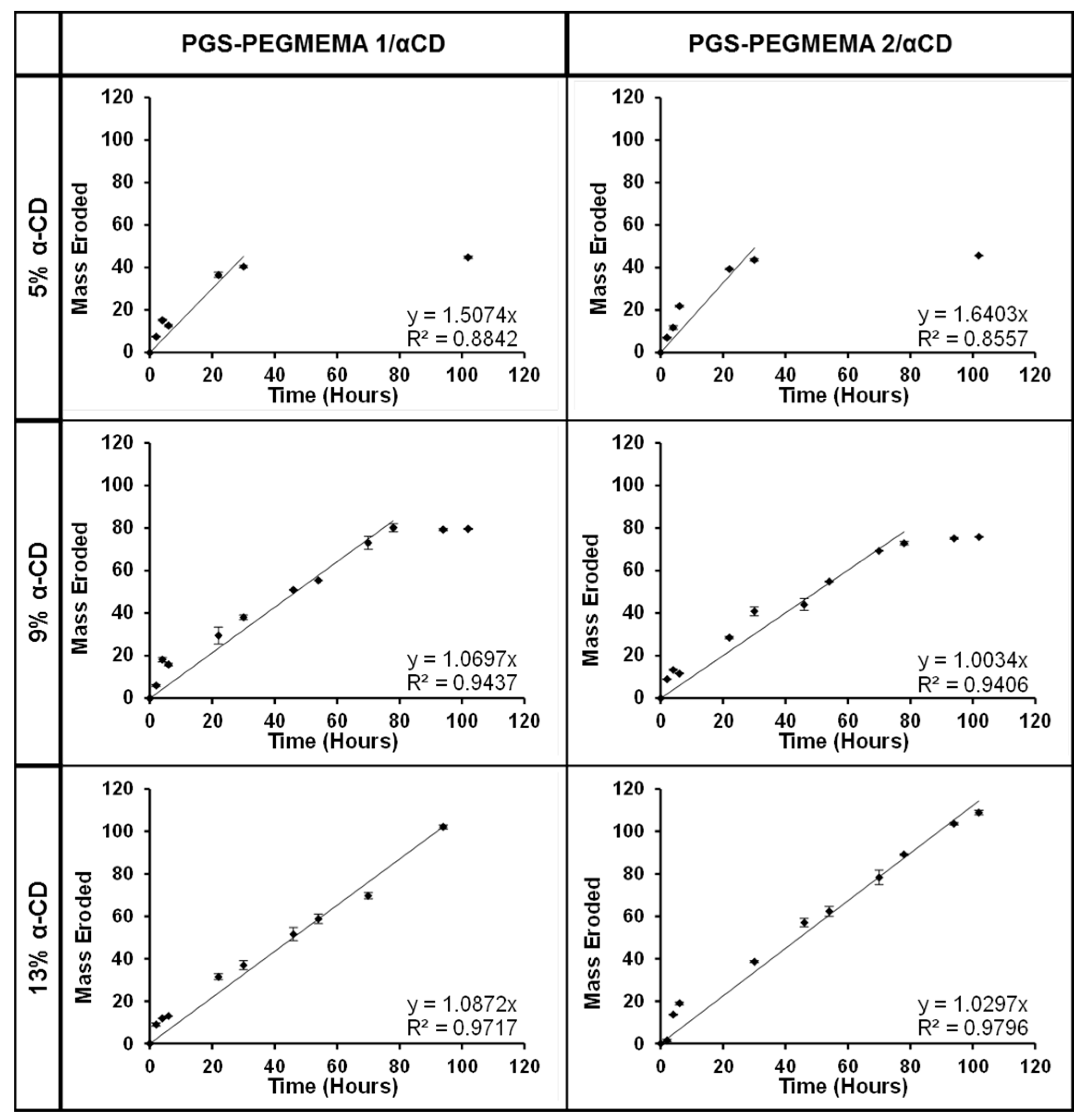
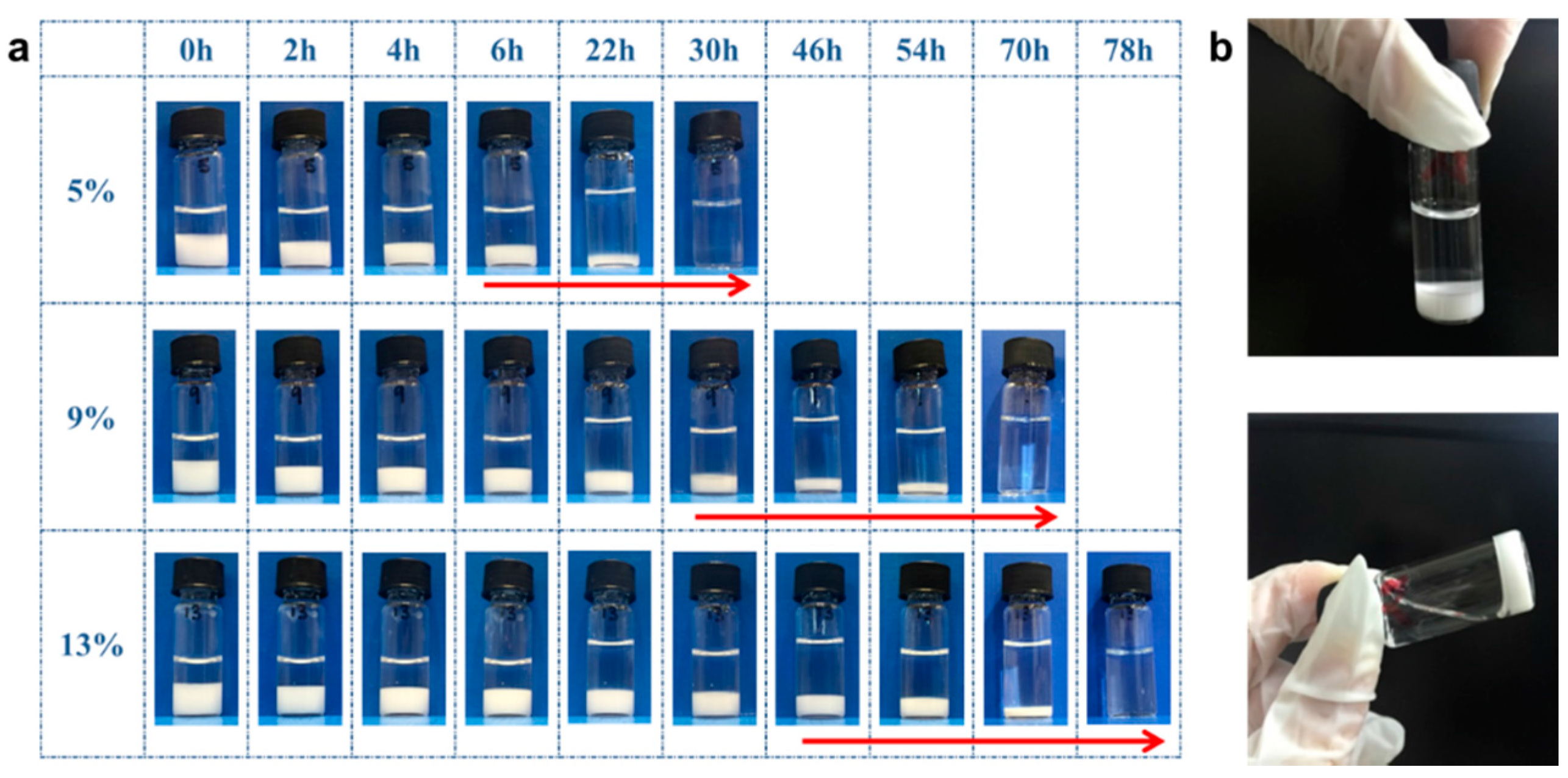
3.6. Hydrogel Erosion of the PGS-PEGMEMA/αCD Hydrogel
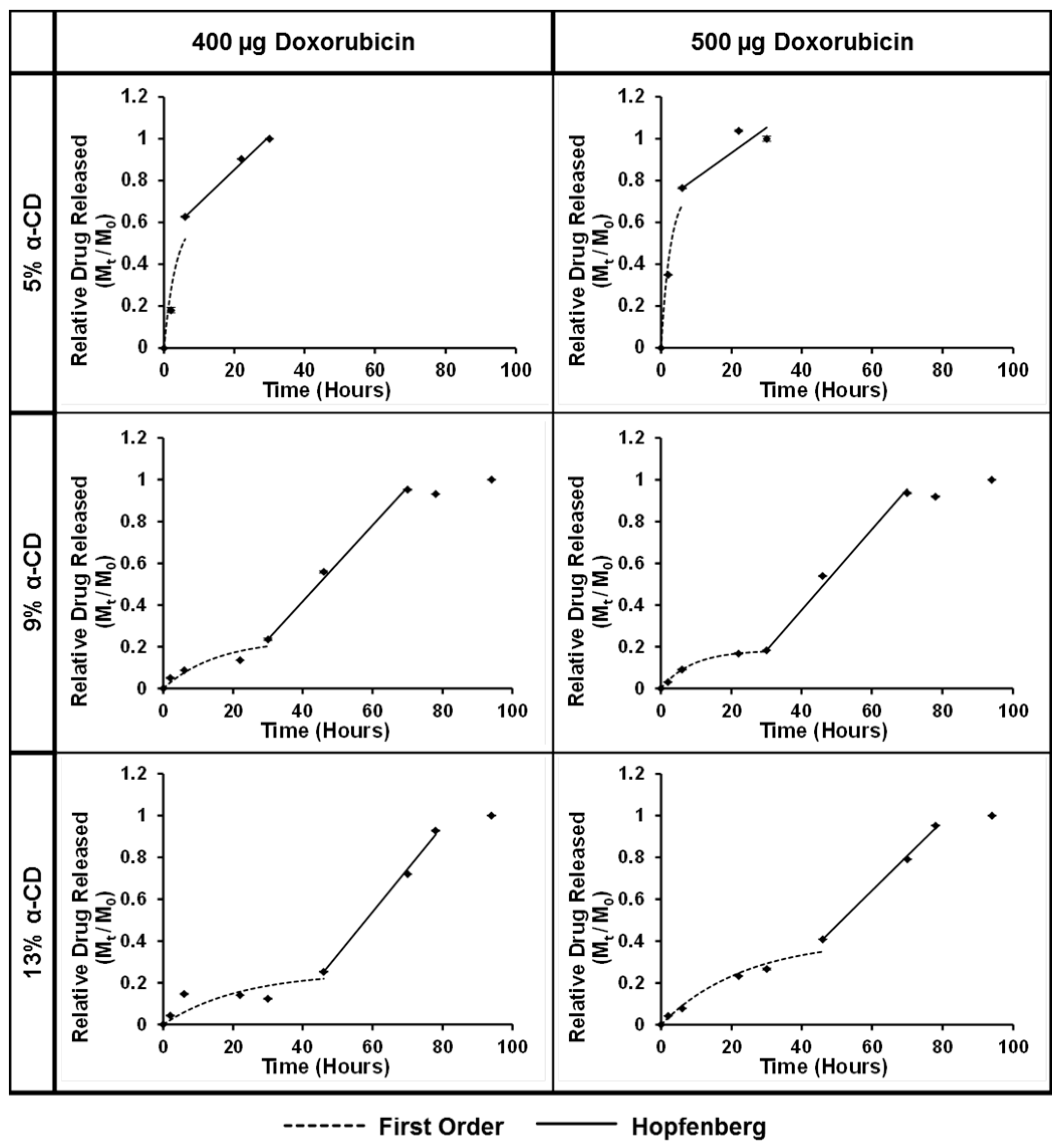
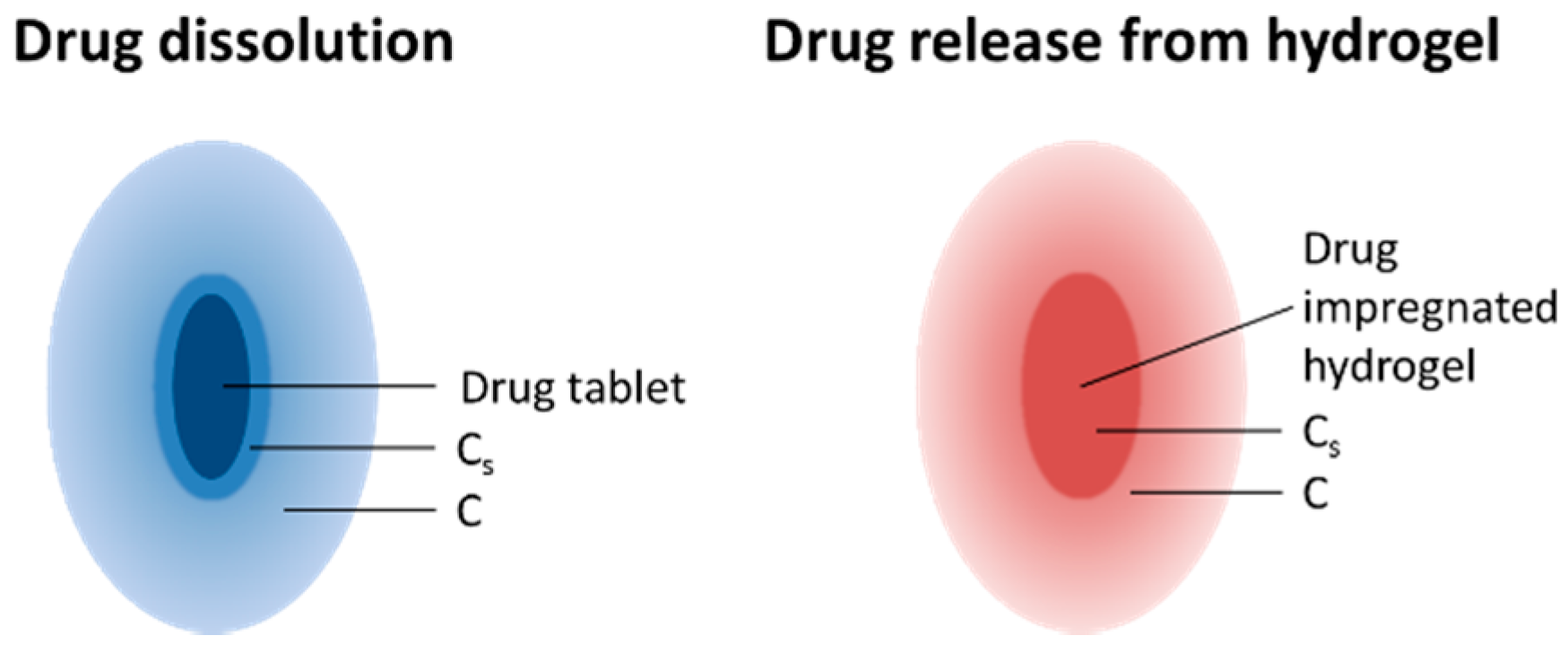
3.7. Drug Release from the PGS-PEGMEMA/αCD Hydrogel
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, Y.-L.; Chen, X.; Wang, W.; Loh, X.J. Engineering bioresponsive hydrogels toward healthcare applications. Macromol. Chem. Phys. 2016, 217, 175–188. [Google Scholar] [CrossRef]
- Liow, S.S.; Dou, Q.; Kai, D.; Karim, A.A.; Zhang, K.; Xu, F.; Loh, X.J. Thermogels: In situ gelling biomaterial. ACS Biomater. Sci. Eng. 2016, 2, 295–316. [Google Scholar] [CrossRef]
- Karim, A.A.; Dou, Q.; Li, Z.; Loh, X.J. Emerging supramolecular therapeutic carriers based on host-guest interactions. Chem. Asian J. 2016. [Google Scholar] [CrossRef]
- Zheng, J.Y.; Tan, M.J.; Thoniyot, P.; Loh, X.J. Unusual thermogelling behaviour of poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA)-based polymers polymerized in bulk. RSC Adv. 2015, 5, 62314–62318. [Google Scholar] [CrossRef]
- Ye, H.; Owh, C.; Loh, X.J. A thixotropic polyglycerol sebacate-based supramolecular hydrogel showing UCST behavior. RSC Adv. 2015, 5, 48720–48728. [Google Scholar] [CrossRef]
- Loh, X.J.; Karim, A.A.; Owh, C. Poly(glycerol sebacate) biomaterial: Synthesis and biomedical applications. J. Mater. Chem. B 2015, 3, 7641–7652. [Google Scholar] [CrossRef]
- Karim, A.A.; Loh, X.J. Design of a micellized alpha-cyclodextrin based supramolecular hydrogel system. Soft Matter 2015, 11, 5425–5434. [Google Scholar] [CrossRef] [PubMed]
- Kai, D.; Low, Z.W.; Liow, S.S.; Karim, A.A.; Ye, H.Y.; Jin, G.R.; Li, K.; Loh, X.J. Development of lignin supramolecular hydrogels with mechanically responsive and self-healing properties. Acs Sustain. Chem. Eng. 2015, 3, 2160–2169. [Google Scholar] [CrossRef]
- Chee, P.L.; Lakshmanan, L.; Jiang, S.; Ye, H.; Kai, D.; Loh, X.J. An injectable double-network hydrogel for cell encapsulation. Aust. J. Chem. 2015, 25, 1344–1351. [Google Scholar] [CrossRef]
- Ye, E.Y.; Chee, P.L.; Prasad, A.; Fang, X.T.; Owh, C.; Yeo, V.J.J.; Loh, X.J. Supramolecular soft biomaterials for biomedical applications. Mater. Today 2014, 17, 194–202. [Google Scholar] [CrossRef]
- Toh, W.S.; Loh, X.J. Advances in hydrogel delivery systems for tissue regeneration. Mater. Sci. Eng. C 2014, 45, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Loh, X.J.; Gan, H.X.; Wang, H.; Tan, S.J.E.; Neoh, K.Y.; Tan, S.S.J.; Diong, H.F.; Kim, J.J.; Lee, W.L.S.; Fang, X.T.; et al. New thermogelling poly(ether carbonate urethane)s based on pluronics F127 and poly(polytetrahydrofuran carbonate). J. Appl. Polym. Sci. 2014. [Google Scholar] [CrossRef]
- Su, X.Y.; Tan, M.J.; Li, Z.B.; Wong, M.H.; Rajamani, L.; Lingam, G.; Loh, X.J. Recent progress in using biomaterials as vitreous substitutes. Biomacromolecules 2015, 16, 3093–3102. [Google Scholar] [CrossRef] [PubMed]
- Loh, X.J.; Lee, T.C.; Dou, Q.; Deen, G.R. Utilising inorganic nanocarriers for gene delivery. Biomater. Sci. 2015, 4, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Kai, D.; Loh, X.J. Polyhydroxyalkanoates: Chemical modifications toward biomedical applications. ACS Sustain. Chem. Eng. 2014, 2, 106–119. [Google Scholar] [CrossRef]
- Kai, D.; Liow, S.S.; Loh, X.J. Biodegradable polymers for electrospinning: Towards biomedical applications. Mater. Sci. Eng. C 2014, 45, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Ye, E.Y.; Loh, X.J. Polymeric hydrogels and nanoparticles: A merging and emerging field. Aust. J. Chem. 2013, 66, 997–1007. [Google Scholar] [CrossRef]
- Loh, X.J.; Yee, B.J.H.; Chia, F.S. Sustained delivery of paclitaxel using thermogelling poly(PEG/PPG/PCL urethane)s for enhanced toxicity against cancer cells. J. Biomed. Mater. Res. Part A 2012, 100A, 2686–2694. [Google Scholar] [CrossRef] [PubMed]
- Loh, X.J.; Guerin, W.; Guillaume, S.M. Sustained delivery of doxorubicin from thermogelling poly(PEG/PPG/PTMC urethane)s for effective eradication of cancer cells. J. Mater. Chem. 2012, 22, 21249–21256. [Google Scholar] [CrossRef]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Euro. Poly. J. 2015, 65, 252–267. [Google Scholar] [CrossRef]
- Wolffsohn, J.; Hall, L.; Mroczkowska, S.; Hunt, O.A.; Bilkhu, P.; Drew, T.; Sheppard, A. The influence of end of day silicone hydrogel daily disposable contact lens fit on ocular comfort, physiology and lens wettability. Contact Lens Anterior Eye 2015, 38, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Abidian, M.; Martin, D.C. Conducting polymers grown in hydrogel scaffolds coated on neural prosthetic devices. J. Biomed. Mater. Res. Part A 2004, 71A, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Hatefi, A.; Amsden, B. Biodegradable injectable in situ forming drug delivery systems. J. Controll. Release 2002, 80, 9–28. [Google Scholar] [CrossRef]
- Chang, J.H.-C. Basic science of oncology hyperthermia and photodynamic therapy; McGraw-Hill: New York, NY, USA, 1998. [Google Scholar]
- Zhang, X.; Wong, W.; Min, W.; Cruz, T.; Hunter, W.L.; Burt, H.M. Development of biodegradable polymeric paste formulations for taxol: An in-vivo and in-vitro study. Int. J. Pharm. 1996, 137, 199–208. [Google Scholar] [CrossRef]
- Liu, J.H.; Li, L.; Liu, E. Release of theophylline from polymer blend hydrogels. Intl. J. Pharm. 2005, 298, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Molly, C.H.T.; Shoichet, S. Fast-gelling injectable blend of hyaluronan and methylcellulose for intrathecal, localized delivery to the injured spinal cord. Biomaterials 2006, 27, 2370–2379. [Google Scholar] [CrossRef] [PubMed]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Cui, H.; Messersmith, P.B. Thermally triggered gelation of alginate for controlled release, in tailored polymeric materials for controlled delivery systems. Amer. Chem. Soc. 1998, 203–211. [Google Scholar]
- Westhaus, E.; Messersmith, P.B. Triggered release of calcium from lipid vesicles: A bioinspired strategy for rapid gelation of polysaccharide and protein hydrogels. Biomaterials 2001, 22, 453–462. [Google Scholar] [CrossRef]
- Cui, H.; Messersmith, P.B. Thermally triggered gelation of alginate for controlled release. In Tailored Polymeric Materials For Controlled Delivery Systems; American Chemical Society: Washington, DC, USA, 1998. [Google Scholar]
- Lambert, W.J.; Peck, K.D. Development of an in situ forming biodegradable poly-lactide-coglycolide system for the controlled release of proteins. J. Controll. Release 1995, 33, 189–195. [Google Scholar] [CrossRef]
- Kranz, H.; Brazeaub, G.A.; Napapornb, J.; Martinb, R.L.; Millardb, W.; Bodmeiera, R. Myotoxicity studies of injectable biodegradable in-situ forming drug delivery systems. Int. J. Pharm. 2001, 212, 11–18. [Google Scholar] [CrossRef]
- Royals, M.A.; Fujita, S.M.; Yewey, G.L.; Rodriguez, J.; Schultheiss, P.C.; Dunn, R.L. Biocompatibility of a biodegradable in situ forming implant system in rhesus monkeys. J. Biomed. Mater. Res. 1999, 45, 231–239. [Google Scholar] [CrossRef]
- Li, J. Self-assembled supramolecular hydrogels based on polymer–cyclodextrin inclusion complexes for drug delivery. NPG Asia Mater. 2010, 2, 112–118. [Google Scholar] [CrossRef]
- Li, J.; Harada, A.; Kamachi, M. Sol-gel transition during inclusion complex formation between [α]-cyclodextrin and high molecular weight poly(ethylene glycol)s in aqueous solution. Polym. J. 1994, 26, 1019–1026. [Google Scholar] [CrossRef]
- Travelet, C.; Schlatter, G.; Hébraud, P.; Brochon, C.; Lapp, A.; Hadziioannou, G. Formation and self-organization kinetics of αCD/PEO-based pseudo-polyrotaxanes in water. A specific behavior at 30 °C. Langmuir 2009, 25, 8723–8734. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; He, L.H.; Sun, T.; Dong, X.; Chen, Y.; Huang, J.; Wang, C. Dual-responsive supramolecular hydrogels from water-soluble peg-grafted copolymers and cyclodextrin. Macromol. Biosci. 2009, 9, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Pomerantseva, I.; Krebs, N.; Hart, A.; Neville, C.M.; Huang, A.Y.; Sundback, C.A. Degradation behavior of poly(glycerol sebacate). J. Biomed. Mater. Res. Part A 2009, 91A, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Kim, Y.M.; Langer, R. In vivo degradation characteristics of poly(glycerol sebacate). J. Biomed. Mater. Res. Part A 2003, 66A, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Nath, M.P.; Chowdhury, L.P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Euro. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Cohen, S.; Yoshioka, T.; Lucarelli, M.; Hwang, L.H.; Langer, R. Controlled delivery systems for proteins based on poly(lactic/glycolic acid) microspheres. Pharm. Res. 1991, 8, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Hopfenberg, H.B. Controlled release from erodible slabs, cylinders, and spheres, in controlled release polymeric formulations. Amer. Chem. Soc. 1976, 26–32. [Google Scholar]
- Akhtar, R.; Sherrattc, M.J.; Cruickshankb, K.J.; Derbya, B. Characterizing the elastic properties of tissues. Mater. Today 2011, 14, 96–105. [Google Scholar] [CrossRef]
- Tatyana, G.; Kuznetsova, M.N.S.; Nicolai, I.; Yegorenkov, S.A.; Chizhik, R.; Zhdanov, I. Atomic force microscopy probing of cell elasticity. Micron 2007, 38, 824–833. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mw (g/mol) | PDI |
|---|---|---|
| PGS prepolymer | 17,700 | 4.34 |
| PGS-PEGMEMA 1 | 118,000 | 3.17 |
| PGS-PEGMEMA 2 | 123,000 | 3.46 |
| Degradation duration | Neutral degradation | Acid degradation | Base degradation | |||
|---|---|---|---|---|---|---|
| Weeks | Mw (g/mol) | PDI | Mw (g/mol) | PDI | Mw (g/mol) | PDI |
| 0 | 123,000 | 3.46 | 123,000 | 3.46 | 123,000 | 3.46 |
| 2 | 92,000 | 2.68 | 57,000 | 2.03 | 50,000 | 1.86 |
| 3 | 123,000 | 2.68 | 54,000 | 2.19 | 47,000 | 1.99 |
| 6 | 148,000 | 2.49 | 57,000 | 1.99 | 47,000 | 2.11 |
| Polymers | 5% α-cyclodextrin | 9% α-cyclodextrin | 13% α-cyclodextrin |
|---|---|---|---|
| PGS-PEGMEMA 1 |  | 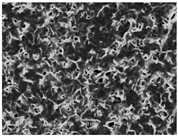 |  |
| PGS-PEGMEMA 2 | 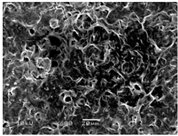 |  |  |
| Phase I | PGS-PEGMEMA 2 (400 µg) | k1 | R2 | Phase II | PGS-PEGMEMA 2 (400 µg) | k2 | R2 |
| 5% | 0.295 | 0.899 | 5% | 0.016 | 0.993 | ||
| 9% | 0.065 | 0.889 | 9% | 0.018 | 0.995 | ||
| 13% | 0.045 | 0.659 | 13% | 0.020 | 0.996 | ||
| PGS-PEGMEMA 2 (500 µg) | PGS-PEGMEMA 2 (500 µg) | ||||||
| 5% | 0.375 | 0.968 | 5% | 0.012 | 0.785 | ||
| 9% | 0.113 | 0.997 | 9% | 0.019 | 0.990 | ||
| 13% | 0.042 | 0.963 | 13% | 0.017 | 0.997 | ||
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, H.; Owh, C.; Jiang, S.; Ng, C.Z.Q.; Wirawan, D.; Loh, X.J. A Thixotropic Polyglycerol Sebacate-Based Supramolecular Hydrogel as an Injectable Drug Delivery Matrix. Polymers 2016, 8, 130. https://doi.org/10.3390/polym8040130
Ye H, Owh C, Jiang S, Ng CZQ, Wirawan D, Loh XJ. A Thixotropic Polyglycerol Sebacate-Based Supramolecular Hydrogel as an Injectable Drug Delivery Matrix. Polymers. 2016; 8(4):130. https://doi.org/10.3390/polym8040130
Chicago/Turabian StyleYe, Hongye, Cally Owh, Shan Jiang, Cavin Zhen Quan Ng, Daniel Wirawan, and Xian Jun Loh. 2016. "A Thixotropic Polyglycerol Sebacate-Based Supramolecular Hydrogel as an Injectable Drug Delivery Matrix" Polymers 8, no. 4: 130. https://doi.org/10.3390/polym8040130
APA StyleYe, H., Owh, C., Jiang, S., Ng, C. Z. Q., Wirawan, D., & Loh, X. J. (2016). A Thixotropic Polyglycerol Sebacate-Based Supramolecular Hydrogel as an Injectable Drug Delivery Matrix. Polymers, 8(4), 130. https://doi.org/10.3390/polym8040130