
A 13C-NMR Study on the 1,3-Dimethylolurea-Phenol Co-Condensation Reaction: A Model for Amino-Phenolic Co-Condensed Resin Synthesis

 ,
, 
Abstract
:1. Introduction
2. Experimental
2.1. Sample Preparation
2.1.1. Samples Prepared under Alkaline and Weak Acidic Conditions
2.1.2. Sample Prepared under Strong Acidic Conditions
2.2. 13C Nuclear Magnetic Resonance
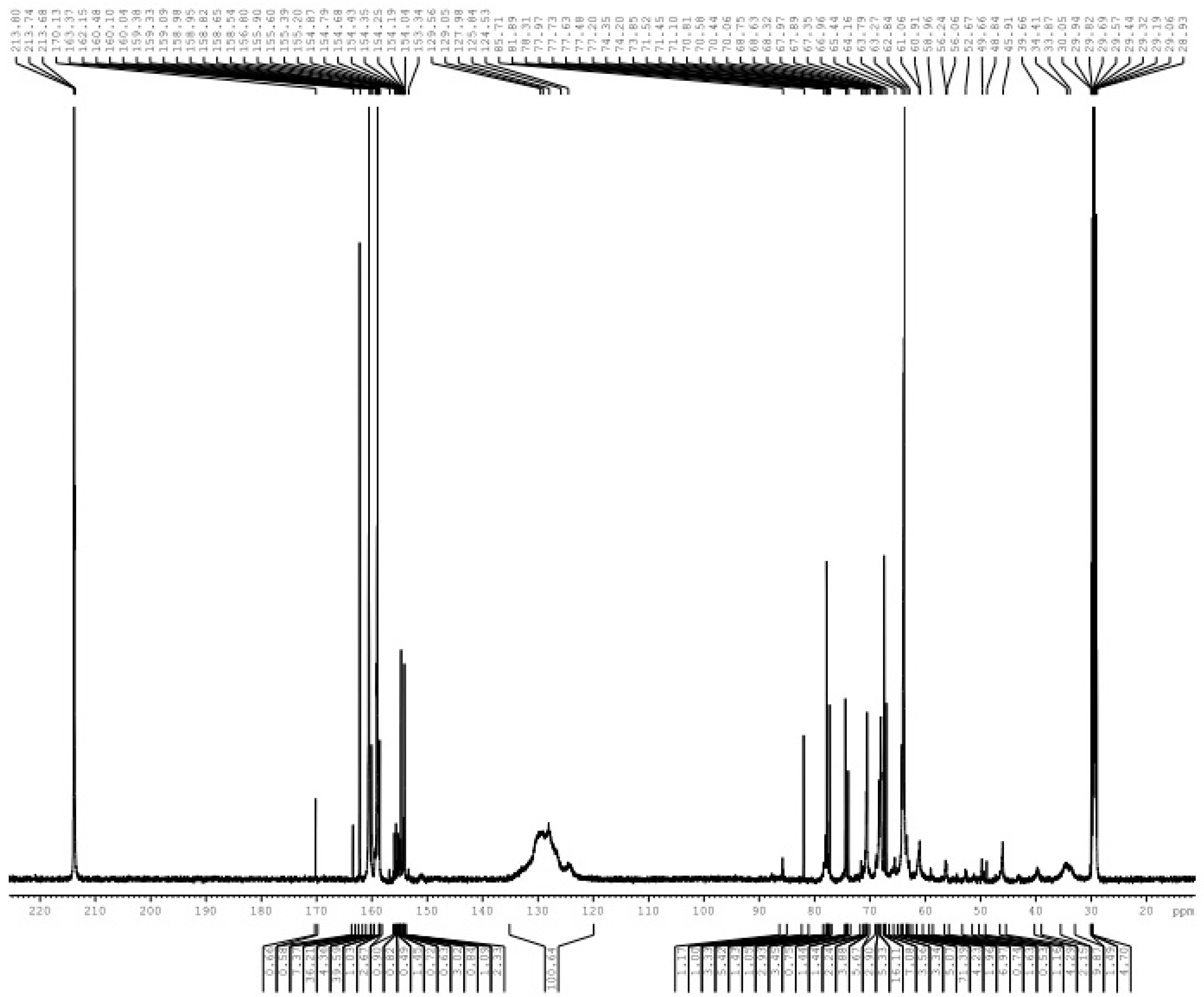
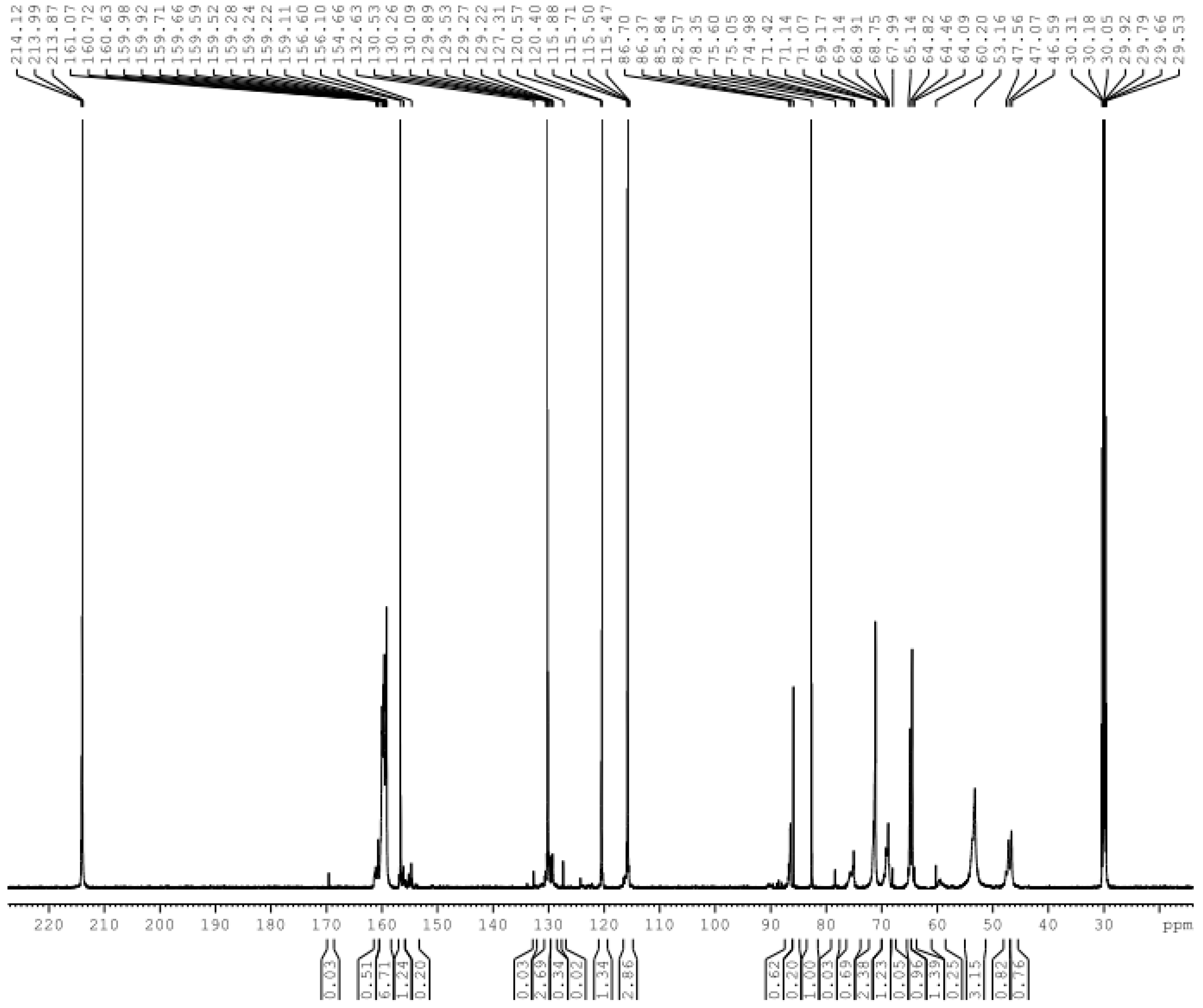
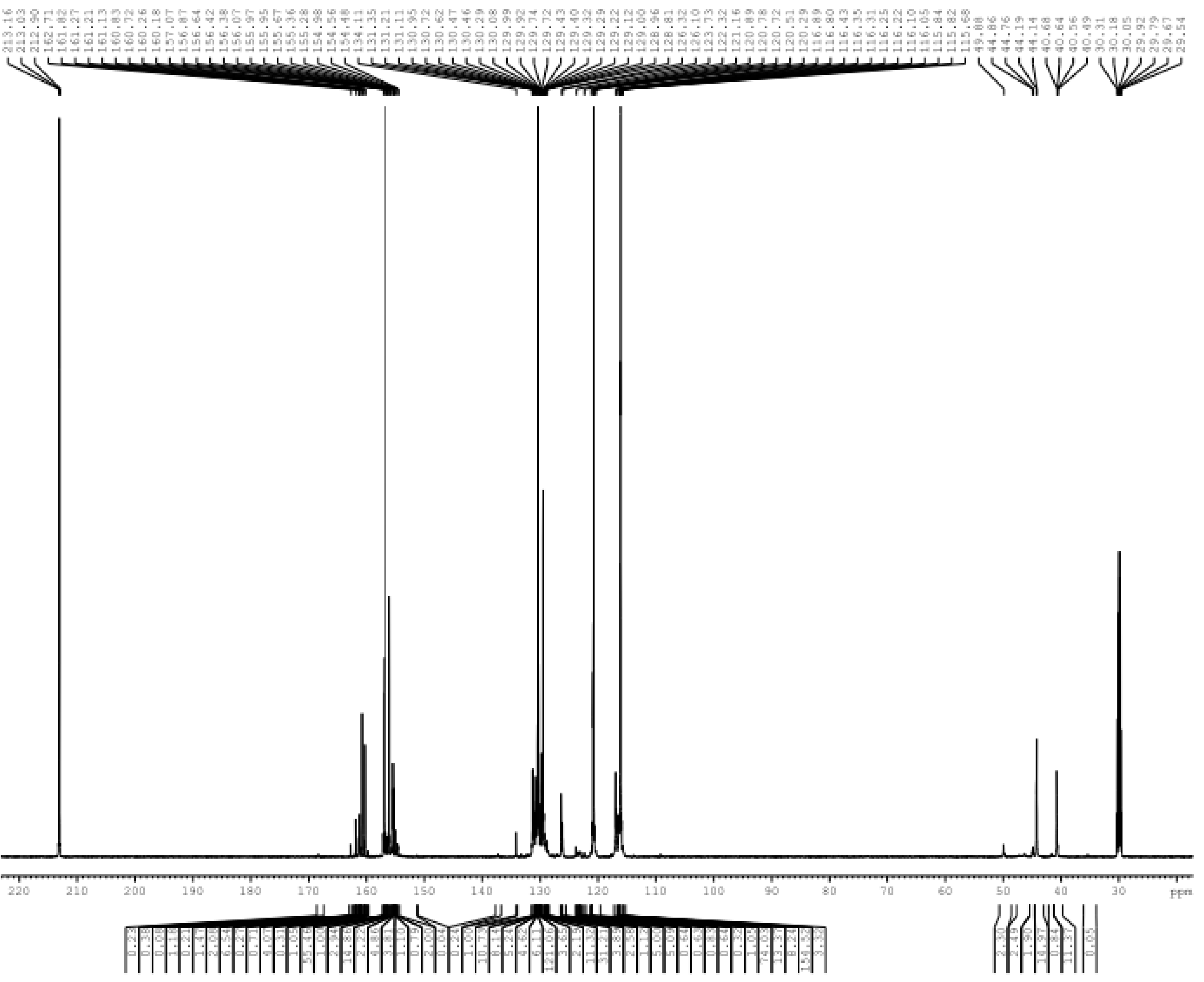
3. Results and Discussion
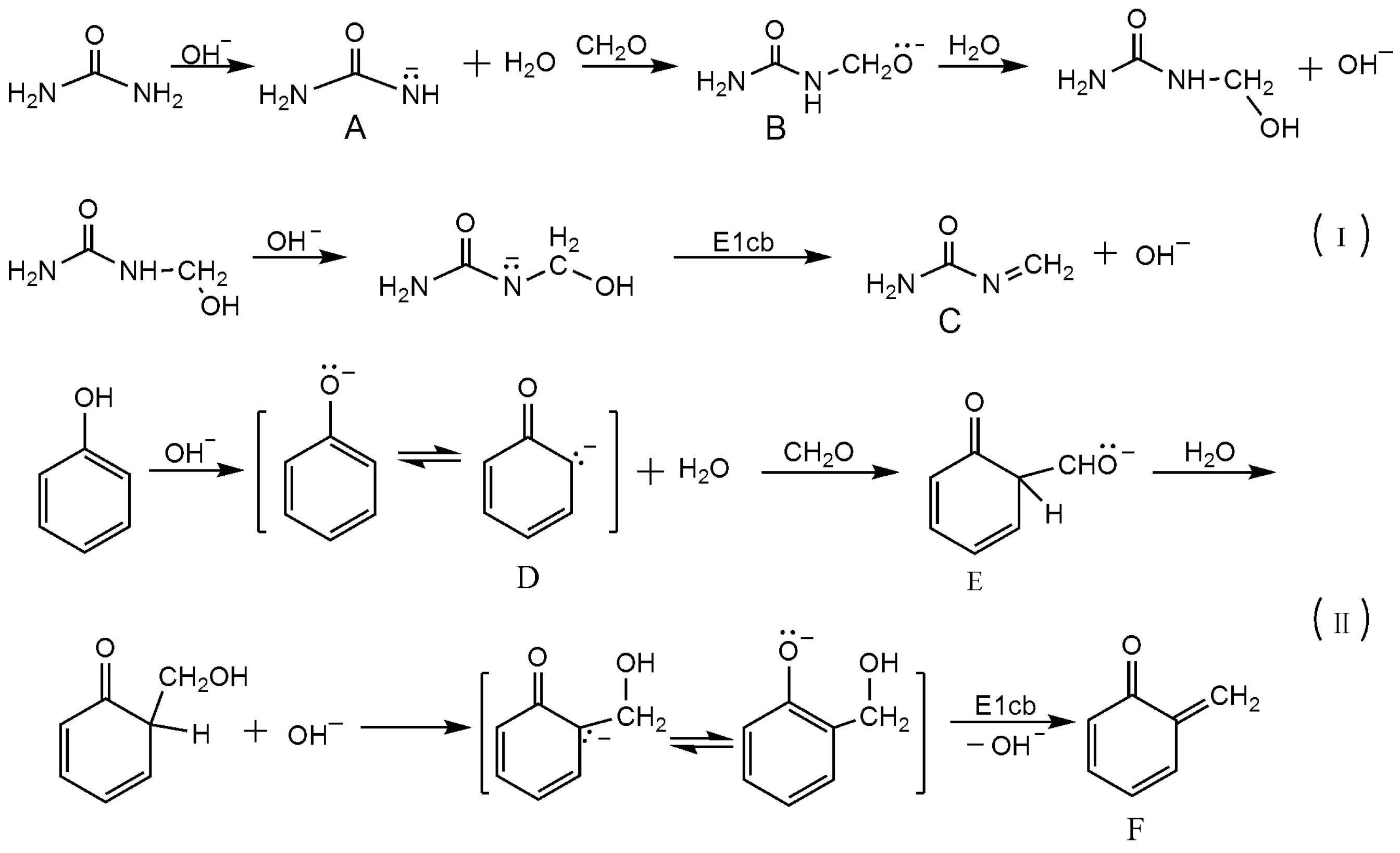
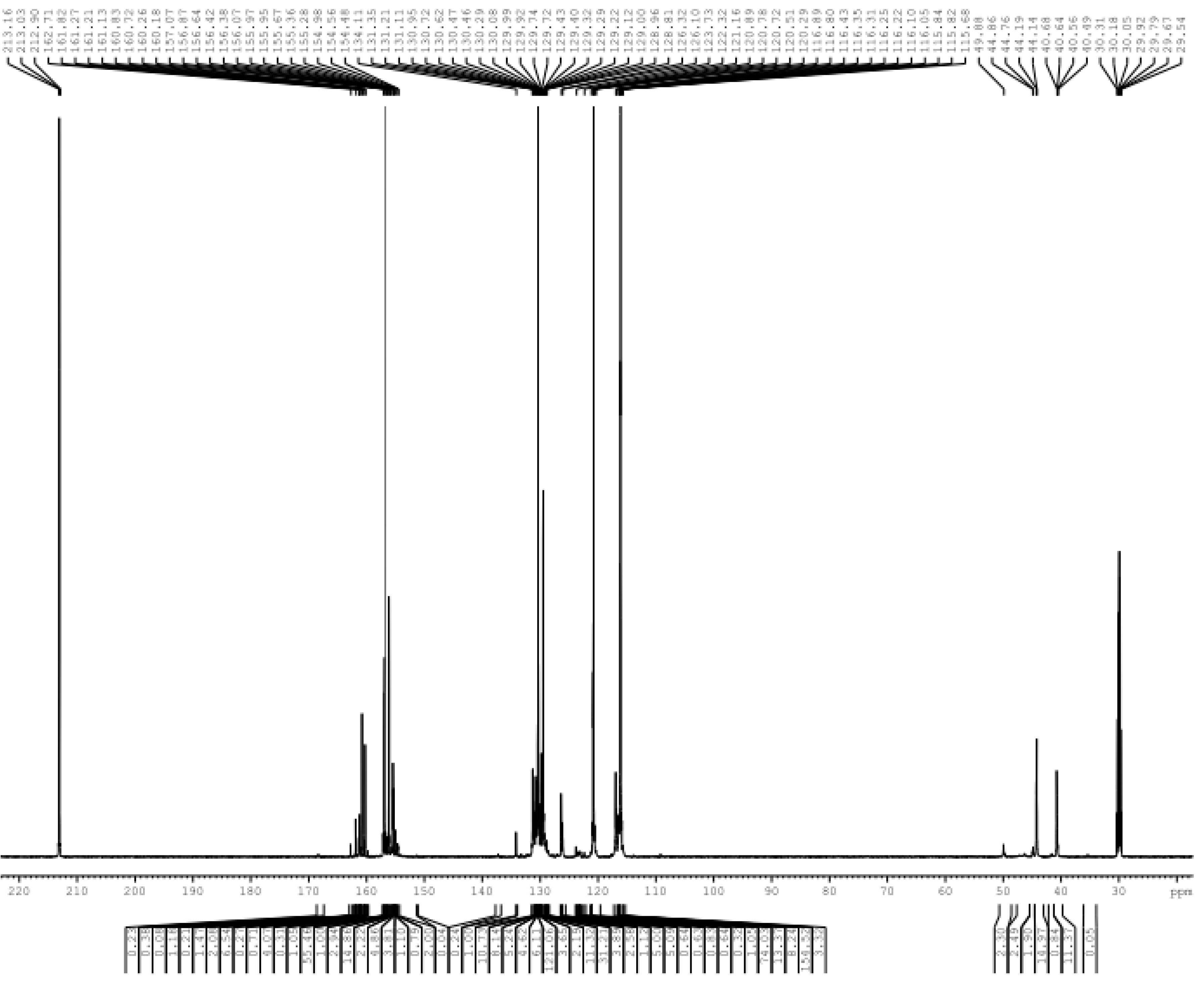
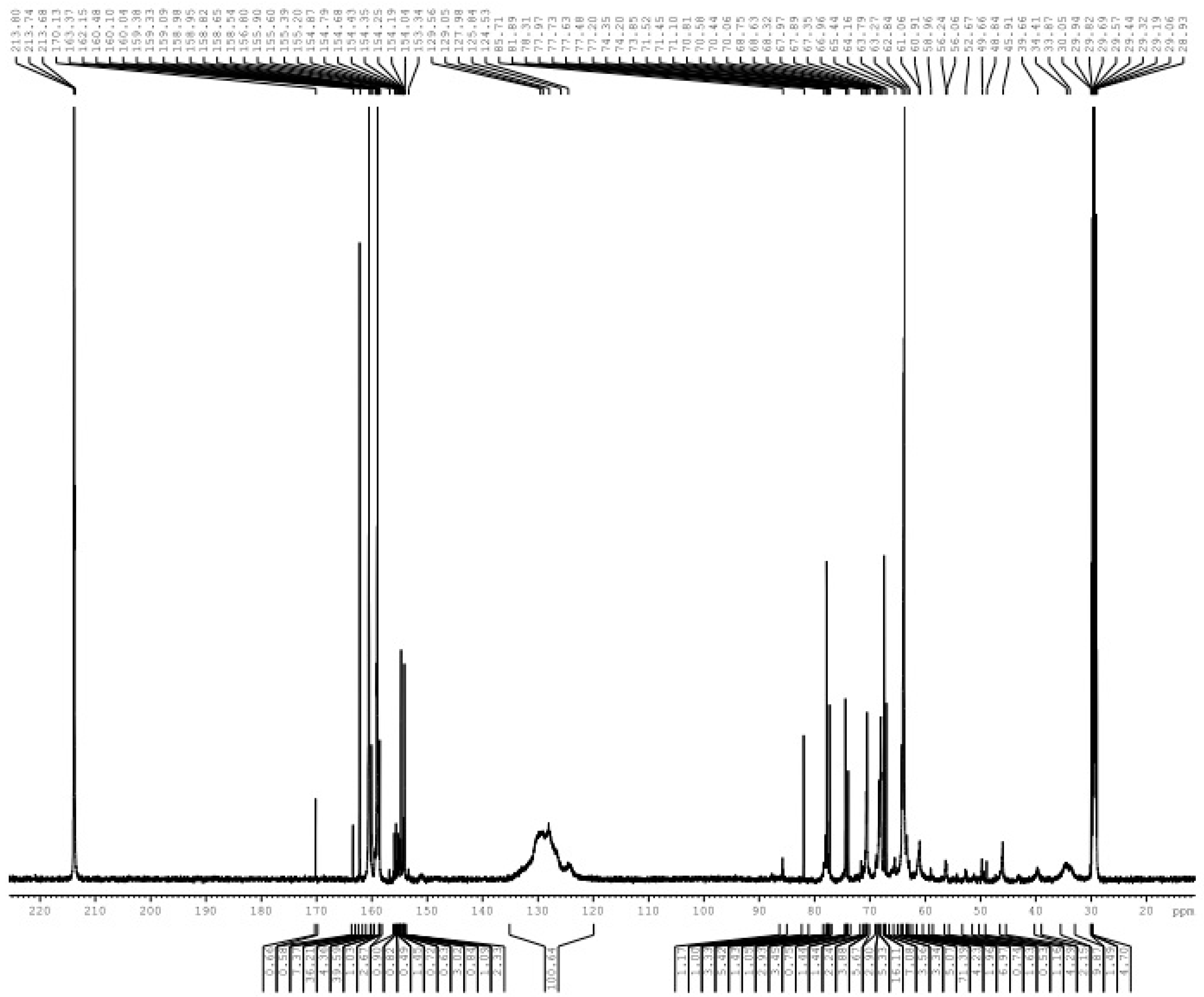
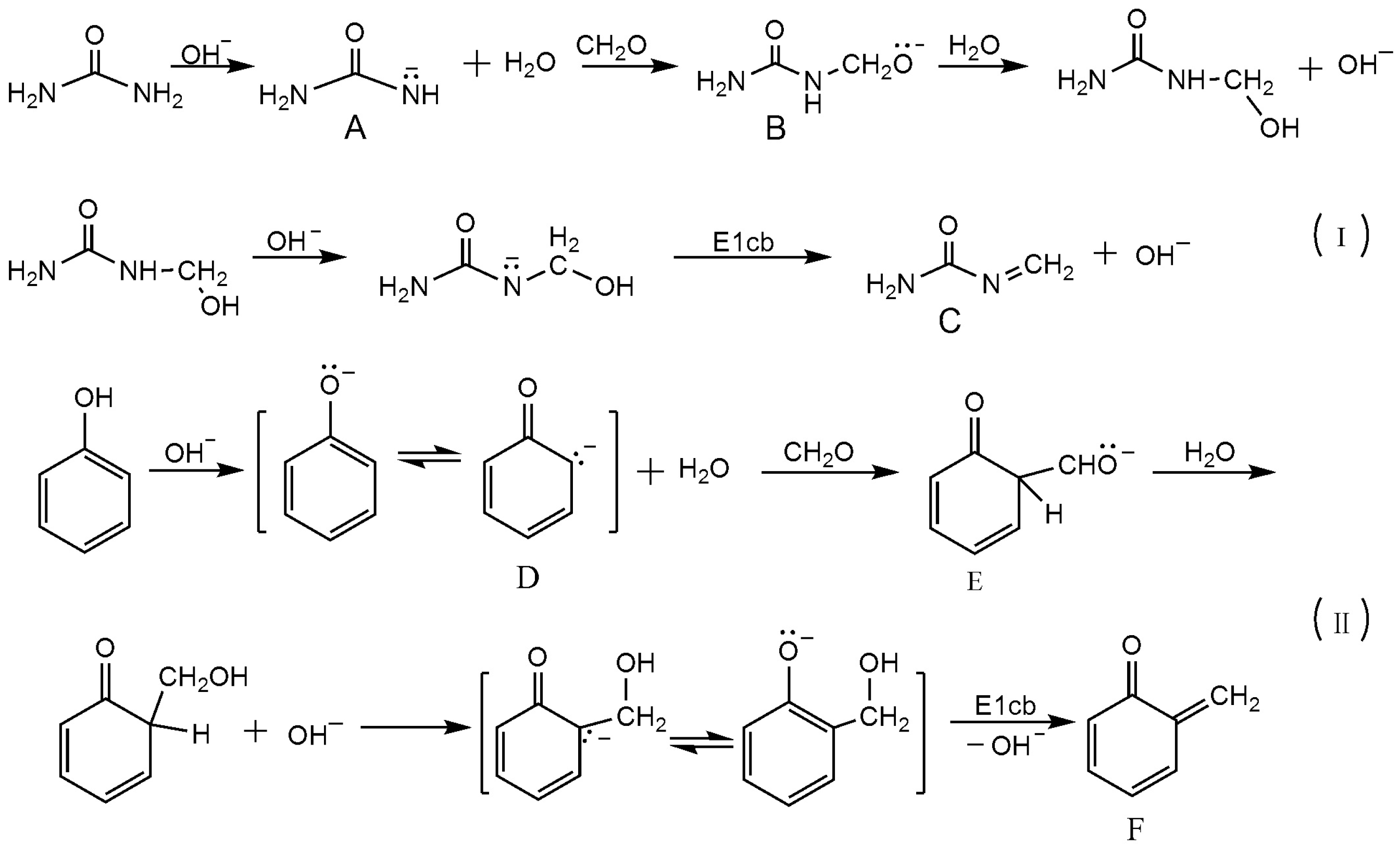
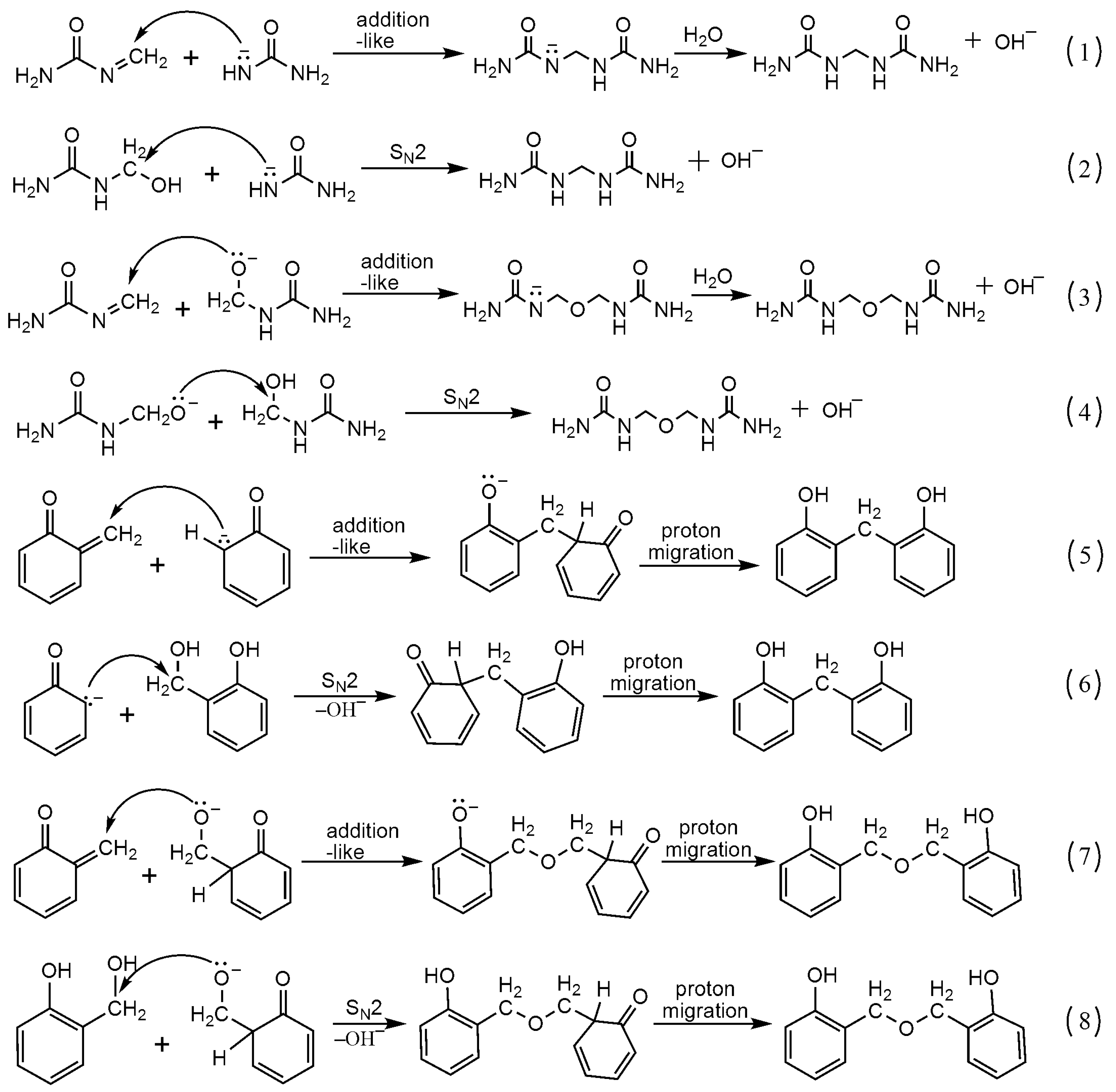
3.1. Reactions of UF2 and Phenol under Alkaline Conditions
3.2. Reactions of UF2 and Phenol under Weak Acidic Conditions
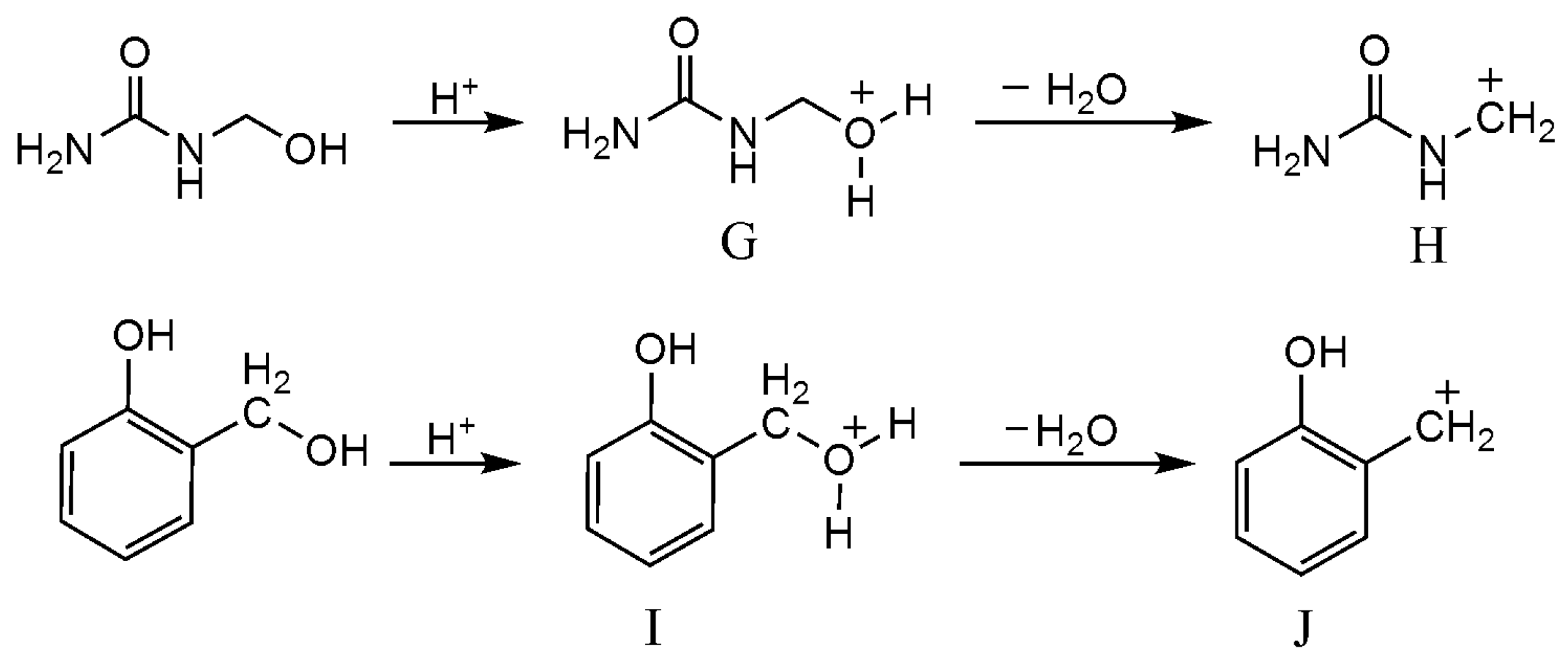
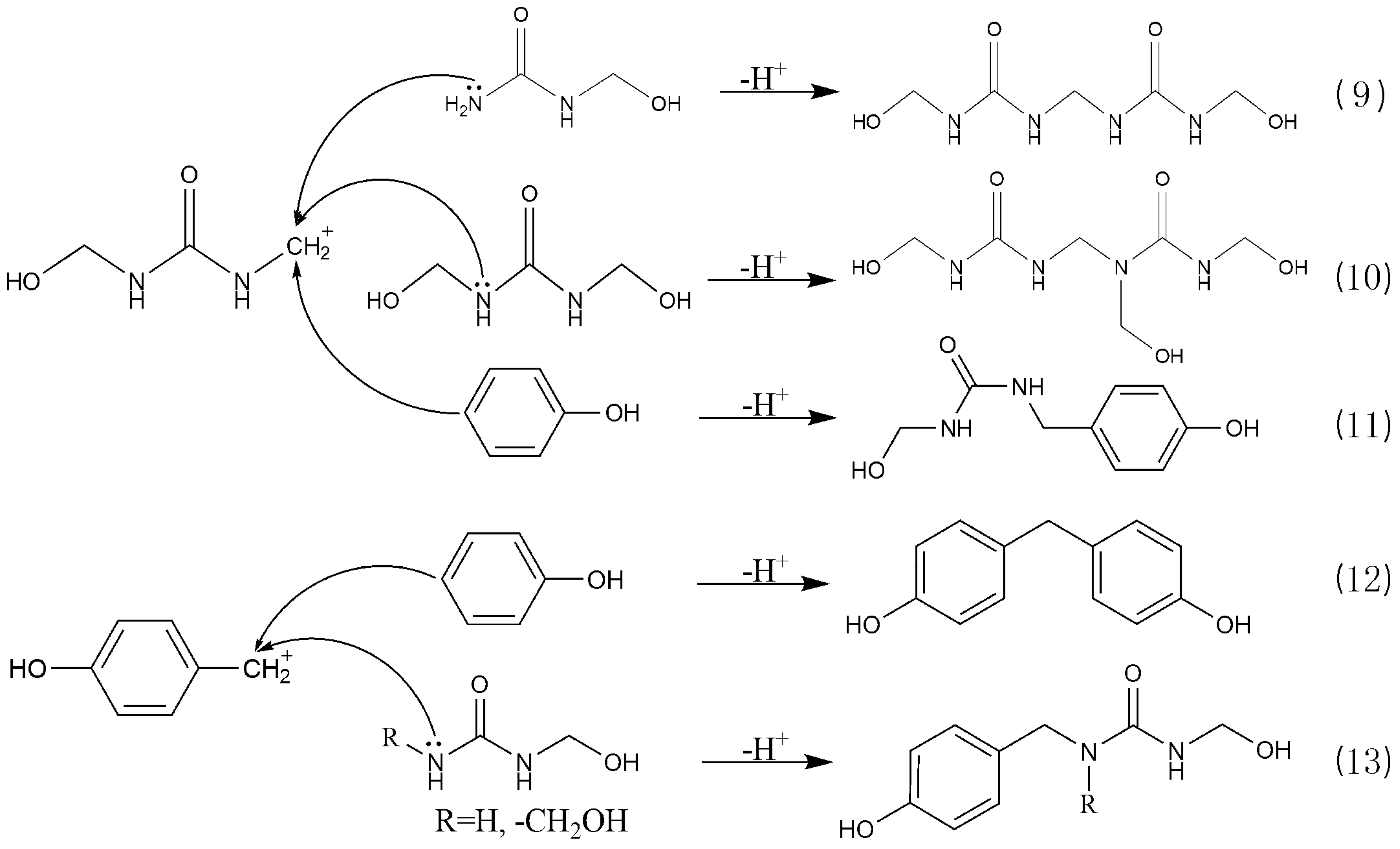
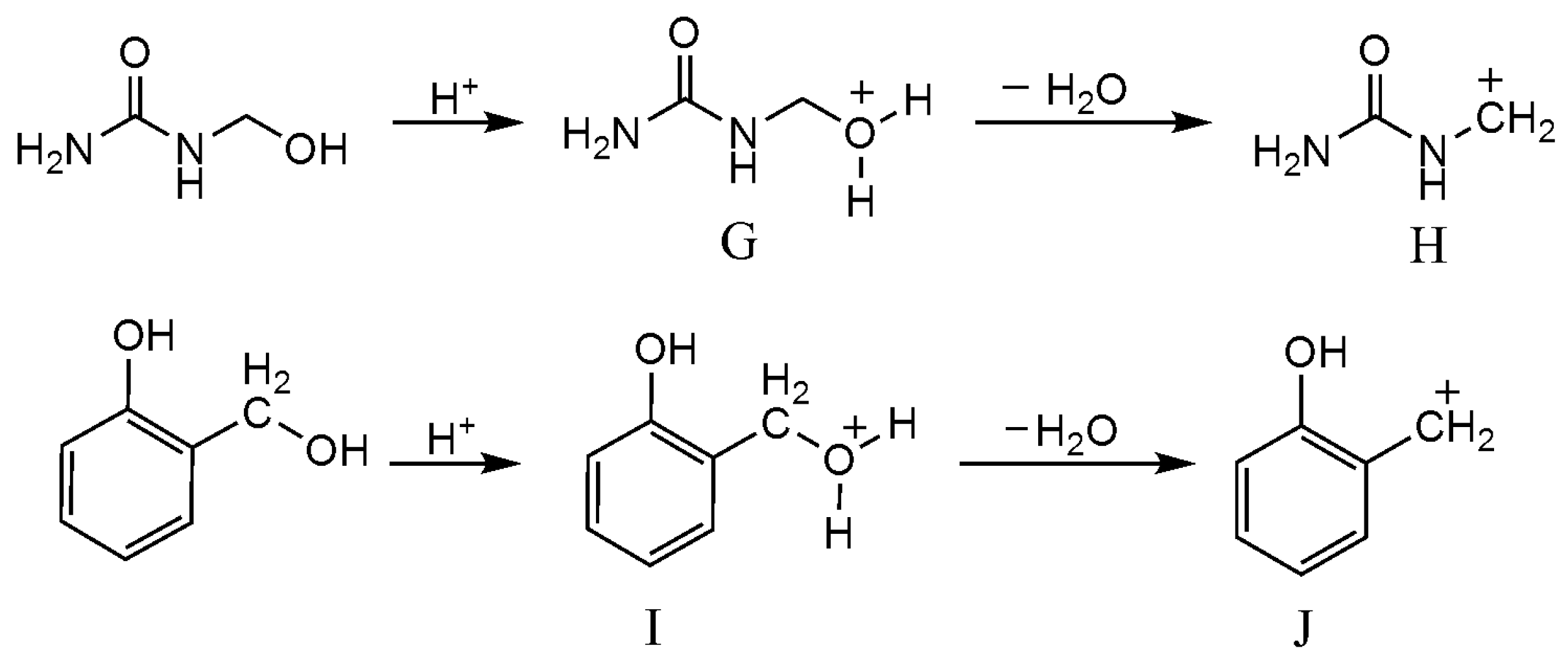
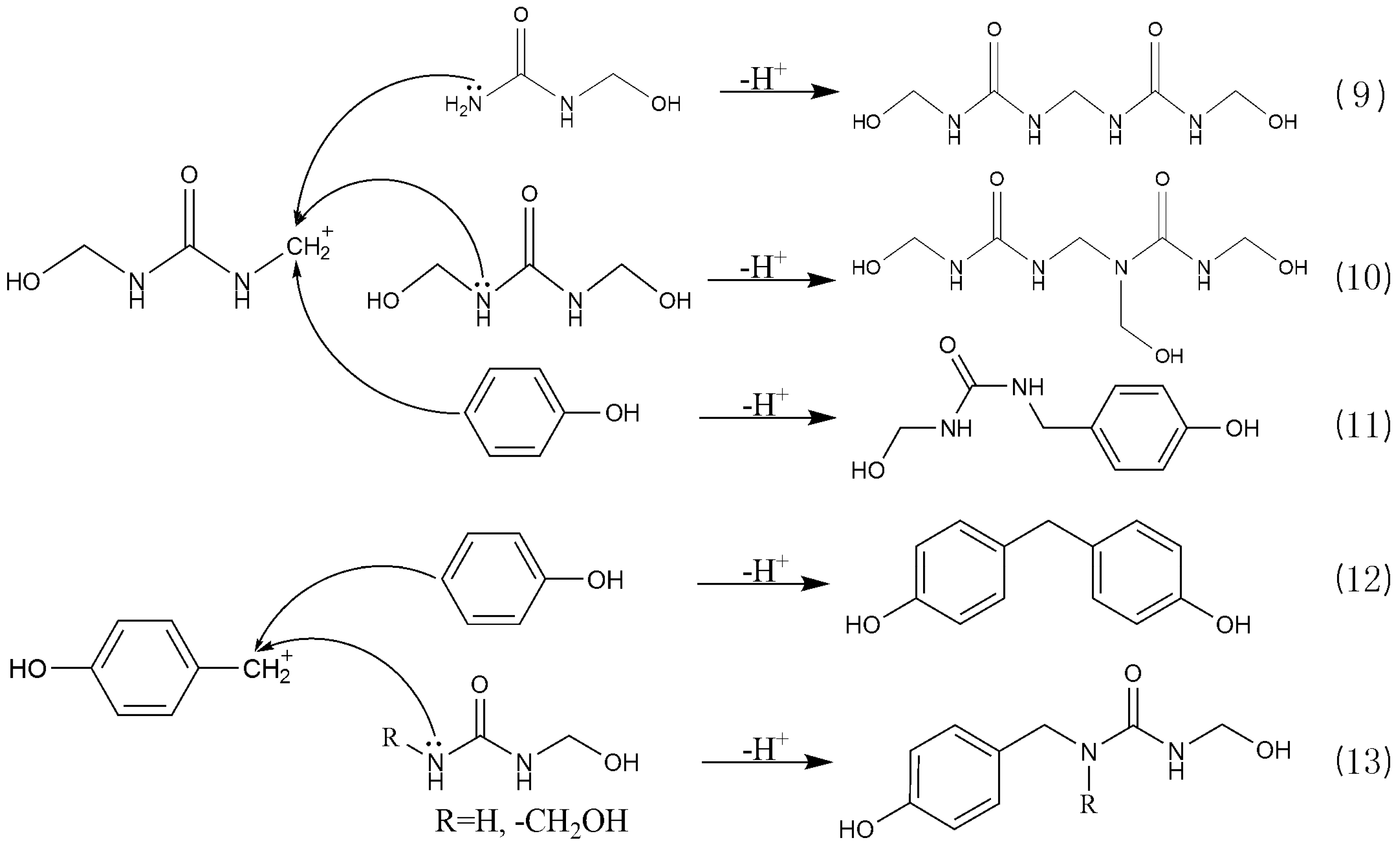
3.3. Reactions between UF2 and Phenol under Strong Acidic Conditions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dunky, M. Urea–formaldehyde (UF) adhesive resins for wood. Int. J. Adhes. Adhes. 1998, 18, 95–107. [Google Scholar] [CrossRef]
- He, G.; Riedl, B. Phenol-urea-formaldehyde cocondensedresol resins: Their synthesis, curing kinetics, and network properties. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 1929–1938. [Google Scholar] [CrossRef]
- Mo, X.; Fan, D.; Qin, T.; Chu, F. 13C-NMR study on the structure of ZnO-catalyzed phenol-urea-formaldehyde resin during its synthesis process. J. Adhes. Sci. Technol. 2014, 28, 2316–2326. [Google Scholar] [CrossRef]
- Fan, D.; Chang, J.; Li, J.; Mao, A.; Zhang, L. 13C-NMR study on the structure of phenol-urea-formaldehyde resins prepared by methylolureas and phenol. J. Appl. Polym. Sci. 2009, 112, 2195–2202. [Google Scholar] [CrossRef]
- Fan, D.; Li, J.; Chang, J.; Gou, J.; Jiang, J. Chemical structure and curing behavior of phenol-urea-formaldehyde cocondensed resins of high urea content. J. Adhes. Sci. Technol. 2009, 23, 1787–1797. [Google Scholar] [CrossRef]
- Du, G.; Lei, H.; Pizzi, A.; Pasch, H. Synthesis-structure-performance relationship of cocondensed phenol-urea-formaldehyde resins by MALDI-TOF and 13C-NMR. J. Appl. Polym. Sci. 2008, 110, 1182–1194. [Google Scholar] [CrossRef]
- Ida, P.; Urška, Š.; Matjaž, K. Characterization of phenol-urea-formaldehyde resin by inline FTIR spectroscopy. J. Appl. Polym. Sci. 2006, 99, 2016–2028. [Google Scholar]
- Tomita, B.; Hse, C.H. Syntheses and structural analyses of cocondensed resins from urea and methylolphenols. Mokuzai Gakkaishi 1993, 39, 1276–1284. [Google Scholar]
- Tomita, B.; Ohyama, M.; Hse, C.Y.; Tomita, B.; Ohyama, M.; Hse, C.Y. Synthesis of phenol-urea-formaldehyde cocondehsed resins from UF-concentrate and phenol. Holzforschung 1994, 48, 522–526. [Google Scholar] [CrossRef]
- Tomita, B.; Hse, C.Y. Phenol-urea-formaldehyde (PUF) co-condensed wood adhesives. Int. J. Adhes. Adhes. 1998, 18, 69–79. [Google Scholar] [CrossRef]
- He, G.; Yan, N. 13C-NMR study on structure, composition and curing behavior of phenol-urea-formaldehyde resole resins. Polymer 2004, 45, 6813–6822. [Google Scholar] [CrossRef]
- Kerstin, S.; Dirk, G.; Harald, P. Preparation of phenol-urea-formaldehyde copolymer adhesives under heterogeneous catalysis. J. Appl. Polym. Sci. 2006, 102, 2946–2952. [Google Scholar]
- Zhao, C.; Pizzi, A.; Garnier, S. Fast advancement and hardening acceleration of low-condensation alkaline PF resins by esters and copolymerized urea. J. Appl. Polym. Sci. 1999, 74, 359–378. [Google Scholar] [CrossRef]
- Zhao, C.; Pizzi, A.; Kühn, A.; Garnier, S. Fast advancement and hardening acceleration of low condensation alkaline phenol-formaldehyde resins by esters and copolymerized urea. II. Esters during resin reaction and effect of guanidine salts. J. Appl. Polym. Sci. 2000, 77, 249–259. [Google Scholar] [CrossRef]
- Kim, M.G. Examination of selected synthesis parameters for typical wood adhesive-type urea-formaldehyde resins by 13C-NMR spectroscopy. I. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 995–1007. [Google Scholar] [CrossRef]
- Kim, M.G. Examination of selected synthesis parameters for typical wood adhesive-type urea-formaldehyde resins by 13C-NMR spectroscopy. II. J. Appl. Polym. Sci. 2000, 75, 1243–1254. [Google Scholar] [CrossRef]
- Kim, M.G. Examination of selected synthesis parameters for wood adhesive-type urea-formaldehyde resins by 13C-NMR spectroscopy. III. J. Appl. Polym. Sci. 1999, 80, 2800–2814. [Google Scholar] [CrossRef]
- Despres, A.; Pizzi, A.; Pasch, H.; Kandelbauer, A. Comparative 13C-NMR and matrix-assisted laser desorption/ionization time-of-flight analyses of species variation and structure maintenance during melamine-urea-formaldehyde resin preparation. J. Appl. Polym. Sci. 2007, 106, 1106–1128. [Google Scholar] [CrossRef]
- Grenier-Loustalot, M.F.; Larroque, S.; Grenier, P.; Leca, J.P.; Bedel, D. Phenolic resins: 1. Mechanisms and kinetics of phenol and of the first polycondensates towards formaldehyde in solution. Polymer 1994, 35, 3046–3054. [Google Scholar] [CrossRef]
- Kibrik, É.J.; Steinhof, O.; Scherr, G.; Thiel, W.R.; Hasse, H. On-line NMR spectroscopic reaction kinetic study of urea–formaldehyde resin synthesis. Ind. Eng. Chem. Res. 2014, 53, 12602–12613. [Google Scholar] [CrossRef]
- Costa, N.A.; Martins, D.; Pereira, J.; Martins, J.; Ferra, J.; Cruz, P.; Mendes, A.; Magalhães, F.D.; Carvalho, L.H. 13C-NMR study of presence of uron structures in amino adhesives and relation with wood-based panels performance. J. Appl. Polym. Sci. 2013, 130, 4500–4507. [Google Scholar] [CrossRef]
- Gao, W.; Li, J. Influence of uron resins on the performance of uf resins as adhesives for plywood. Maderas Ciencia y Tecnología 2012, 14, 3–12. [Google Scholar] [CrossRef]
- Pizzi, A.; Pasch, H.; Celzard, A.; Szczurek, A. Oligomer distribution at the gel point of tannin-resorcinol-formaldehyde cold-set wood adhesives. J. Adhes. Sci. Technol. 2012, 26, 79–88. [Google Scholar]
- Christjanson, P.; Pehk, T.; Paju, J. Structure and curing mechanism of resol phenol-formaldehyde prepolymer resins/resoolsete fenoolformaldehuudvaikude analuus. Proc. Estonian Acad. Sci. 2010, 59, 225–232. [Google Scholar] [CrossRef]
- Ying, Y.G.; Pan, Y.P.; Rui, R.; Dang, J.M.; Liu, C.L. Effect of the molecular structure of phenolic novolac precursor resins on the properties of phenolic fibers. Mater. Chem. Phys. 2013, 143, 455–460. [Google Scholar] [CrossRef]
- Naruyuki, K.; Tetsuo, K.; Mitsuhiro, M. Condensation reactions of phenolic resins. VI. Dependence of the molecular association of 2,4,6-trihydroxymethylphenol on the concentration in an aqueous alkaline medium. J. Appl. Polym. Sci. 2007, 103, 2849–2854. [Google Scholar]
- Singh, M. Preparation, molecular weight determination and structure elucidation of melamine-urea-formaldehyde, melamine-methylureaformaldehyde and melamine-dimethylureaformaldehyde polymer resins with IR spectroscopy. Indian J. Chem. Sect. A 2004, 43, 1696–1700. [Google Scholar]
- Li, T.; Guo, X.; Liang, J.; Wang, H.; Xie, X.; Du, G. Competitive formation of the methylene and methylene ether bridges in the urea-formaldehyde reaction in alkaline solution: A combined experimental and theoretical study. Wood Sci. Technol. 2015, 49, 475–493. [Google Scholar] [CrossRef]
- Pizzi, A.; Horak, R.M.; Ferreira, D.; Roux, D.G. Condensates of phenol, resorcinol, phloroglucinol, and pyrogallol as model compounds of flavonoid A- and B-rings with formaldehyde. J. Appl. Polym. Sci. 1979, 24, 1571–1578. [Google Scholar] [CrossRef]
- Pizzi, A. Pine tannin adhesives for particleboard. Holz als Roh- und Werkstoff 1982, 40, 293–301. [Google Scholar] [CrossRef]
- Pizzi, A.; Stephano, A. Fast vs. Slow-reacting non-modified tannin extracts for exterior particleboard adhesives. Holz als Roh- und Werkstoff 1994, 52, 218–222. [Google Scholar] [CrossRef]
- Pizzi, A.; Tekel, P. 13C-NMR of Zn2+ acetate-induced autocondensation of polyflavonoid tannins for phenolic polycondensates. J. Appl. Polym. Sci. 1995, 56, 633–636. [Google Scholar] [CrossRef]
- Lagel, M.C.; Pizzi, A.; Giovando, S. Matrix-assisted laser desorption-ionization time of flight (MALDI-TOF) mass spectrometry of phenol-formaldehyde-chestnut tannin resins. J. Renew. Mater. 2014, 2, 207–219. [Google Scholar] [CrossRef]
- Lagel, M.C.; Pizzi, A.; Basso, M.C.; Abdalla, S. Development and characterization of abrasive grinding wheels with a tannin-furanic resins matrix. Ind. Crop. Prod. 2015, 65, 343–348. [Google Scholar] [CrossRef]
- Pizzi, A. The chemistry and development of tannin/urea-formaldehyde condensates for exterior wood adhesives. J. Appl. Polym. Sci. 1979, 23, 2777–2792. [Google Scholar] [CrossRef]
- Moubarik, A.; Pizzi, A.; Allal, A.; Charrier, F.; Khoukh, A.; Charrier, B. Cornstarch-mimosa tannin-urea formaldehyde resins as adhesives in the particleboard production. Starch Starke 2010, 62, 131–138. [Google Scholar] [CrossRef]
- Moubarik, A.; Pizzi, A.; Charrier, F.; Allal, A.; Badia, M.; Mansouri, H.R.; Charrier, B. Mechanical characterization of industrial particleboard panels glued with cornstarch-mimosa tannin-urea formaldehyde resins. J. Adhes. Sci. Technol. 2013, 27, 423–429. [Google Scholar] [CrossRef]
- Lei, H.; Wu, Z.; Cao, M.; Du, G. Study on the soy protein-based wood adhesive modified by hydroxymethyl phenol. Polymers 2016, 8, 256–265. [Google Scholar] [CrossRef]
- No, B.Y.; Kim, M.G. Syntheses and properties of low-level melamine-modified urea-melamine-formaldehyde resins. J. Appl. Polym. Sci. 2004, 93, 2559–2569. [Google Scholar] [CrossRef]
- Higuchi, M.; Urakawa, T.; Morita, M. Condensation reactions of phenolic resins. 1. Kinetics and mechanisms of the base-catalyzed self-condensation of 2-hydroxymethylphenol. Polym. J. 2001, 42, 4563–4567. [Google Scholar] [CrossRef]
- Higuchi, M.; Urakawa, T.; Morita, M. Kinetics and mechanisms of the condensation reactions of phenolic resins II. Base-catalyzed self-condensation of 4-hydroxymethylphnenol. Polym. J. 2001, 33, 799–806. [Google Scholar] [CrossRef]
- Kamo, N.; Higuchi, M.; Yoshimatsu, T.; Morita, M. Condensation reactions of phenolic resins IV: Self-condensation of 2,4-dihydroxymethylphenol and 2,4,6-trihydroxymethylphenol (2). J. Wood Sci. 2004, 50, 68–76. [Google Scholar] [CrossRef]
- Conner, A.H.; Lorenz, L.F.; Hirth, K.C. Accelerated cure of phenol-formaldehyde resins: Studies with model compounds. J. Appl. Polym. Sci. 2002, 86, 3256–3263. [Google Scholar] [CrossRef]
- Nair, B.R.; Francis, D.J. Kinetics and mechanism of urea-formaldehyde reaction. Polymer 1983, 24, 626–630. [Google Scholar] [CrossRef]
- Li, T.; Liang, J.; Cao, M.; Guo, X.; Xie, X.; Du, G. Re-elucidation of the acid-catalyzed urea-formaldehyde reactions: A theoretical and 13C-NMR study. J. Appl. Polym. Sci. 2016, 133, 44339–44356. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Chemical shift (ppm) | A1 (%) | A2 (%) | A3 (%) | Structure | Chemical shift (ppm) | A1 (%) | A2 (%) | A3 (%) |
|---|---|---|---|---|---|---|---|---|---|
| Φ–CH2–Φ o–o′ | 30–31 | 3.3 | - | - | HO–CH2–OH | 81–83 | 0.6 | 7.4 | - |
| Φ–CH2–Φ o–p | 33–35 | 5.3 | - | 0.2 | HOCH2–O–CH2–OCH2OH | 85–87 | 0.6 | 6.1 | - |
| Φ–CH2–Φ p–p′ | 39–41 | 1.2 | - | 38.6 | HOCH2–O–CH2–OCH2OH | 90–91 | - | - | - |
| o-Ph–CH2–NHCO– | H(CH2O)nOCH2OCH3 | 94–95 | - | - | - | ||||
| –NH–CH2–NH– (I) | 46–48 | 2.3 | 11.6 | 7.9 | Total | 1.2 | 13.5 | - | |
| o-Ph–CH2–N(–CH2–)CO– | |||||||||
| –NH–CH2–N= (II) | 52–53 | 1.2 | 23.3 | - | –NH–CH2–O–CH3 | 72–73 | - | - | - |
| =N–CH2–N= (III) | 59–61 | 0.4 | 1.7 | - | NH2–CO–NH2 | 163–164 | 7.4 | - | 2.2 |
| p-Ph–CH2–NHCO– | 44–45 | 0.4 | - | 53.3 | NH2–CO–NH– | 161–162 | 40.4 | - | 29.0 |
| p-Ph–CH2–N(–CH2–)CO– | 49–50 | - | - | - | –NH–CO–NH–/–NH–CO–N= | 158–160 | 43.2 | 97.4 | 68.8 |
| CH3OH a | Uron | 154–156 | 9.0 | 2.6 | - | ||||
| Total | 14.1 | 36.6 | 100.0 | Total | 100.0 | 100.0 | 100.0 | ||
| –NH–CH2OCH2NH– (I) | 68–70 | 16.7 | 9.4 | - | Unsubstituted ortho | 115–119 | 100.0 | 39.2 | 44.1 |
| –NH–CH2OCH2N= (II)/Uron | 74–76 | 3.0 | 5.1 | - | Unsubstituted para | 120–124 | - | 18.4 | 1.0 |
| =N–CH2OCH2N= (III) | 77–79 | 7.7 | 0.2 | - | Substituted ortho | 127–130 | - | 5.0 | 16.3 |
| Uron | meta Carbon | 129–133 | - | 37.0 | 38.3 | ||||
| Total | 27.4 | 14.7 | Substituted para | 132–135 | - | 0.4 | 0.3 | ||
| o-Ph-CH2OH | 60–62 | 3.8 | 0.2 | - | Total | 100.0 | 100.0 | 100.0 | |
| p-Ph-CH2OH | 63–65 | 46.4 | 17.4 | - | ortho and para substitution | 151–153 | - | - | 0.1 |
| –NH–CH2OH (I) | |||||||||
| –NH(–CH2)–CH2OH (II) | 70–72 | 7.1 | 17.6 | - | ortho substitution | 153–157 | 72.1 | 8.0 | 3.1 |
| Ph-CH2OCH2-Ph | para substitution | 155–158 | 27.9 | 92.0 | 33.0 | ||||
| Total | 57.3 | 35.2 | P | 157–158 | - | - | 63.8 | ||
| Total | 100.0 | 100.0 | 100.0 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, M.; Li, T.; Liang, J.; Wu, Z.; Zhou, X.; Du, G. A 13C-NMR Study on the 1,3-Dimethylolurea-Phenol Co-Condensation Reaction: A Model for Amino-Phenolic Co-Condensed Resin Synthesis. Polymers 2016, 8, 391. https://doi.org/10.3390/polym8110391
Cao M, Li T, Liang J, Wu Z, Zhou X, Du G. A 13C-NMR Study on the 1,3-Dimethylolurea-Phenol Co-Condensation Reaction: A Model for Amino-Phenolic Co-Condensed Resin Synthesis. Polymers. 2016; 8(11):391. https://doi.org/10.3390/polym8110391
Chicago/Turabian StyleCao, Ming, Taohong Li, Jiankun Liang, Zhigang Wu, Xiaojian Zhou, and Guanben Du. 2016. "A 13C-NMR Study on the 1,3-Dimethylolurea-Phenol Co-Condensation Reaction: A Model for Amino-Phenolic Co-Condensed Resin Synthesis" Polymers 8, no. 11: 391. https://doi.org/10.3390/polym8110391
APA StyleCao, M., Li, T., Liang, J., Wu, Z., Zhou, X., & Du, G. (2016). A 13C-NMR Study on the 1,3-Dimethylolurea-Phenol Co-Condensation Reaction: A Model for Amino-Phenolic Co-Condensed Resin Synthesis. Polymers, 8(11), 391. https://doi.org/10.3390/polym8110391