Abstract
2-Dimethylamino)ethyl methacrylate/styrene statistical copolymers (poly(DMAEMA-stat-styrene)) with feed compositions fDMAEMA = 80–95 mol%, (number average molecular weights Mn = 9.5–11.2 kg mol−1) were synthesized using succinimidyl ester-functionalized BlocBuilder alkoxyamine initiator at 80 °C in bulk. Polymerization rate increased three-fold on increasing fDMAEMA = 80 to 95 mol%. Linear Mn increases with conversion were observed up to about 50% conversion and obtained copolymers possessed monomodal, relatively narrow molecular weight distributions (polydispersity = 1.32–1.59). Copolymers with fDMAEMA = 80 and 90 mol% were also cleanly chain-extended with DMAEMA/styrene mixtures of 95 and 90 mol% DMAEMA, respectively, confirming the livingness of the copolymers. Copolymer phase behavior in aqueous solutions was examined by dynamic light scattering and UV-Vis spectroscopy. All copolymers exhibited lower critical solution temperature (LCST)-type behavior. LCST decreased with increasing styrene content in the copolymer and with increasing solution concentration. All copolymers were completely water-soluble and temperature insensitive at pH 4 but were more hydrophobic at pH 10, particularly copolymers with fDMAEMA = 80 and 85 mol%, which were water-insoluble. At pH 10, LCST of copolymers with fDMAEMA = 90 and 95 mol% were more than 10 °C lower compared to their solutions in neutral, de-ionized water. Block copolymers with two statistical blocks with different DMAEMA compositions exhibited a single LCST, suggesting the block segments were not distinct enough to exhibit separate LCSTs in water.1. Introduction
Stimuli-responsive polymers have sharp and reversible responses to small changes in environmental conditions such as temperature, pH, light, ionic strength, electric and magnetic fields, and they have emerged as a class of materials known as “smart” materials [,]. Water-soluble polymers that can respond to one or more stimuli include poly(N-isopropylacrylamide) (PNIPAM), poly(hydroxypropyl acrylate) (PHPA), poly(oligo(ethylene glycol)methyl ether methacrylate) (POEGMEA) and poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA). These are being extensively studied for numerous potential applications, particularly in the biomedical field, such as drug/gene delivery systems, bioseparations, biosensors, and tissue engineering [-].
PDMAEMA is a water-soluble polymer sensitive to both temperature and pH changes. It exhibits a lower critical solution temperature (LCST)-type phase separation upon heating at neutral or basic conditions and is completely water-soluble in acidic media [-]. To fine-tune the transition conditions and polymer properties, DMAEMA has been incorporated into block copolymers with various counter blocks such as the pH-sensitive poly(2-vinyl pyrindine) to obtain pH-dependent micelles [], a temperature-responsive block such as POEGMA [] and PNIPAM [] to alter the LCST, and a hydrophobic block such as poly(2-(N-carbazolyl)ethyl methacrylate) to make amphiphilic diblock copolymers with hole-transport properties [].
In order to synthesize well-defined polymers and block copolymers that are crucial to obtain the desired performance, precise controls of molecular weight, block sequence length and molecular weight distribution are required. To achieve these goals with relatively robust experimental conditions, controlled radical polymerization (CRP) techniques like atom transfer radical polymerization (ATRP), reversible addition-fragmentation transfer (RAFT) and nitroxide mediated polymerization (NMP) have emerged []. For DMAEMA specifically, ATRP [-] and RAFT [,,] have been successfully applied to obtain well-defined (co)polymers. NMP, being a solely thermal process, does not involve metal catalysts that ATRP requires or bimolecular exchange with sulfur-based agents as in RAFT, which is potentially advantageous for synthesis of non-cytotoxic polymers for biomedical applications [-] or electronic applications where metal contamination is undesirable []. However, controlled polymerization of methacrylates like DMAEMA is still challenging for NMP because methacrylates have very high equilibrium constant K resulting from slow recombination of nitroxides and sterically-hindered poly(methacrylate) radicals, leading to high concentration of propagating radicals that can promote self-termination, resulting in “dead” polymers at low conversions [,].
First-generation nitroxide mediators such as TEMPO (2,2,6,6-tetramethylpiperidinyl-1-oxy) limited NMP to mostly styrenic-based monomers [-]. Important monomers like acrylates and methacrylates could not be controlled with TEMPO. More recently developed alkoxyamine initiators have opened up possibilities towards accessing (meth)acrylic-based polymers. Aside from the alkoxyamines synthesized specifically for methacrylates [,], a commercially available SG1-based unimolecular alkoxyamine called BlocBuilder (N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-O-(2-carboxylprop-2-yl) hydroxylamine; molecular structure of SG1 and BlocBuilder shown in Scheme 1(a)) has achieved controlled polymerization of a range of methacrylates, including methyl [,], ethyl [], butyl [], benzyl [] and poly(ethylene glycol) methyl ether methacrylates [], with a small amount (4–10 mol%) of styrene in the feed and in the presence of about 10% excess of free SG1 relative to the BlocBuilder initiator. The success of such a copolymerization approach was attributed to the low cross-propagation rate constant (kp/reactivity ratio) and much lower equilibrium constant K of styrene compared to methacrylates [,,], which dramatically lowers the product of the average propagation rate constant, <kp>, and average equilibrium constant, <K>, <kp><K>, which is often used to assess control of such NMP polymerization processes.
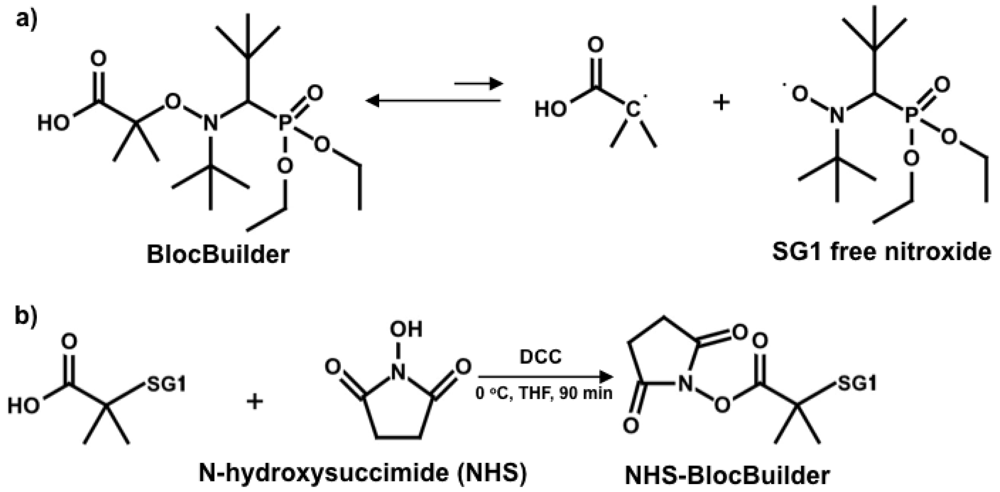
Besides controlling polymer microstructure, the alkoxyamine initiator fragment can permit functionalization and derivatization, such as using an initiator bearing an activated ester moiety. N-hydroxysuccinimidyl (NHS) esters readily react with nucleophiles in single-step reactions [] and precursors functionalized by NHS have been reported for ATRP [] and RAFT [] to yield α-functional polymers. Vinas et al. synthesized the first NHS-functionalized initiator for NMP using BlocBuilder []. The NHS-BlocBuilder (Scheme 1(b)) has been used to effectively control the homopolymerization of styrene and n-butyl acrylate, as well as statistical copolymerization of glycidyl methacrylate and styrene without aid of additional free SG1 nitroxide [,].
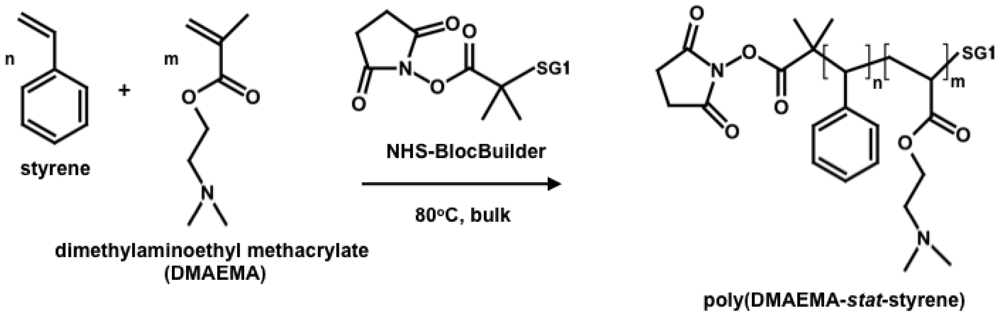
In this study, the NHS-BlocBuilder was used as an initiator to statistically copolymerize DMAEMA with a small amount of styrene as the controlling co-monomer (80–95 mol% DMAEMA in the feed) at 80 °C in bulk (Scheme 2). The combination of NHS-BlocBuilder with DMAEMA-rich feeds was envisioned to enable the development of a water-soluble PDMAEMA with LCST behavior that could also later be conjugated to biological compounds or other amine-containing materials. Further, NMP with NHS-BlocBuilder will be tested to see if no additional free SG1 nitroxide was required, as has recently been shown for other methacrylate-rich feeds []. The following study details results concerning the NMP synthesis of DMAEMA-rich copolymerizations (kinetics, level of control and livingness of the copolymers) followed by the responses of the copolymers to temperature and pH changes in aqueous media.
2. Experimental Section
2.1. Materials
Styrene (99%), 2-(dimethylamino)ethyl methacrylate (DMAEMA, 98%), N,N′-dicyclohexylcarbodiimide (DCC, 99.9%), N-hydroxysuccinimide (NHS, 98%), basic alumina (Brockmann, Type I, 150 mesh), and calcium hydride (90-95% reagent grade) were purchased from Aldrich. Tetrahydrofuran (THF, 99.9%), pentane and hexane were obtained from Fisher; deuterated chloroform (CDCl3) was obtained from Cambridge Isotope Laboratories Inc. N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-O-(2-carboxylprop-2-yl) hydroxylamine, also known as BlocBuilder (99%), was obtained from Arkema and was used as received. Styrene and DMAEMA monomers were purified by passing through a column of basic alumina mixed with 5 wt% calcium hydride; they were stored in a sealed flask under a head of nitrogen in a refrigerator until needed. All other compounds were used as received.
2.2. Synthesis of N-Hydroxysuccinimide Functionalized BlocBuilder (NHS-BlocBuilder)
The following procedures for the synthesis of NHS-BlocBuilder was first described by Vinas et al. and adapted for use here as follows (Scheme 1(b)) []. BlocBuilder (15.0 g, 38 mmol) and N-hydroxysuccinimide (NHS, 5.43 g, 47 mmol) were dissolved in dry THF (60 ml) and placed in an ice-water bath. In a separate flask, N,N′-dicyclohexylcarbodiimide (DCC) was dissolved in 15 mL of dry THF. Both solutions were degassed by bubbling ultra-pure N2 gas for 30 min. The degassed DCC solution was then added to the NHS/BlocBuilder solution. After stirring the mixed solution at 0 °C for 90 min, the precipitated N,N′-dicyclohexylurea (DCU) was removed by filtration. The filtrate was concentrated under reduced pressure to about one third of its volume. The residual DCU was precipitated at −20 °C overnight and removed by filtration. Precipitation of NHS-BlocBuilder was done in pentane and the obtained white solid was washed with de-ionized water to remove residual NHS. After drying under vacuum at room temperature, NHS-BlocBuilder was obtained as a white powder (10.6 g, 22 mmol, 56% yield). The structure of the NHS-BlocBuilder was confirmed by 1H NMR (400 MHz, CDCl3, δ, ppm): 1.1–1.4 (24H), 1.6–1.9 (6H), 2.7–2.9 (4H), 3.2–3.4 (1H), 3.9–4.4 (4H).
2.3. Synthesis of Statistical Copolymers of 2-(Dimethylamino)ethyl Methacrylate and Styrene (Poly(DMAEMA-stat-styrene)) by NMP
Statistical copolymerizations of DMAEMA and styrene were performed in a 50 mL three-neck round-bottom glass flask equipped with a reflux condenser and a thermal well. Target molecular weight (number average molecular weight Mn at 100% conversion) of 30 kg mol−1 was used for all copolymerizations. A typical procedure is given as an illustration. The initiator, N-hydroxysuccinimidyl-functionalized BlocBuilder (NHS-BlocBuilder, 0.097 g, 0.20 mmol), was dissolved in the monomer mixture (6.0 g) with initial molar compositions with respect to DMAEMA (fDMAEMA) varied from 85 to 95 mol% (Table 1). Dissolved oxygen in the mixture was removed by purging with an ultra-pure nitrogen flow for 30 min prior to heating. The mixture was then heated by a heating mantle to 80 °C at a rate of about 8 °C min−1 while maintaining a nitrogen purge. The time when the mixture reached 80 °C was taken as the start of the reaction (t = 0). Samples were taken periodically to determine conversion and molecular weight. Polymerization was stopped when the reaction mixture became too viscous to withdraw samples. The copolymers were precipitated in hexane, decanted and dried in vacuum at room temperature overnight; the copolymers were then re-dissolved in THF, re-precipitated in hexane, decanted and dried under vacuum to obtain the final purified copolymers.

Two chain extension experiments were performed using the copolymers synthesized. The copolymers DMAEMA/S-90/10 and DMAEMA/S-80/20 were used as the macroinitiators (0.35 g, 0.03 mmol). The chain-extensions were performed using the same methods as described for the statistical copolymerizations. The macroinitiator was chain-extended with a mixture of DMAEMA and styrene (a total of 3.0 g) after purging with nitrogen for 30 min followed by heating up to 80 °C. The work-up was identical to that used for statistical copolymerizations. The detailed experimental conditions are listed in Table 2.
2.4. Polymer Characterization Using Gel Permeation Chromatography (GPC) and Nuclear MagneticResonance (NMR)
Monomer conversion and copolymer composition were determined by 1H NMR with a 400 MHz Varian Gemini 2000 spectrometer using CDCl3 as solvent. Conversion of DMAEMA was calculated using the two protons adjacent to the ester oxygen of the monomer (δ = 3.7–3.8 ppm) and those corresponding to the polymer (δ = 3.9–4.1 ppm). Styrene conversion was determined using the three vinyl protons (δ = 6.6–6.7, 5.6–5.7, and 5.1–5.2 ppm) of the monomers and the five aromatic protons of both monomers and polymers (δ = 6.9–7.2 ppm). Average conversion was then calculated using the composition of the copolymers with respect to DMAEMA (FDMAEMA, mol%) determined by the same NMR spectra (Conv.ave = FDMAEMA × Conv.DMAEMA + (1 − FDMAEMA) × Conv.styrene). Molecular weight and polydispersity index (PDI) were characterized by GPC (Waters Breeze) using THF as the mobile phase. The GPC was equipped with three Waters Styragel HR columns (molecular weight measurement ranges: HR1: 102 − 5 × 103 g mol−1, HR2: 5 × 102 − 2 × 104 g mol-1, HR3: 5 × 103 − 6 × 105 g mol−1) and a guard column. The columns were operated at 40 °C and a mobile phase flow rate of 0.3 mL min−1 during analysis. The GPC was also equipped with both ultraviolet (UV 2487) and differential refractive index (RI 2410) detectors. The molecular weight measurements were calibrated relative to poly(styrene) narrow molecular weight standards in THF at 40 °C. The percentage of copolymer chains capped by SG1 was determined using 31P NMR with a 200 MHz Varian Gemini 2000 spectrometer using CDCl3 as solvent and diethyl phosphite (DEP) as internal standard. In each sample, known amounts of copolymer and DEP were dissolved in CDCl3. The ratio of SG1-capped chains to DEP was calculated using the 31P resonance at δ = 24–27 ppm for SG1 and δ = 8–9 ppm for DEP. This ratio was then compared to the molar ratio of copolymer to DEP in the sample to calculate the percentage of SG1-capped chains.
2.5. Transition Temperature Characterization Using UV-Visible Spectrometry and Dynamic Light Scattering (DLS)
The statistical copolymers and block copolymers were dissolved in de-ionized water and buffer solutions of pH 4 and 10 with 0.1, 0.3 and 0.5 wt% concentrations. Light transmittance of the solutions at various temperatures was measured using a Cary 5000 UV-Vis-NIR Spectrophotometer at 600 nm. The solutions were equilibrated for 10 min at the starting temperature and then heated at a rate of 0.5 °C min−1. The transmittance was recorded every 0.5 °C.
The DLS measurements for these solutions were performed using a Malvern ZetaSizer (Nano-ZS). The instrument was equipped with a He-Ne laser operating at 633 nm and an avalanche photodiode detector. In a typical DLS measurement, the solution was filtered using a 0.2 μm pore size filter into a plastic cuvette. The cuvette was then placed in the temperature-controlled measurement cell and equilibrated at the starting temperature for at least 30 min. Measurements were then performed automatically for every degree Celsius after an equilibration time of 2 min. The first-order intensity correlation function, g1(t), was measured at a scattering angle of 173° using Non-Invasive Back-Scatter (NIBS) and analyzed by the method of cumulants [] to estimate the average decay rate, Γ, and the width of the decay, μ2/Γ2.
The average decay rate Γ is given by:
In the limit of low concentrations, D can be approximated as the diffusion coefficient for spherical particles, D0, which is related to the hydrodynamic radii of the particles, Rh, by the Stokes-Einstein equation:
3. Results and Discussion
3.1. Synthesis of Statistical Copolymers of 2-(dimethylamino)ethyl Methacrylate (DMAEMA) and Styrene
3.1.1. Kinetic Results of the Statistical Copolymerizations
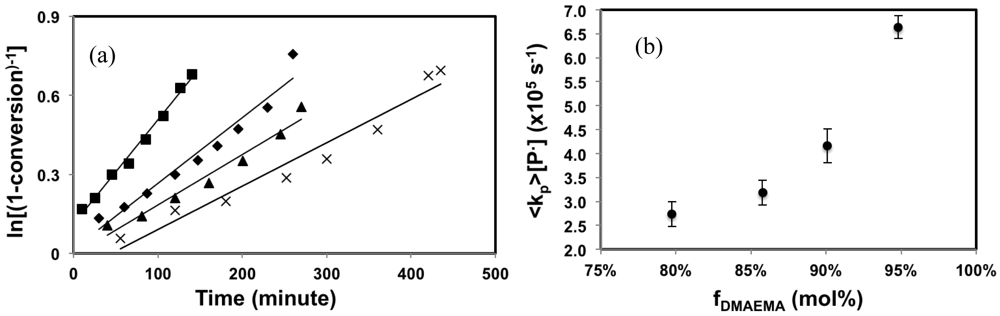
The kinetic results of the copolymerizations of DMAEMA and styrene using N-hydroxysuccinimide functionalized BlocBuilder (NHS-BlocBuilder) as initiator are presented in Figure 1. All copolymerizations obeyed first order kinetics as described by Equation 5 and generated good fits to linear kinetic plots as shown in Figure 1(a).
In Equation 5, [M]0 and [M]t are concentrations of monomer at time zero and subsequent later time t, respectively; <kp> is the average propagation rate constant; and [P•] is the propagating radical concentration.
The apparent rate constant represented by the slopes of the kinetic plots, <kp>[P•], increased rapidly with the initial DMAEMA feed composition (fDMAEMA), especially when fDMAEMA ≥ 90 mol%, as shown in Figure 1(b). This result is expected as methacrylates have generally higher kp and much higher K (defined by Equation 6 when SG1 is the nitroxide) compared to styrene [,,].
The result is consistent with similar copolymerization studies of methacrylates with styrene using BlocBuilder [,-]. The kinetic results showed that the feed composition had a strong effect on the polymerization rate of the DMAEMA-rich mixtures and the polymerization rate of DMAEMA using NHS-BlocBuilder as the initiator can be controlled with as little as 5 mol% styrene.
3.1.2. Level of Control of the Copolymerizations and the Livingness of the Copolymers
Linear molecular weight increase with conversion and narrow molecular weight distribution of the polymers are characteristics generally associated with controlled polymerizations. In Figure 2(a), number-average molecular weight (Mn) was plotted against conversion for the various copolymerizations. Mn increased linearly with conversion for all copolymerizations up to about 50% conversion. However, the slopes of the Mnversus conversion with different compositions varied and differed from the theoretical one as shown in Figure 2(a). For copolymerizations with fDMAEMA = 80–90 mol%, the slopes were very similar but slightly smaller than the theoretical predicted line. The small deviation in slopes can be attributed to the differences in hydrodynamic volumes of the copolymers and that of the poly(styrene) standards that used to calibrate the GPC. The copolymerization with fDMAEMA = 95 mol%, on the other hand, had a significantly flatter slope compared to the others, where the difference in hydrodynamic volumes cannot be the sole reason. The copolymerization with fDMAEMA = 95 mol% exhibited the most rapid kinetic behavior as shown in Figure 1. The fast kinetics was the result of not only higher <kp>, but also likely a comparatively higher <K> from the highly methacrylate-rich mixtures []. The latter point is particularly important for the level of control because high concentration of propagating radicals favors self-termination and the large K of methacrylates such as methyl methacrylate was claimed to be the main cause for its uncontrolled homopolymerization using nitroxides []. Such irreversible termination reactions can lower the overall Mn and lead to the decrease in slope, as was observed for the copolymerization with fDMAEMA = 95 mol% as observed in Figure 2(a).
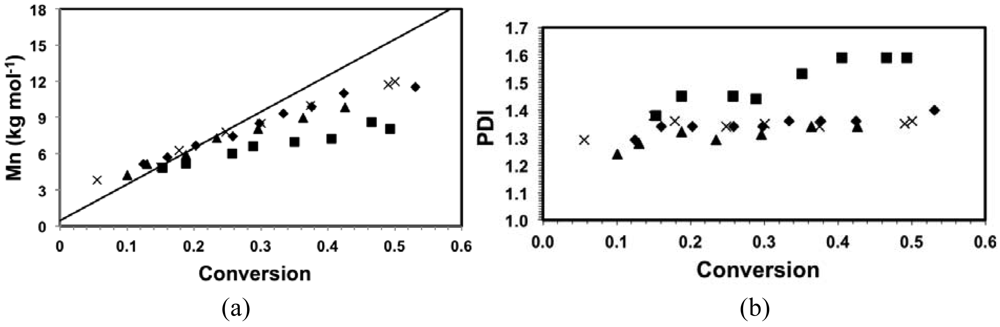
The change in control as a function of feed composition was also reflected in the observed PDIs of the copolymers. The copolymer PDIs with fDMAEMA = 80–90 mol% remained relatively constant at about 1.3–1.4 throughout the polymerizations. In contrast, PDIs of copolymers with fDMAEMA = 95 mol% started at about 1.4 and continued to increase to about 1.6 at 49% conversion. The broadening of the molecular weight distribution is a sign of chain termination events, which is consistent with the results of the Mn trend with conversion.
The results of the DMAEMA/styrene statistical copolymerizations using NHS-BlocBuilder as initiator are summarized in Table 3. Statistical copolymers had compositions very similar to the feed compositions and copolymers with higher styrene content possessed lower PDIs. The percentage of SG1-capped polymer chains (fLC, Table 3) was also examined since the reversible SG1 termination is necessary for the re-initiation of polymer chains. This was the measure of the living fraction of the chains. From the results shown in Table 3, copolymers with FDMAEMA up to 89 mol% had more than 80% living chains, whereas the copolymer FDMAEMA = 96 mol% had only 69% living chains. The results of fLC can vary by about ±10% because of the uncertainties in Mn due to different hydrodynamic volumes between the copolymers and poly(styrene) standards. Nonetheless, the fLC results confirm the high level of livingness of the copolymers with FDMAEMA up to 89 mol% and decrease in livingness for the copolymer with FDMAEMA = 96 mol%, in agreement with the kinetic data.
To further investigate the ability of the copolymers to re-initiate and form block copolymers, two chain-extension experiments were performed using the statistical copolymer synthesized in DMAEMA/S-80/20 (FDMAEMA = 81 mol%, Mn = 11.2 kg mol−1, PDI = 1.32) and DMAEMA/S-90/10 (FDMAEMA = 89 mol%, Mn = 11.1 kg mol−1, PDI = 1.39) as the macroinitiator. The experimental conditions and results of the chain extension experiments are summarized in Table 4. With a fresh mixture of 95 mol% DMAEMA, the DMAEMA/S-80/20 macroinitiator with FDMAEMA = 81 mol% was extended with a more DMAEMA-rich block with Mn of approximately 16.5 kg mol−1 and the overall DMAEMA molar composition of the block copolymer was increased from 81 mol% o f the macroinitiator to 90 mol%. On the other hand, the DMAEMA/S-90/10 macroinitiator with FDMAEMA = 89 mol% was chain-extended with a DMAEMA/styrene mixture of similar composition (fDMAEMA = 90 mol%) to obtain a block copolymer that was essentially a statistical copolymer. The two chain extended copolymers were also used to examine the effect of polymer microstructure and molecular weight on LCST behavior, which will be discussed in Section 3.2.5.
The PDIs of the block copolymers (1.91 and 1.70) were significantly higher than that of the macroinitiator (1.32 and 1.39), indicating either termination occurred during the chain extension or some macroinitiator was not initiated.
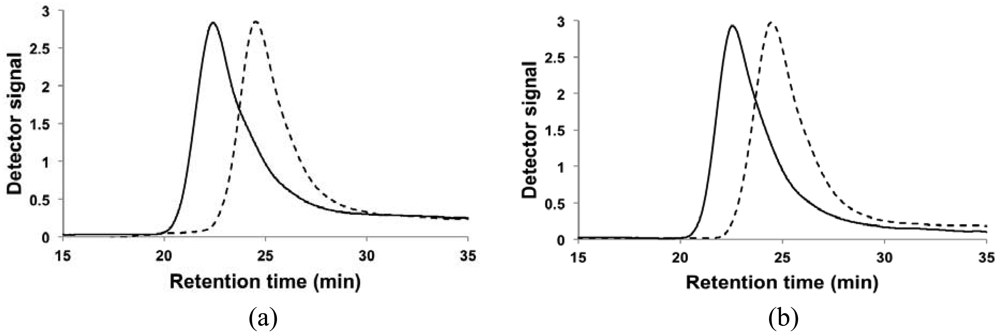
The results of the chain-extension experiments can be more clearly observed by examining the GPC trace of the macroinitiator compared to the chromatogram of the chain-extended polymer in Figure 3. The trace of the chain-extended polymer retained generally its mono-modal nature and has a clear shift to the left, indicating most of the macroinitiators were extended and the level of livingness was high. The tails of chain-extended copolymers overlap to some degree with the associated macroinitiator traces in both cases, but the tailing was slightly more apparent for the chain extension of the DMAEMA/S-80/20 macroinitiator. From the fLC results listed in Table 3, DMAEMA/S-80/20 macroinitiator had a slightly lower percentage of SG1-capped chains than the DMAEMA/S-90/10 macroinitiator but the second batch of monomer was richer in DMAEMA. As noted previously for the statistical copolymer, control was poorer with broader molecular weight distributions when the feed was very rich in DMAEMA (fDMAEMA = 95 mol%). This suggested a small amount of “dead” macroinitiator combined with a richer DMAEMA second feed contributed to the broadening of the molecular weight distribution after chain-extension. Baseline drifts immediately after the main peaks were also observed in all traces. To investigate the potential reasons for the drifts, the copolymers were fractionated to remove any low Mn chains and were re-examined by GPC. The baseline drift persisted for the fractionated copolymers, indicating adsorption of the polar amine group of PDMAEMA to the GPC column is likely the cause for the baseline drift observed [,].
In summary, well-defined DMAEMA-rich copolymers capable of initiating a second batch of monomer relatively cleanly were synthesized using NHS-BlocBuilder. To our knowledge, this is the first time such a synthesis using nitroxide-mediated polymerization is reported. Lokaj et al. [] attempted to synthesize a block copolymer of styrene and DMAEMA using TEMPO-terminated poly(styrene) macroinitiators. However, conversion stopped increasing after 2 h of reaction and the final copolymer contained only about 40 mol% DMAEMA. Low conversion and early termination can be results of the slow initiation of poly(styrene) macroinitiator [,] and/or β-hydrogen transfer from the propagating radical to the initiator [,].
SG1-base initiators such as BlocBuilder have lower activation energy and much higher dissociation rate constant (kd) compared to TEMPO-based initiators []. Additionally, Dire et al. [] confirmed that the rate constant for β-hydrogen transfer (kβtr) of methyl methacrylate/SG1 system was three orders of magnitude lower compared to a n-butyl methacrylate/TEMPO system. As a result, SG1-based initiators can be activated at lower temperatures and allow controlled polymerizations of a wider range of monomers, such as methacrylates, compared to TEMPO. However, when polymerizing non-styrenic monomers using BlocBuilder, additional SG1 nitroxide was required to further reduce polymerization rate and produce well-defined copolymers [,-].
The kd of NHS-BlocBuilder measured by Vinas et al. was about 15 times higher than that of BlocBuilder (5 s−1 for NHS-BlocBuilder and 0.32 s−1 for BlocBuilder) at 120 °C in tert-butyl benzene []. Under the same experimental conditions, they also found that the activation energy of NHS-BlocBuilder (Ea = 105 kJ mol−1) was lower than that of BlocBuilder (Ea = 112 kJ mol−1). The high dissociation rate and lower activation energy of NHS-BlocBuilder led to fast initiation and quick release of the regulating SG1 nitroxide, which mimics the situation when additional SG1 free nitroxide is added at the onset to the solution to improve control. Consequently, controlled polymerization of the DMAEMA-rich mixtures without the requirement of additional SG1, as is typical for methacrylate-rich polymerizations by BlocBuilder, was largely accomplished. The results of the DMAEMA/styrene statistical copolymerizations demonstrated that NHS-BlocBuilder was not only an effective initiator for synthesizing DMAEMA-rich copolymers, but also simplifies polymerization procedures and provides opportunities for further functionalization of the copolymers. Our group earlier noticed similar performance for NHS-BlocBuilder controlled copolymerizations of glycidyl methacrylate with styrene [].
3.2. Studies of Temperature and pH Sensitivities of Copolymers in Aqueous Solutions
The sensitivity to both temperature and pH of PDMAEMA in aqueous solution is a hallmark of such materials for applications requiring it as a stimuli-responsive material. The LCST of such a polymer can be affected by many parameters, including molecular weight [,], polymer composition [,,], solution concentration [], and pH [,] in the case of PDMAEMA. Because the DMAEMA/styrene copolymerizations using NHS-BlocBuilder were relatively controlled as demonstrated earlier, final copolymers obtained have similar Mn (10.4 ± 0.9 kg mol−1) and PDI (1.42 ± 0.12). Therefore, the second part of this study was to determine the LCST of the copolymers as a function of polymer composition, solution concentration and pH. This is necessary as the small amount of styrene required as a controlling comonomer for the BlocBuilder-mediated NMP may affect the LCST compared to that of pure PDMAEMA.
3.2.1. LCST-Type Phase Separation Illustrated by UV-Visible Spectrometry and Dynamic Light Scattering
UV-Visible spectrometry (UV-Vis) and dynamic light scattering (DLS) were used to characterize the phase behavior of the copolymers. UV-Vis monitored the light transmittance of the solutions as temperature varied. DLS measured the hydrodynamic radius (Rh) distribution of the polymers in solution. Each copolymer was dissolved in de-ionized water at concentrations of 0.1, 0.3 and 0.5 wt% and buffer solutions of pH 4 and 10 at 0.5 wt%. Figures 4 and 5 show the changes of light transmittance and Rh with temperature of solutions in de-ionized water, respectively. Copolymers with up to 16 mol% styrene (DMAEMA/S-85/15) were soluble in de-ionized water at all three concentrations. Copolymer with 19 mol% styrene (DMAEMA/S-80/20) was only soluble in de-ionized water at 0.1 wt%.
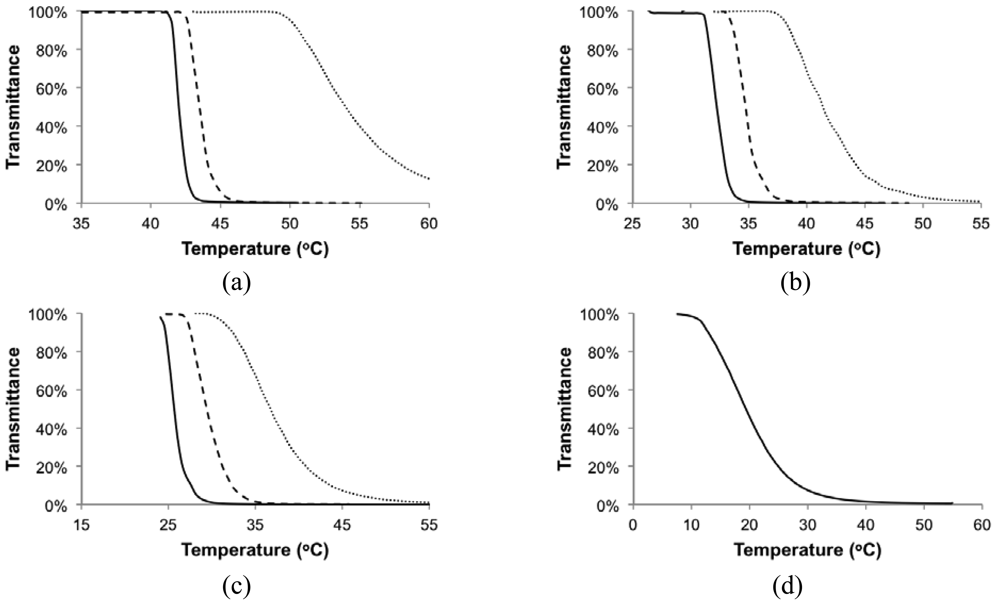
The light transmittance profiles reflected typical behavior of LCST-type phase separation, where solutions were 100% transparent at low temperature and became cloudy (0% transmittance) quickly above the critical temperature, illustrating the sudden solubility drop of the polymer in water. The transition temperature decreased as concentration increased. The shapes of the profiles were similar for different copolymers at the same concentration, where the transitions became broader as the concentration decreased from 0.5 wt% to 0.1 wt%.
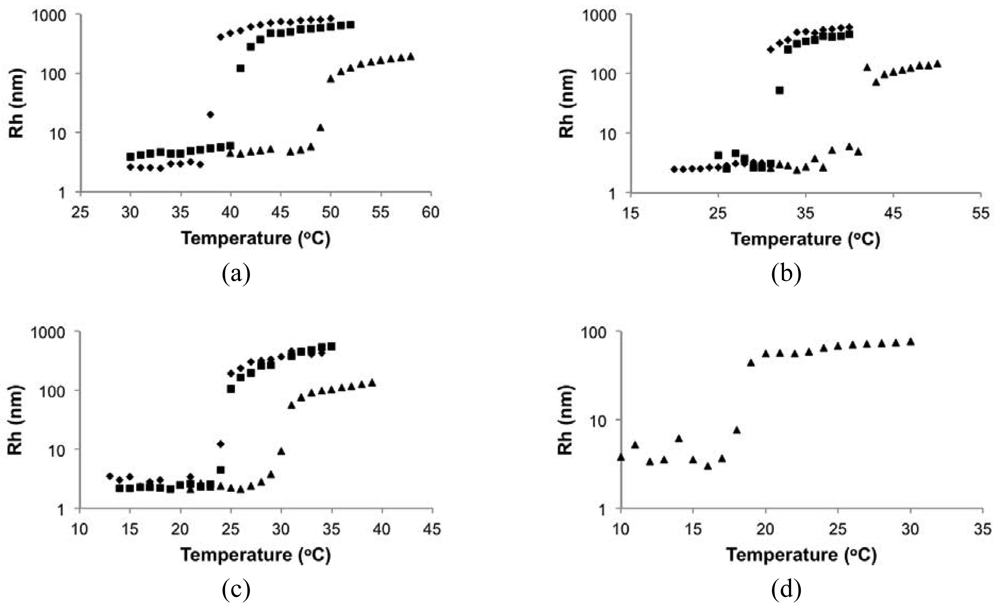
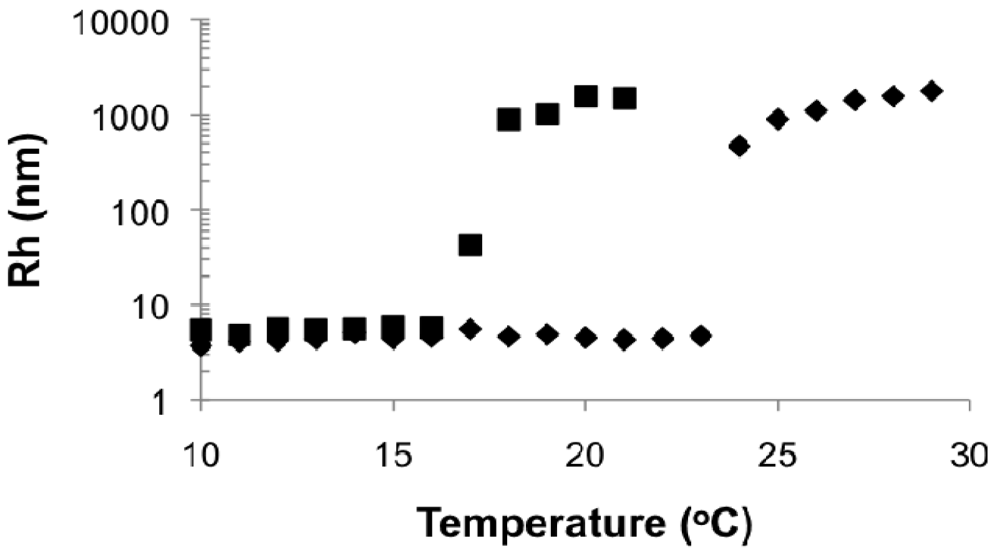
In the DLS measurements, Rh was below 10 nm at low temperature for all cases and rapidly rose up to about 500 nm for 0.3 and 0.5 wt% solutions and about 100 nm for 0.1 wt% solutions when the critical temperature was reached. It demonstrated the transformation of polymers from small, dispersed unimers [] to large aggregated particles upon heating. The shifts of critical temperature with concentration were similar to those observed by UV-Vis.
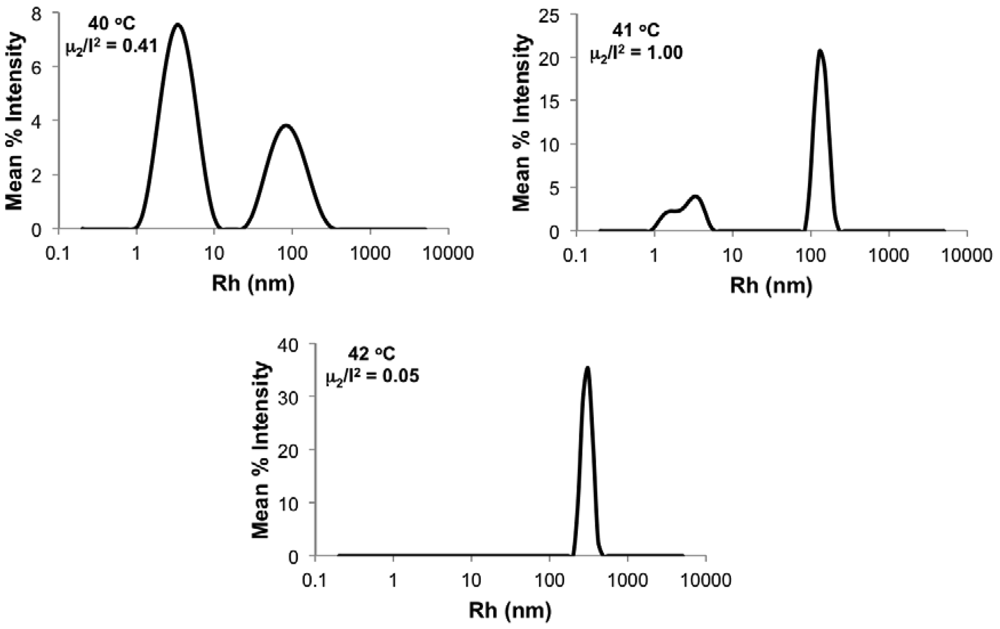
Figure 6 shows typical Rh distributions near the critical temperature using the 0.3 wt% solution of poly(DMAEMA-stat-styrene) with FDMAEMA = 96 mol% as an example. Below LCST (temperature = 40 °C in Figure 6), a unimer peak of Rh < 10 nm can be observed with a trace amount of loose aggregates. As temperature approached the LCST (temperature = 41 °C in Figure 6), the amount of aggregates increased and the width of the overall distribution (μ2/Γ2) became much broader as phase separation started. At temperature = 42 °C in Figure 6, the peak corresponding to unimers completely disappeared, leaving only the narrow peak representing large aggregates with μ2/Γ2 below 0.1. Because the distributions were intensity-based, the height of the peak is proportional to the corresponding size to the sixth order, according to the Rayleigh approximation. Therefore, the distribution was heavily biased toward larger sizes as long as peaks of different sizes coexisted. Given this, the LCST was defined as the temperature where the large aggregate peak (Rh ≥ 100 nm) became the only peak in the distribution (42 °C in Figure 6). For UV-Vis, LCST was defined as the temperature where the transmittance dropped to 80%.
3.2.2. Effect of Polymer Composition on LCST
Composition plays an important role on LCST of statistical copolymers with comonomers of different hydrophilicity []. Higher content of the less hydrophilic comonomer decreases LCST whereas higher content of the more hydrophilic comonomer increases LCST. Many researchers have fine-tuned the LCST of different copolymers by varying their composition [,,]. In this study, styrene is a hydrophobic comonomer and it was expected that with more styrene incorporated into the copolymer, the LCST would be lower.
Indeed, the LCST of the statistical copolymers decreased with the higher styrene content in the copolymer as shown in Figure 7. The results from DLS and UV-Vis were very similar with the largest difference of 5 °C for the 0.1 wt% solution of the least soluble copolymer with 81 mol% DMAEMA (DMAEMA/S-80/20). Since the copolymer with 81 mol% DMAEMA was not soluble at 0.3 wt% concentration, 0.1 wt% was likely close to its saturation limit at the storage temperature (5 °C). As temperature increased, small amounts of polymer may precipitate before large aggregates could be formed due to a decreasing saturation concentration with temperature. This could introduce errors in its LCST measurements. For copolymers with 84 mol% or more DMAEMA, LCST changed linearly with composition and the slopes of the trends for different concentrations were very similar, which is consistent with the results of other studies [,,].
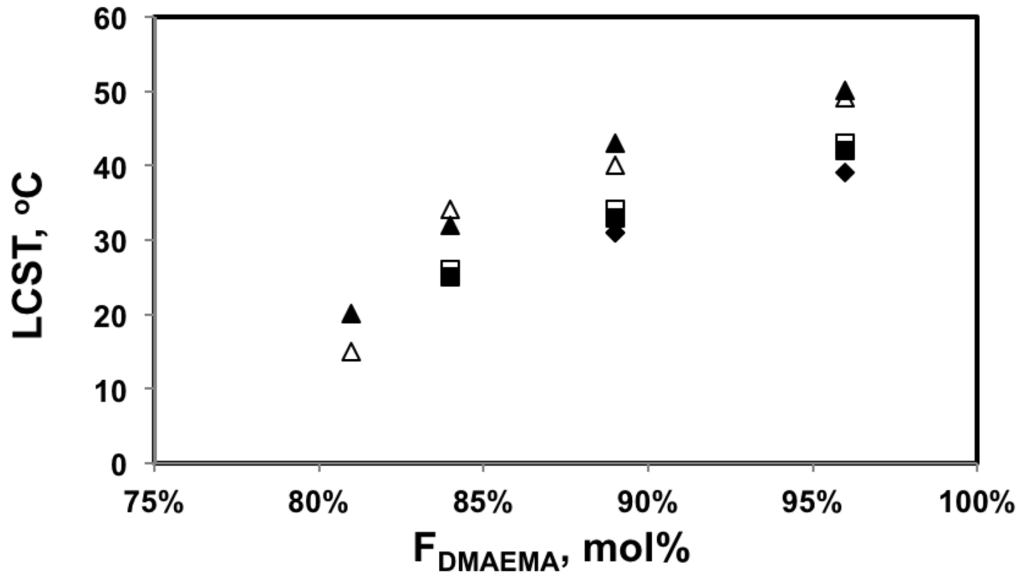
3.2.3. Effect of Concentration on LCST
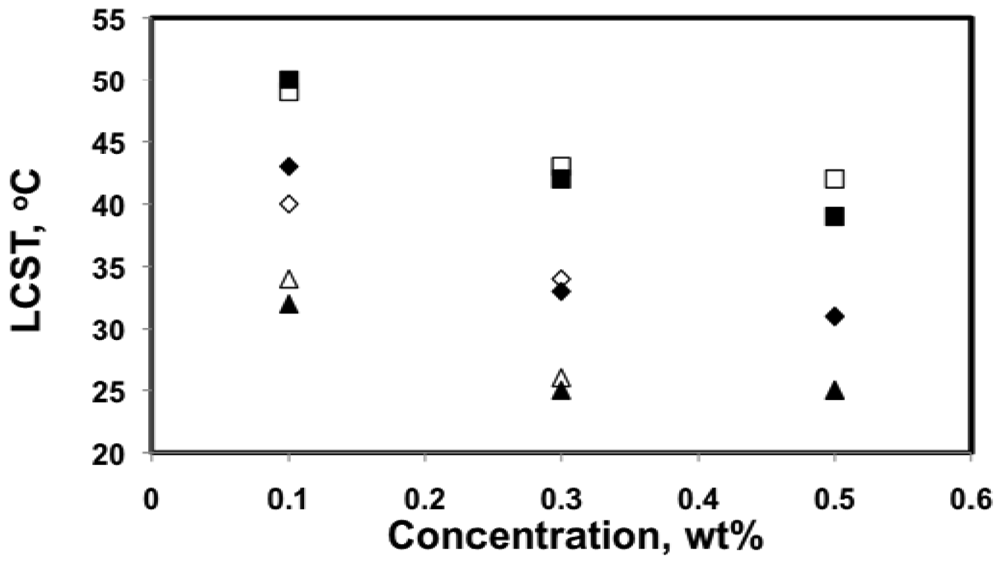
The LCST of poly(DMAEMA-stat-styrene) copolymer solutions in de-ionized water at 0.1, 0.3 and 0.5 wt% were summarized in Figure 8. Overall, LCST decreased as concentration increased, which is consistent with the literature [,]. The LCST decreased by 7–10 °C when concentration increased from 0.1 to 0.3 wt%, whereas the LCST for 0.5 wt% solutions were very similar to those for 0.3 wt% solutions, with much smaller differences (0–3 °C).
From a thermodynamic perspective, when both the enthalpy (ΔHm) and entropy (ΔSm) become negative upon mixing with a dominant entropic term (ΔSmT), the Gibb's energy of mixing (ΔGm) becomes negative and a polymer with some solvatophobic structures may be solvated below the LCST []. In aqueous solutions, hydrogen bonding between water molecules and polymer chains can lead to formation of a layer of clathrate-like structures around a hydrophobic structure in a polymer chain which lowers the entropy of mixing and increases the solubility of the polymer in water [,]. As temperature increases above a critical temperature, the ordered water layer is disrupted and the polymer chains collapse and form large aggregates.
It was found that LCST in aqueous solutions was not very concentration-sensitive as long as the solution was not too dilute (typically above 1 wt%) [,,]. For dilute solutions, large amounts of water molecules are present in between each of the polymer chains, which can increase the energy needed to bring polymer chains together to form aggregates. This explains the broader transitions and smaller aggregate size for 0.1 wt% solutions shown in Figures 4 and 5. Because the transitions for 0.3 and 0.5 wt% solutions were very similar in terms of LCST, sharpness of the transition and the size of the aggregates, 0.5 wt% is likely close to the critical concentration, above which LCST is no longer sensitive to concentration.
3.2.4. Effect of pH on the Solubility of Copolymers in Aqueous Solution
It is known that PDMAEMA is pH sensitive with a pKa of about 7.3 in water []. To investigate the pH sensitivity of the statistical copolymers, solutions at pH 4 and 10 were prepared and their phase behavior was compared to that in de-ionized water solutions. The results are summarized in Table 5.
All copolymers, including the one with 81 mol% DMAEMA (DMAEMA/S-80/20), which was only soluble at 0.1 wt% in de-ionized water, were soluble at pH 4 at 0.5 wt% and no LCST was observed up to 90 °C. At pH 4, PDMAEMA is completely ionized [] into a polyelectrolyte. As a result, the solubility of PDMAEMA in water greatly increases and becomes insensitive to temperature [,]. Because the charged copolymer is highly hydrophillic, even with as much as 19 mol% styrene, the copolymer was completely water-soluble at 0.5 wt%.
At pH 10, LCSTs were observed and decreased with DMAEMA content in the copolymer, similar to the results in de-ionized water solutions. However, the LCST is more than 10 °C lower when comparing the copolymers in the pH = 10 solutions to the neutral, de-ionized water solutions. Additionally, the copolymer with 84 mol% DMAEMA (DMAEMA/S-85/15) that was soluble in de-ionized water was not soluble at pH = 10. In Figure 9, the hydrodynamic radii of the copolymers with 96 and 89 mol% DMAEMA (DMAEMA/S-95/5 and DMAEMA/S-90/10, respectively) increased rapidly from below 10 nm to ∼μm at their LCST. Because pH 10 is higher than the pKa of PDMAEMA, the copolymers were totally deprotonated. As a result, the copolymers became more hydrophobic and easier to precipitate, indicated by the lower LCST at pH 10 than that in neutral water (Table 5). These results demonstrated the DMAEMA/styrene statistical copolymers with 89 and 96 mol% DMAEMA possessed similar pH sensitivity to that expected for PDMAEMA homopolymer.
3.2.5. Effect of Polymer Microstructure and Molecular Weight on LCST
DMAEMA has been incorporated into various block copolymers to obtain pH and/or temperature induced micelles in aqueous solutions because of the sensitivity to both pH and temperature of PDMAEMA [,,,]. In this study, a diblock copolymer (D/S80/20-b-D/S95/5) was synthesized by chain-extending a macroinitiator with 81 mol% DMAEMA (DMAEMA/S-80/20, Mn = 11.2 kg mol−1) with a fresh mixture of DMAEMA and styrene (fDMAEMA = 95 mol%, Mn,2nd block ≈ 16.6 kg mol−1, Mn relative to poly(styrene) standards, Table 4) and its phase behavior in water was studied. Since the two blocks have different hydrophilicities as demonstrated earlier, it was anticipated that micellization with a D/S80/20 core and D/S95/5 corona might occur at temperatures above the LCST of D/S80/20 and below the LCST of D/S95/5, followed by aggregation of entire polymer chains above the LCST of D/S95/5.
In Figure 10, the Rh of the block copolymer as a function of temperature was compared to its macroinitiator (DMAEMA/S-80/20, FDMAEMA = 81 mol%, Mn = 11.2 kg mol−1) and the statistical polymers with similar composition and molecular weight as the second block (DMAEMA/S-95/5, FDMAEMA = 96 mol%, Mn = 9.6 kg mol−1). The Rh of the block copolymer remained relatively constant at below 10 nm up to 23 °C with no obvious change at the LCST of the copolymer with FDMAEMA = 81 mol% at 20 °C. Above 23 °C, Rh increased quickly to about 100 nm and stayed constant even above the LCST of the copolymer with FDMAEMA = 96 mol% at 49 °C. These results showed a single step phase separation of the block copolymer similar to the observations of the statistical copolymers was occurring and microphase separation between the two blocks was not observed (i.e., two distinct LCST transitions).
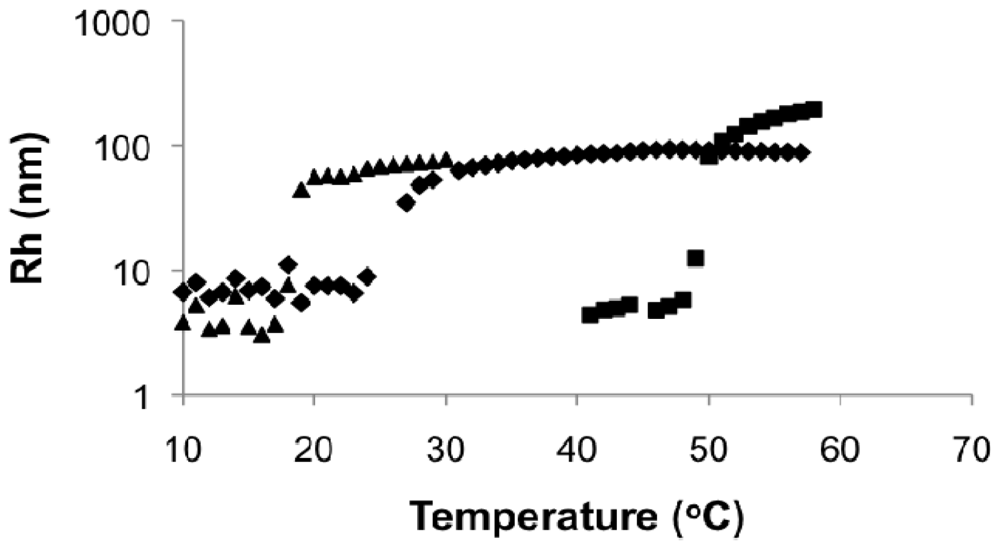
The reason for the absence of two LCSTs was most likely the lack of sufficient solubility difference between the two blocks in aqueous solution. The constituents of the two blocks were essentially the same except for the slightly different compositions. It is obvious in this case that the solubility parameters of the blocks were not sufficiently different to induce specific micellizations corresponding to each block. The slightly larger Rh below the LCST of the block copolymer was due to the higher molecular weight (Mn = 27.8 kg mol−1) of the block copolymer compared to the statistical copolymers (Mn = 9.6 and 11.2 kg mol−1). The higher LCST of the block copolymer compared to its DMAEMA/S-80/20 macroinitiator was a result of increased water solubility from the more DMAEMA-rich second block.
Since the D/S80/20-b-D/S95/5 block copolymer synthesized had an overall composition (FDMAEMA,overall) of 90 mol% DMAEMA, its phase behavior was also compared with the statistical copolymers with FDMAEMA ≈ 90 mol% that only differ by molecular weight (DMAEMA/S-95/5 and “block copolymer” D/S80/20-b-D/S-90/10, which was essentially as statistical copolymer since the chain extension was done with the same feed composition as the macroinitiator). As shown in Figure 11, the block copolymer had a considerably lower LCST compared to the statistical copolymers with similar overall composition. The two statistical copolymers with FDMAEMA ≈ 90 mol% had the same LCST despite the difference in Mn, indicating Mn did not influence the LCST of the statistical copolymers significantly in the range of 10–30 kg mol−1 and was not the cause for decreased LCST of the block copolymer.
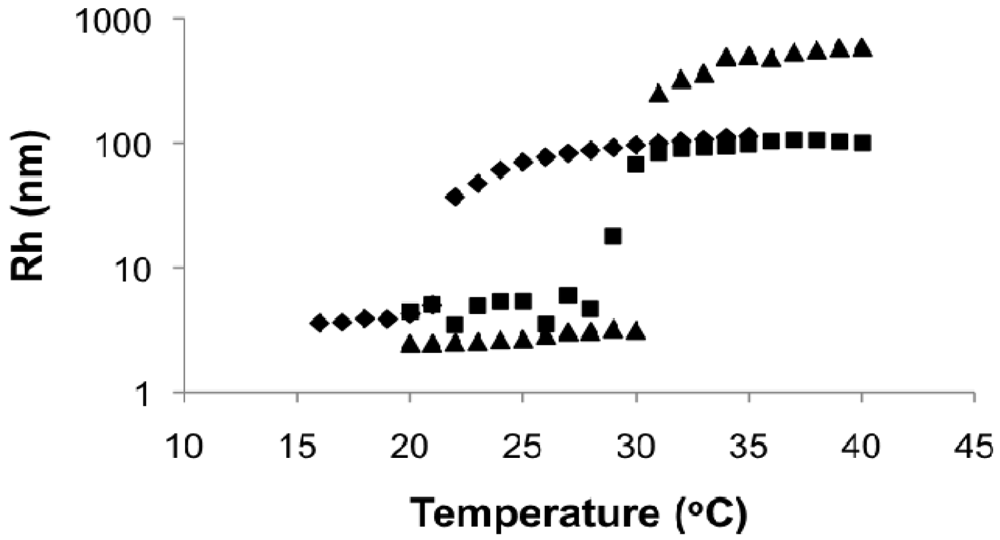
The change of LCST probably resulted from the differences in polymer microstructure. As reactivity ratios of methacrylates and styrene are generally similar and less than unity [], the distribution of DMAEMA and styrene in the statistical polymers should be relatively random. In the block copolymer, although distinctive microscopic phase separation was not observed, the LCST behavior became more complicated because DMAEMA was more concentrated in one block than the other.
Park et al. reported that gradient copolymers of poly(2-alkyl-2-oxazoline)s showed sharp transitions at their LCST and did not find any evidence of micelle formation [,]. Okabe et al. observed gradual phase separation of the gradient copolymer of 2-ethoxyethyl vinyl ether with 2-methoxyethyl vinyl ether, in contrast to the sharp transitions of the corresponding block copolymer []. Moreover, the gradient copolymer started phase separation just 3 °C higher than the LCST of the block copolymer. From Figure 10, the block copolymer D/S80/20-b-D/S95/5 had a slightly broader transition compared to the statistical copolymers. Additionally, the LCST of the block copolymer was lower than the statistical copolymers with similar overall composition and close to the micellization temperature (LCST of the D/S80/20 block) if there was one. These observations implied the phase behavior of D/S80/20-b-D/S95/5 resembled more of what would be expected of a gradient polymer. The exact mechanism of phase separation for such a copolymer was still not clear and should be examined in a more detailed study in the future.
4. Conclusions
In this study, DMAEMA was statistically copolymerized with 5–20 mol% styrene using NHS-BlocBuilder. Polymerization accelerated with increasing DMAEMA content in the feed as the average propagation rate constant <kp> and average equilibrium constant <K> increased. The linear increase of Mn with conversion and successful chain extensions demonstrated the polymerizations were controlled without aid of additional SG1 and the obtained copolymers were living and relatively narrow in molecular weight distribution (PDI = 1.32–1.59). Slight decreases in level of control and livingness were observed for the most DMAEMA-rich copolymer, which is likely attributed to the higher average equilibrium constant <K>. The copolymers exhibited LCST-type behavior in aqueous solutions, as expected. The LCST was tuned by varying copolymer composition, solution concentration and pH. The block copolymer of two statistical blocks with different DMAEMA compositions had a single LCST and did not exhibit microphase separation, likely due to the lack of sufficient incompatibility between the two blocks. This study marks the first time that DMAEMA could be polymerized with a small amount of comonomer by NMP and without any additional free nitroxide, due to the use of the NHS-BlocBuilder initiator. Such water-soluble DMAEMA-rich statistical copolymers and block copolymers could later be conjugated with various nucleophiles using the NHS-fragment from the initiator.
Acknowledgments
The authors are very grateful for the funding support from the NSERC Discovery Grant 288125 and Canada Foundation for Innovation New Opportunities Fund.
References
- Ahn, S.-K.; Kasi, R.M.; Kim, S.-C.; Sharma, N.; Zhou, Y. Stimuli-responsive polymer gels. Soft Matter 2008, 4, 1151–1157. [Google Scholar]
- Jeong, B.; Gutowska, A. Lessons from nature: Stimuli-responsive polymers and their biomedical applications. Trends Biotechnol. 2002, 20, 305–311. [Google Scholar]
- Alarcon, C.D.L.H.; Pennadam, S.; Alexander, C. Stimuli responsive polymers for biomedical applications. Chem. Soc. Rev. 2005, 34, 276–285. [Google Scholar]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Delivery Rev. 2006, 58, 1655–1670. [Google Scholar]
- Xu, F.-J.; Li, H.; Li, J.; Zhang, Z.; Kang, E.-T.; Neoh, K.-G. Pentablock copolymers of poly(ethylene glycol), poly((2-dimethyl amino)ethyl methacrylate) and poly(2-hydroxyethyl methacrylate) from consecutive atom transfer radical polymerizations for non-viral gene delivery. Biomaterials 2008, 29, 3023–3033. [Google Scholar]
- Hoffman, A.S. “Intelligent” polymers in medicine and biotechnology. Macromol. Symp. 1995, 98, 645–664. [Google Scholar]
- Lutz, J.-F. Polymerization of oligo(ethylene glycol) (meth)acrylates: Towards new generations of smart biocompatible materials. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 3459–3470. [Google Scholar]
- Gohy, J.-F.; Antoun, S.; Jerome, R. pH-Dependent micellization of poly(2-vinylpyridine)-block-poly((dimethylamino)ethyl methacrylate) diblock copolymers. Macromolecules 2001, 34, 7435–7440. [Google Scholar]
- Fournier, D.; Hoogenboom, R.; Thijs, H.M.L.; Paulus, R.M.; Schubert, U.S. Tunable pH- and Temperature-sensitive copolymer libraries by reversible addition-fragmentation chain transfer copolymerizations of methacrylates. Macromolecules 2007, 40, 915–920. [Google Scholar]
- Emileh, A.; Vasheghani-Farahani, E.; Imani, M. Swelling behavior, mechanical properties and network parameters of pH- and temperature-sensitive hydrogels of poly((2-dimethyl amino) ethyl methacrylate-co-butyl methacrylate). Eur. Polym. J. 2007, 43, 1986–1995. [Google Scholar]
- Feil, H.; Bae, Y.H.; Feijen, J.; Kim, S.W. Mutual influence of pH and temperature on the swelling of ionizable and thermosensitive hydrogels. Macromolecules 1992, 25, 5528–5530. [Google Scholar]
- Krasia, T.C.; Patrickios, C.S. Synthesis and aqueous solution characterization of amphiphilic diblock copolymers containing carbazole. Polymer 2002, 43, 2917–2920. [Google Scholar]
- Matyjaszewski, K. Controlled radical polymerization. Curr. Opin. Solid State Mater. Sci. 1996, 1, 769–776. [Google Scholar]
- Zhang, X.; Xia, J.; Matyjaszewski, K. Controlled/“Living” radical polymerization of 2-(dimethylamino)ethyl methacrylate. Macromolecules 1998, 31, 5167–5169. [Google Scholar]
- Lee, S.B.; Russell, A.J.; Matyjaszewski, K. ATRP synthesis of amphiphilic random, gradient, and block copolymers of 2-(dimethylamino)ethyl methacrylate and n-butyl methacrylate in aqueous media. Biomacromolecules 2003, 4, 1386–1393. [Google Scholar]
- Monge, S.; Darcos, V.; Haddleton, D.M. Effect of DMSO used as solvent in copper mediated living radical polymerization. J. Polym. Sci. Part A: Polym. Chem. 2004, 42, 6299–6308. [Google Scholar]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; Rizzardo, E.; Thang, S.H. Living free-radical polymerization by reversible addition-fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar]
- Perrier, S.; Takolpuckdee, P. Macromolecular design via reversible addition-fragmentation chain transfer (RAFT)/xanthates (MADIX) polymerization. J. Polym. Sci. Part A: Polym. Chem. 2005, 43, 5347–5393. [Google Scholar]
- Chenal, M.; Boursier, C.; Guillaneuf, Y.; Taverna, M.; Couvreur, P.; Nicolas, J. First peptide/protein PEGylation with functional polymers designed by nitroxide-mediated polymerization. Polym. Chem. 2011, 2, 1523–1530. [Google Scholar]
- Nicolas, J.; Couvreur, P.; Charleux, B. Comblike polymethacrylates with poly(ethylene glycol) side chains via nitroxide-mediated controlled free-radical polymerization. Macromolecules 2008, 41, 3758–3761. [Google Scholar]
- Grubbs, R.B. Nitroxide-mediated radical polymerization: Limitations and versatility. Polym. Rev. 2011, 51, 104–137. [Google Scholar]
- Ananchenko, G.S.; Souaille, M.; Fischer, H.; LeMercier, C.; Tordo, P. Decomposition of model alkoxyamines in simple and polymerizing systems. II. Diastereomeric N-(2-methylpropyl)-N-(1-diethyl-phosphono-2,2-dimethyl-propyl)-aminoxyl-based compounds. J. Polym. Sci. A: Polym. Chem. 2002, 40, 3264–3283. [Google Scholar]
- Marestin, C.; Noel, C.; Guyot, A.; Claverie, J. Nitroxide mediated living radical polymerization of styrene in emulsion. Macromolecules 1998, 31, 4041–4044. [Google Scholar]
- Gabaston, L.I.; Jackson, R.A.; Armes, S.P. Living free-radical dispersion polymerization of styrene. Macromolecules 1998, 31, 2883–2888. [Google Scholar]
- Alam, M.N.; Zetterlund, P.B.; Okubo, M. TEMPO-mediated radical polymerization of styrene in aqueous miniemulsion: Macroinitiator concentration effects. Polymer 2008, 49, 3428–3435. [Google Scholar]
- Guillaneuf, Y.; Gigmes, D.; Marque, S.R.A.; Astolfi, P.; Greci, L.; Tordo, P.; Bertin, D. First effective nitroxide-mediated polymerization of methyl methacrylate. Macromolecules 2007, 40, 3108–3114. [Google Scholar]
- Greene, A.C.; Grubbs, R.B. Nitroxide-mediated polymerization of methyl methacrylate and styrene with new alkoxyamines from 4-nitrophenyl 2-methylpropionat-2-yl radicals. Macromolecules 2010, 43, 10320–10325. [Google Scholar]
- Charleux, B.; Nicolas, J.; Guerret, O. Theoretical expression of the average activation-deactivation equilibrium constant in controlled/living free-radical copolymerization operating via reversible termination. Application to a strongly improved control in nitroxide-mediated polymerization of methyl methacrylate. Macromolecules 2005, 38, 5485–5492. [Google Scholar]
- Nicolas, J.; Dire, C.; Mueller, L.; Belleney, J.; Charleux, B.; Marque, S.R.A.; Bertin, D.; Magnet, S.; Couvreur, L. Living character of polymer chains prepared via nitroxide-mediated controlled free-radical polymerization of methyl methacrylate in the presence of a small amount of styrene at low temperature. Macromolecules 2006, 39, 8274–8282. [Google Scholar]
- Lessard, B.; Maric, M. Incorporating glycidyl methacrylate into block copolymers using poly(methacrylate-ran-styrene) macroinitiators synthesized by nitroxide-mediated polymerization. J. Polym. Sci. Part A: Polym. Chem. 2009, 47, 2574–2588. [Google Scholar]
- Zhang, C.; Lessard, B.; Maric, M. Synthesis and characterization of benzyl methacrylate/styrene random copolymers prepared by NMP. Macromol. React. Eng. 2010, 4, 415–423. [Google Scholar]
- Beuermann, S.; Buback, M. Rate coefficients of free-radical polymerization deduced from pulsed laser experiments. Prog. Polym.Sci. 2002, 27, 191–254. [Google Scholar]
- Benoit, D.; Grimaldi, S.; Robin, S.; Finet, JP.; Tordo, P.; Gnanou, Y. Kinetics and mechanism of controlled free-radical polymerization of styrene and n-butyl acrylate in the presence of an acyclic β-phosphonylated nitroxide. J.Am.Chem. Soc. 2000, 122, 5929–5939. [Google Scholar]
- Anderson, G.W.; Zimmerman, J.E.; Callahan, F.M. The use of esters of N-hydroxysuccinimide in peptide synthesis. J. Am. Chem. Soc. 1964, 86, 1839–1842. [Google Scholar]
- Lecolley, F.; Tao, L.; Mantovani, G.; Durkin, I.; Lautru, S.; Haddleton, D.M. A new approach to bioconjugates for proteins and peptides (“pegylation”) utilising living radical polymerisation. Chem. Commun. 2004, 2026–2027. [Google Scholar]
- Bathfield, M.; D'Agosto, F.; Spitz, R.; Charreyre, M.-T.; Delair, T. Versatile precursors of functional RAFT agents. Application to the synthesis of bio-related end-functionalized polymers. J. Am. Chem. Soc. 2006, 128, 2546–2547. [Google Scholar]
- Vinas, J.; Chagneux, N.; Gigmes, D.; Trimaille, T.; Favier, A.; Bertin, D. SG1-based alkoxyamine bearing a N-succinimidyl ester: A versatile tool for advanced polymer synthesis. Polymer 2008, 49, 3639–3647. [Google Scholar]
- Moayeri, A.; Lessard, B.; Maric, M. Nitroxide mediated controlled synthesis of glycidyl methacrylate-rich copolymers enabled by SG1-based alkoxyamines bearing succinimidyl ester groups. Polym. Chem. 2011, 2, 2084–2092. [Google Scholar]
- Koppel, D.E. Analysis of macromolecular polydispersity in intensity correlation spectroscopy: The method of cumulants. J. Chem. Phys. 1972, 57, 4814–4820. [Google Scholar]
- Creutz, S.; Teyssié, P.; Jérôme, R. Living anionic homopolymerization and block copolymerization of (dimethylamino)ethyl methacrylate. Macromolecules 1997, 30, 6–9. [Google Scholar]
- Lokaj, J.; Vlcek, P.; Kriz, J. Synthesis of polystyrene-poly(2-(dimethylamino)ethyl methacrylate) block copolymers by stable free-radical polymerization. Macromolecules 1997, 30, 7644–7646. [Google Scholar]
- Zhang, X.; Matyjaszewski, K. Synthesis of well-defined amphiphilic block copolymers with 2-(dimethylamino)ethyl methacrylate by controlled radical polymerization. Macromolecules 1999, 32, 1763–1766. [Google Scholar]
- Burguiere, C.; Dourges, M.-A.; Charleux, B.; Vairon, J.-P. Synthesis and characterization of ω-unsaturated poly(styrene-b-n-butyl methacrylate) block copolymers using TEMPO-mediated controlled radical polymerization. Macromolecules 1999, 32, 3883–3890. [Google Scholar]
- Moad, G.; Anderson Albert, G.; Ercole, F.; Johnson Charles, H.J.; Krstina, J.; Moad Catherine, L.; Rizzardo, E.; Spurling Thomas, H.; Thang San, H. Controlled-growth free-radical polymerization of methacrylate esters: Reversible chain transfer versus reversible termination. ACS Symp. Ser. 1998, 685, 332–360. [Google Scholar]
- Marque, S.; Le Mercier, C.; Tordo, P.; Fischer, H. Factors influencing the C-O-bond homolysis of trialkylhydroxylamines. Macromolecules 2000, 33, 4403–4410. [Google Scholar]
- Dire, C.; Belleney, J.; Nicolas, J.; Bertin, D.; Magnet, S.; Charleux, B. beta-Hydrogen transfer from poly(methyl methacrylate) propagating radicals to the nitroxide SG1: Analysis of the chain-end and determination of the rate constant. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 6333–6345. [Google Scholar]
- Lessard, B.; Graffe, A.; Maric, M. Styrene/tert-butyl acrylate random copolymers synthesized by nitroxide-mediated polymerization: Effect of free nitroxide on kinetics and copolymer composition. Macromolecules 2007, 40, 9284–9292. [Google Scholar]
- Couvreur, L.; Lefay, C.; Belleney, J.; Charleux, B.; Guerret, O.; Magnet, S. First nitroxide-mediated controlled free-radical polymerization of acrylic acid. Macromolecules 2003, 36, 8260–8267. [Google Scholar]
- Lessard, B.; Maric, M. Nitroxide-mediated synthesis of poly(poly(ethylene glycol) acrylate) (PPEGA) comb-like homopolymers and block copolymers. Macromolecules 2008, 41, 7870–7880. [Google Scholar]
- Liu, Q.; Yu, Z.; Ni, P. Micellization and applications of narrow-distribution poly[2-(dimethylamino)ethyl methacrylate]. Colloid Polym. Sci. 2004, 282, 387–393. [Google Scholar]
- Bütün, V.; Armes, S.P.; Billingham, N.C. Synthesis and aqueous solution properties of near-monodisperse tertiary amine methacrylate homopolymers and diblock copolymers. Polymer 2001, 42, 5993–6008. [Google Scholar]
- Lutz, J.-F.; Hoth, A. Preparation of ideal PEG analogues with a tunable thermosensitivity by controlled radical copolymerization of 2-(2-methoxyethoxy)ethyl methacrylate and Oligo(ethylene glycol) methacrylate. Macromolecules 2006, 39, 893–896. [Google Scholar]
- Becer, C.R.; Kokado, K.; Weber, C.; Can, A.; Chujo, Y.; Schubert, U.S. Metal-free synthesis of responsive polymers: Cloud point tuning by controlled “click” reaction. J. Polym. Sci. Part A: Polym. Chem. 2010, 48, 1278–1286. [Google Scholar]
- Okabe, S.; Seno, K.-I.; Kanaoka, S.; Aoshima, S.; Shibayama, M. Micellization study on block and gradient copolymer aqueous solutions by DLS and SANS. Macromolecules 2006, 39, 1592–1597. [Google Scholar]
- Huang, J.; Matyjaszewski, K. Atom transfer radical polymerization of dimethyl(1-ethoxycarbonyl)vinyl phosphate and corresponding block copolymers. Macromolecules 2005, 38, 3577–3583. [Google Scholar]
- Hua, F.; Jiang, X.; Li, D.; Zhao, B. Well-defined thermosensitive, water-soluble polyacrylates and polystyrenics with short pendant oligo(ethylene glycol) groups synthesized by nitroxide-mediated radical polymerization. J. Polym. Sci. A: Polym. Chem. 2006, 44, 2454–2467. [Google Scholar]
- Ueki, T.; Arai, A.A.; Kodama, K.; Kaino, S.; Takada, N.; Morita, T.; Nishikawa, K.; Watanabe, M. Thermodynamic study on phase transitions of poly(benzyl methacrylate) in ionic liquid solvents. Pure Appl. Chem. 2009, 81, 1829–1841. [Google Scholar]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar]
- Winnik, F.M. Cononsolvency of poly(N-isopropylacrylamide) in mixed water-methanol solutions. Macromolecules 1992, 25, 6007–6017. [Google Scholar]
- Taylor, L.D.; Cerankowski, L.D. Preparation of films exhibiting a balanced temperature dependence to permeation by aqueous solutions—a study of lower consolute behavior. J. Polym. Sci.: Polym. Chem. Ed. 1975, 13, 2551–2570. [Google Scholar]
- Bütün, V.; Armes, S.P.; Billingham, N.C. Synthesis and aqueous solution properties of near-monodisperse tertiary amine methacrylate homopolymers and diblock copolymers. Polymer 2001, 42, 5993–6008. [Google Scholar]
- Braun, D.; Czerwinski, W.; Disselhoff, G.; Tüdős, F.; Kelen, T.; Turcsányi, B. Analysis of the linear methods for determining copolymerization reactivity ratios, VII. A critical reexamination of radical copolymerizations of styrene. Die Angew. Makromol. Chem. 1984, 125, 161–205. [Google Scholar]
- Park, J.-S.; Kataoka, K. Precise control of lower critical solution temperature of thermosensitive poly(2-isopropyl-2-oxazoline) via gradient copolymerization with 2-ethyl-2-oxazoline as a hydrophilic comonomer. Macromolecules 2006, 39, 6622–6630. [Google Scholar]
- Park, J.-S.; Kataoka, K. Comprehensive and accurate control of thermosensitivity of poly(2-alkyl-2-oxazoline)s via well-defined gradient or random copolymerization. Macromolecules 2007, 40, 3599–3609. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).