
Application of Computational Studies Using Density Functional Theory (DFT) to Evaluate the Catalytic Degradation of Polystyrene

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Computer Simulations
2.2. Catalysts
2.3. Acid and Base Degradation Mechanisms
3. Results and Discussion
3.1. Mechanism of Product Formation in Alkaline and Acidic Conditions
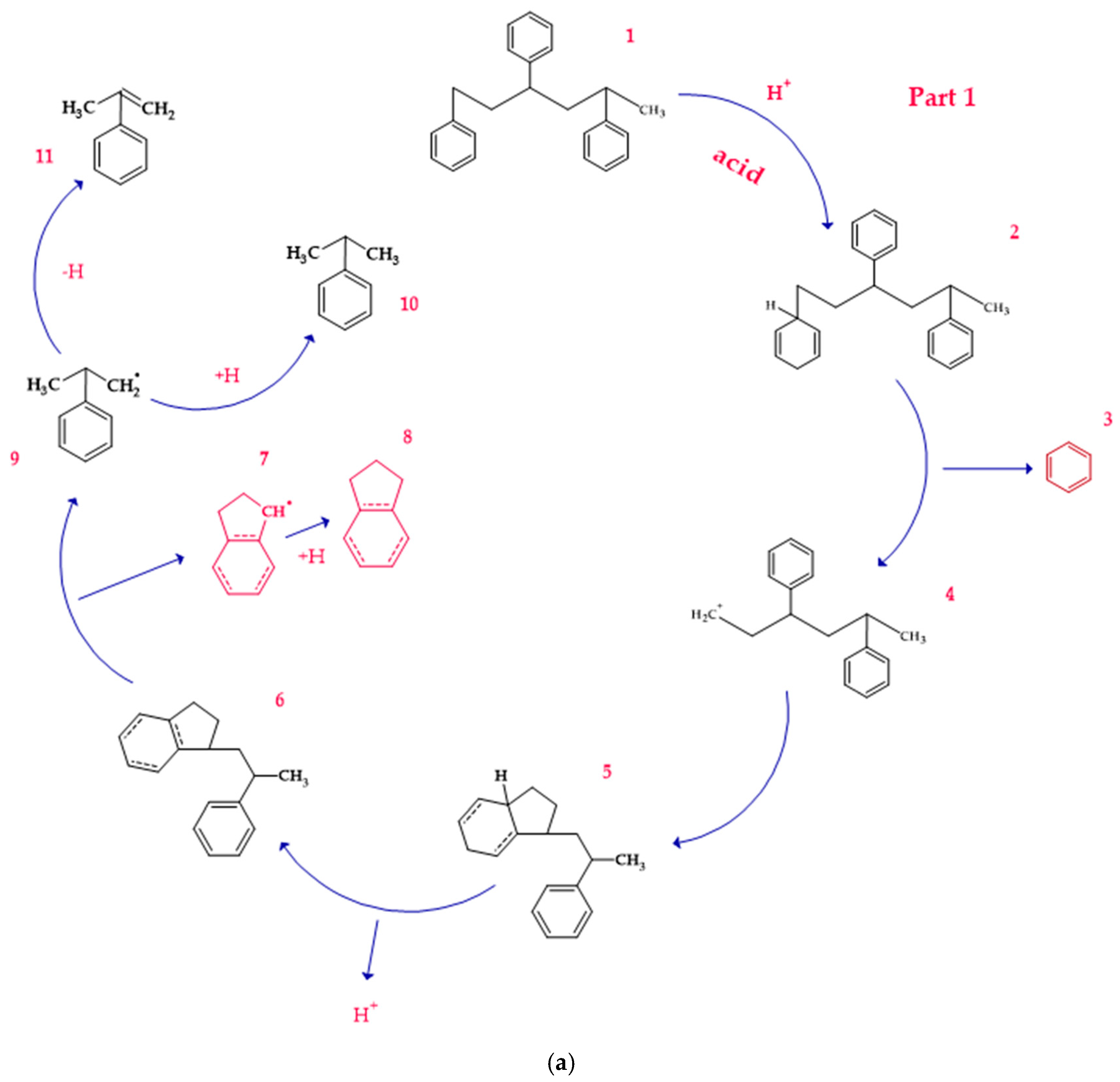
3.1.1. Degradation in the Presence of Acid Catalysts
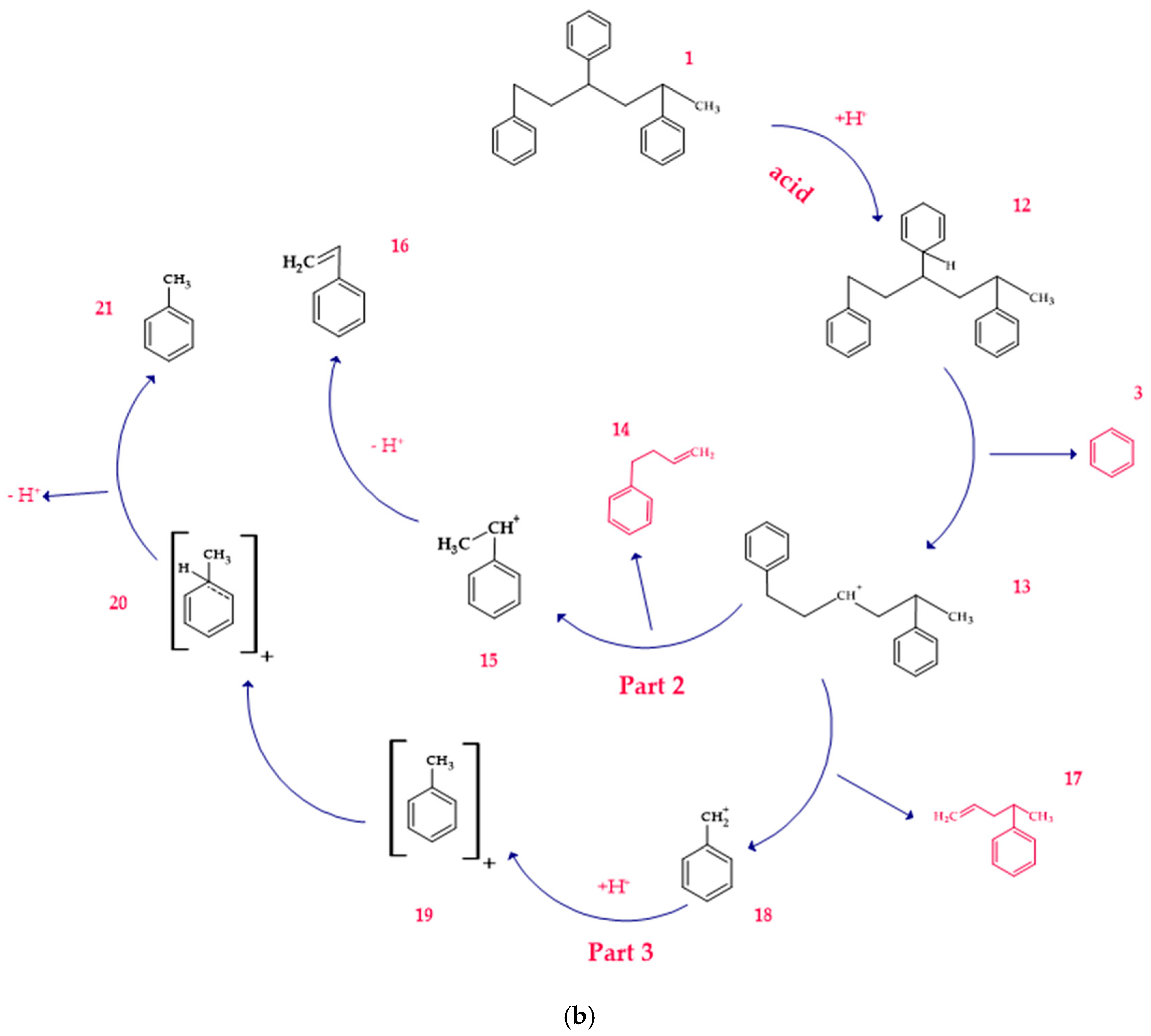
3.1.2. Degradation in the Presence of Basic Catalysts
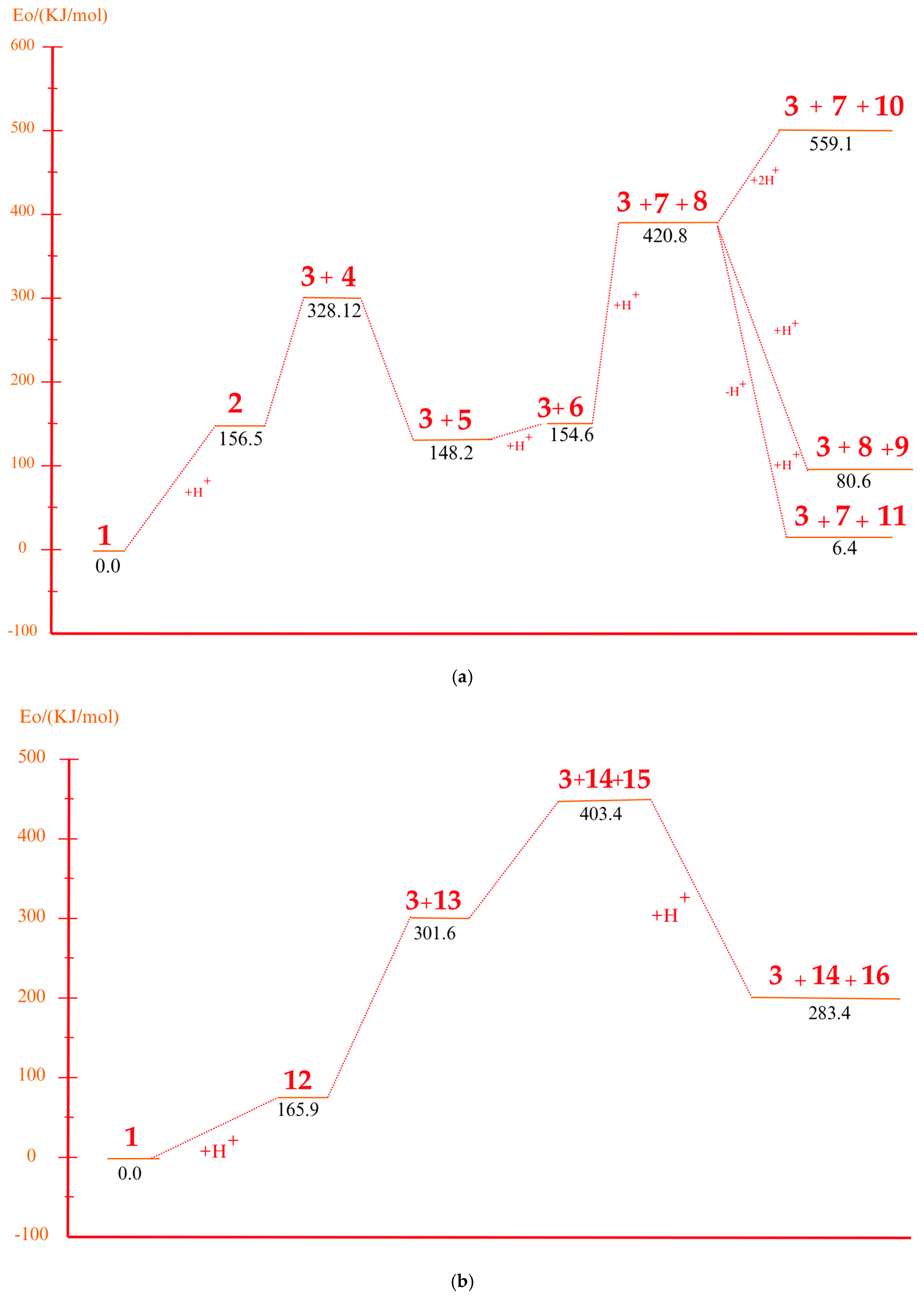
3.2. Bond Dissociation Energy
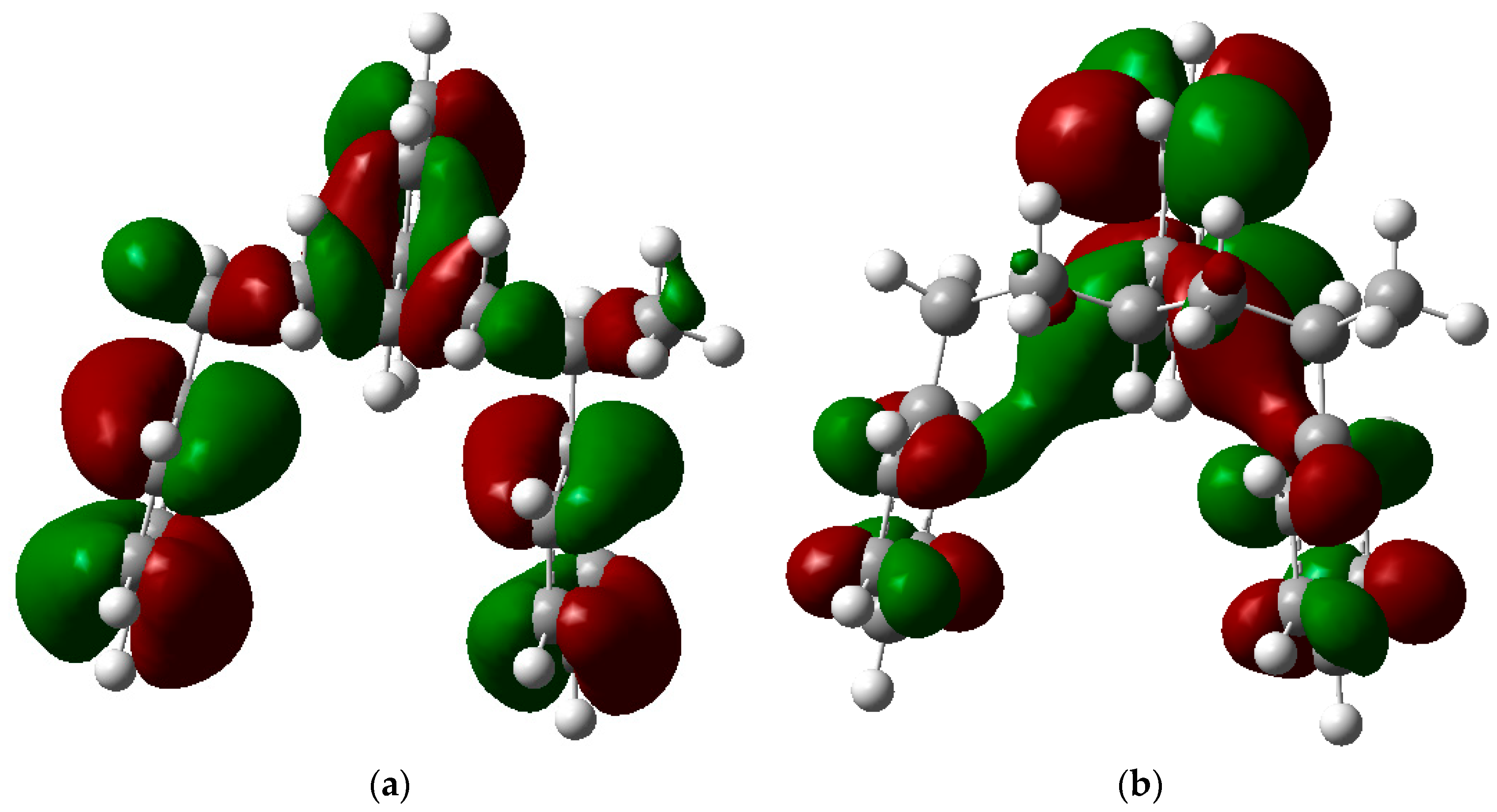
3.3. HOMO-LUMO
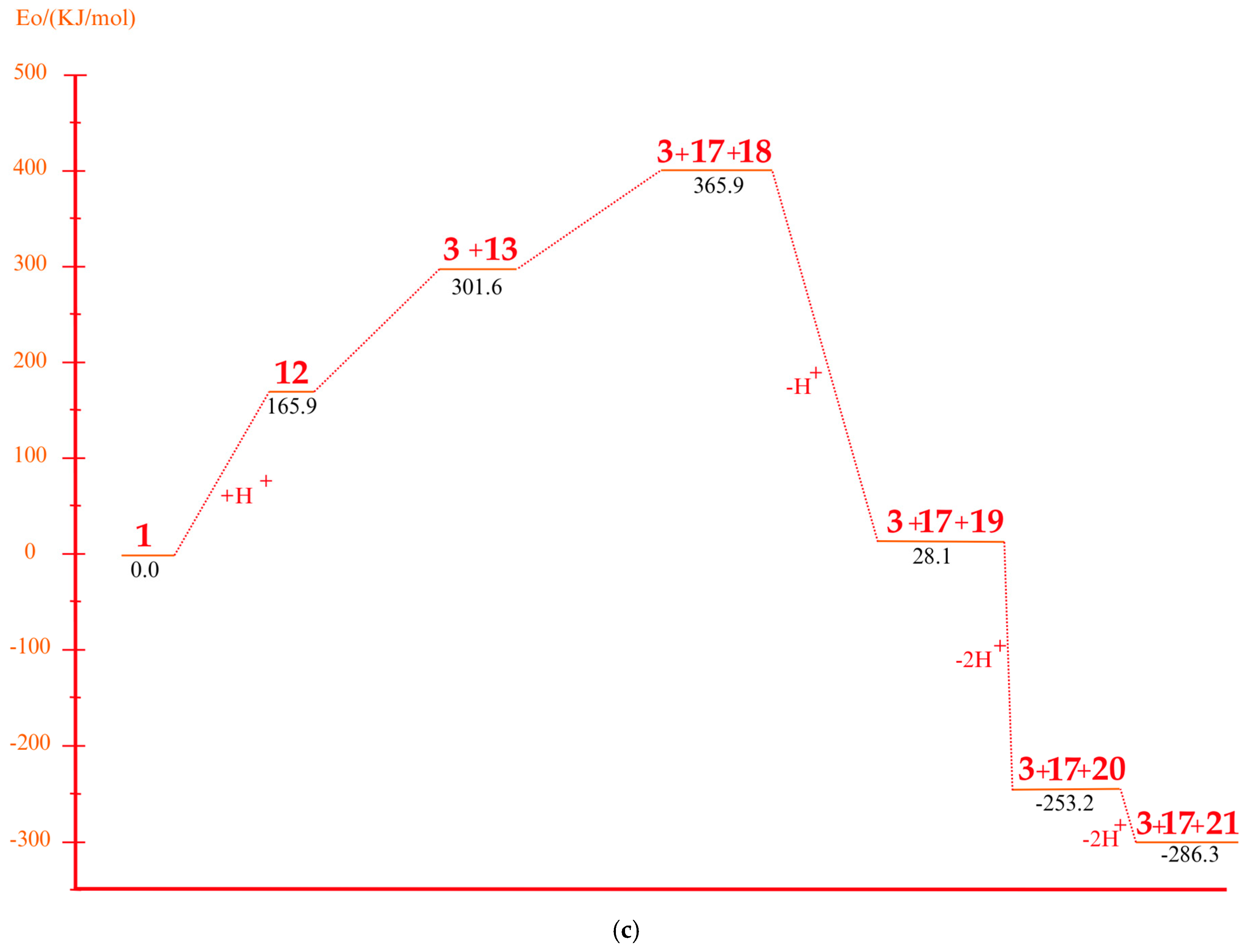
3.4. Mechanism of Product Formation Under Acidic Conditions
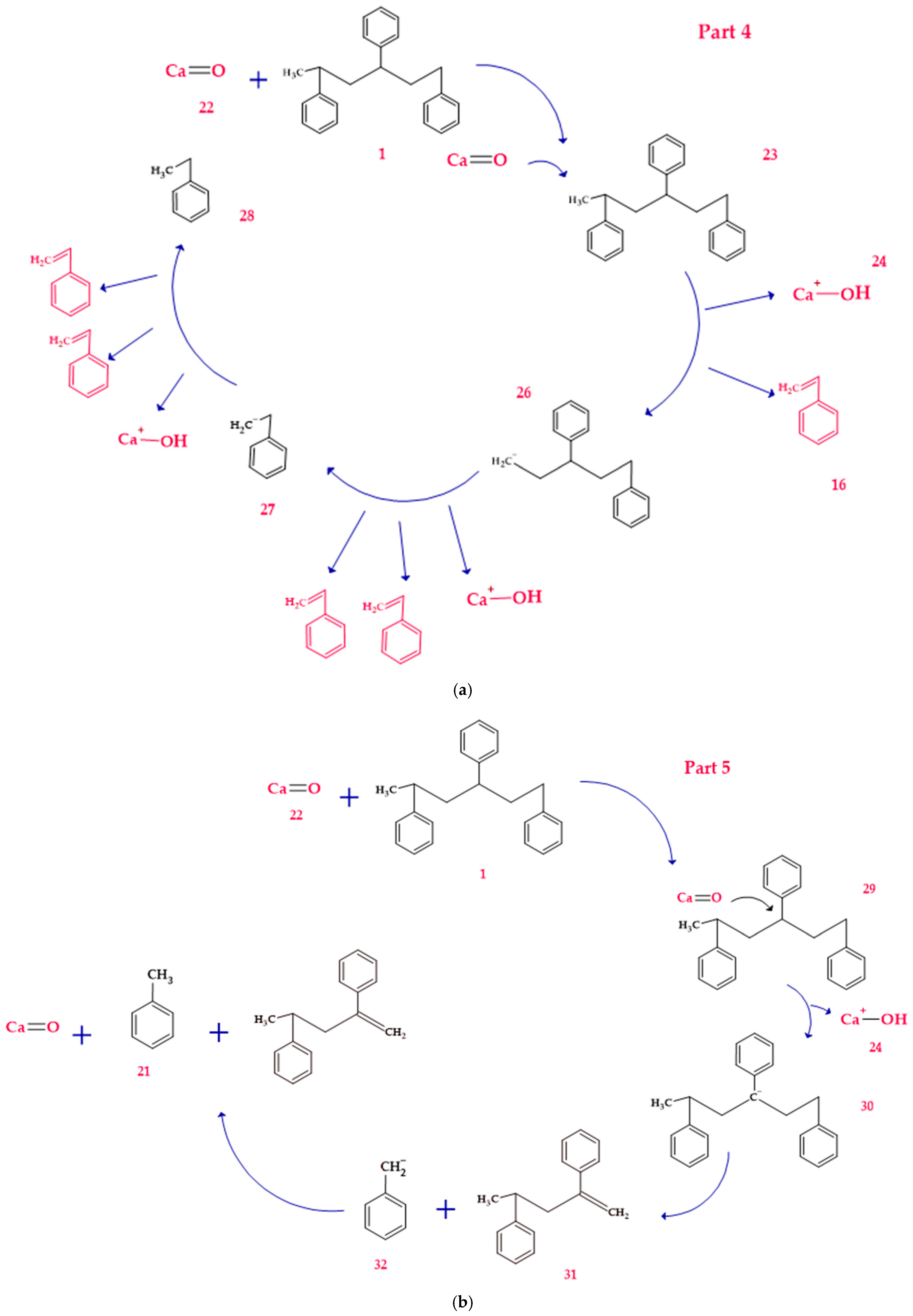
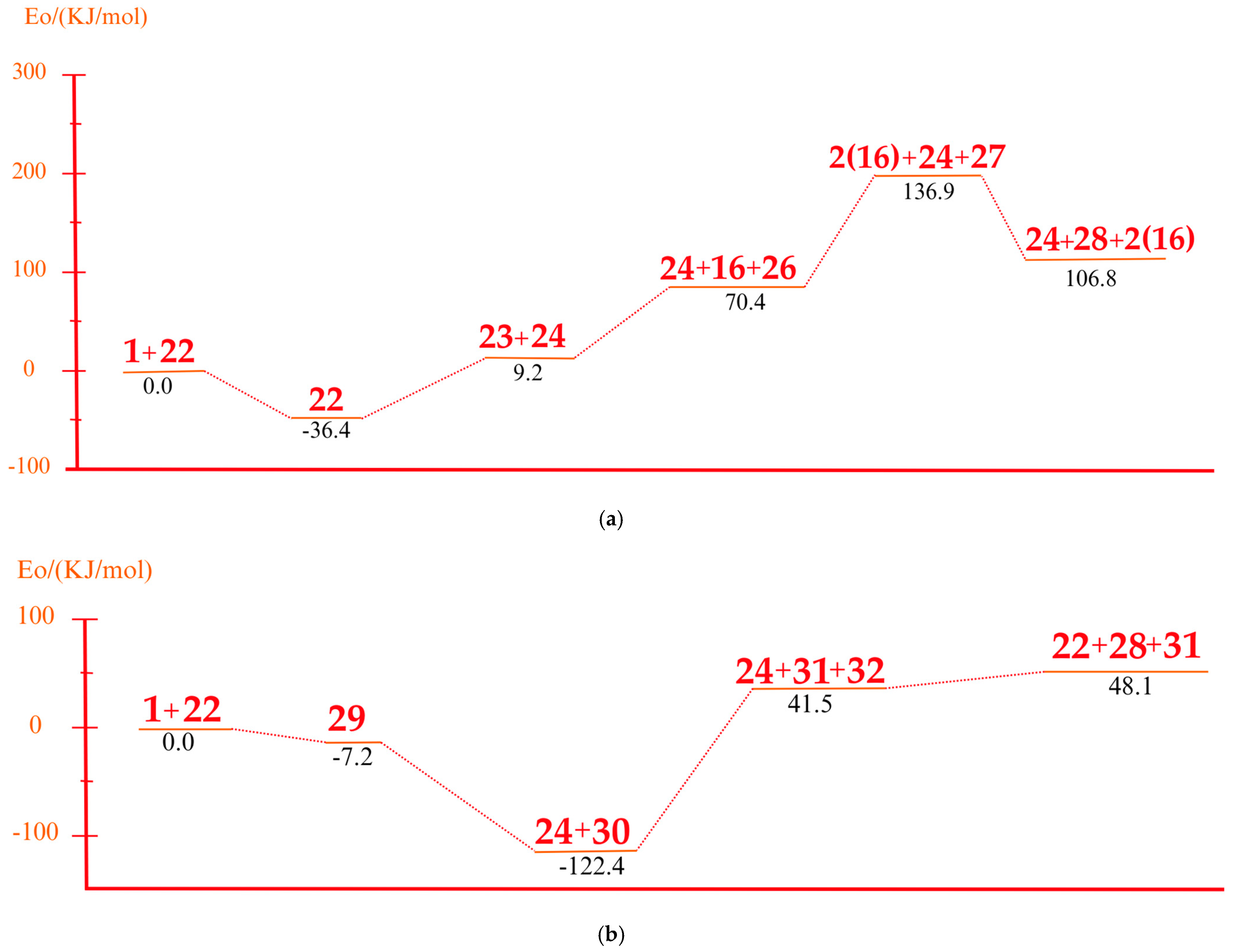
3.5. Mechanism of Product Formation Under Alkaline Conditions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef]
- Lopez, G.; Artetxe, M.; Amutio, M.; Bilbao, J.; Olazar, M. Thermochemical routes for the valorization of waste polyolefinic plastics to produce fuels and chemicals. A review. Renew. Sustain. Energy Rev. 2017, 73, 346–368. [Google Scholar]
- Ogunola, O.S.; Onada, O.A.; Falaye, A.E. Mitigation measures to avert the impacts of plastics and microplastics in the marine environment—A review. Environ. Sci. Pollut. Res. 2018, 25, 9293–9310. [Google Scholar]
- Plastics Europe. Plastics—The Facts 2012. 2021. Available online: https://plasticseurope.org/wp-content/uploads/2021/12/Plastics-the-Facts-2021-web-final.pdf (accessed on 20 January 2025).
- Al-Salem, S.M.; Lettieri, P.; Baeyens, J. The valorization of plastic solid waste (PSW) by primary to quaternary routes: From re-use to energy and chemicals. Prog. Energy Combust. Sci. 2010, 36, 103–129. [Google Scholar] [CrossRef]
- Marczewski, M.; Kamińska, E.; Marczewska, H.; Godek, M.; Rokicki, G.; Sokołowski, J. Catalytic decomposition of polystyrene. The role of acid and basic active centers. Appl. Catal. B Environ. 2013, 129, 236–246. [Google Scholar]
- US EPA. Advancing Sustainable Materials Management: Facts and Figures Report. 2024. Available online: https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/advancing-sustainable-materials-management (accessed on 20 January 2025).
- Ali, S.S.; Elsamahy, T.; Koutra, E.; Kornaros, M.; El-Sheekh, M.; Abdelkarim, E.A.; Zhu, D.; Sun, J. Degradation of conventional plastic wastes in the environment: A review on current status of knowledge and future perspectives of disposal. Sci. Total Environ. 2021, 771, 144719. [Google Scholar] [PubMed]
- Hwang, G.-C.; Choi, J.-H.; Bae, S.-Y.; Kumazawa, H. Degradation of polystyrene in supercritical n-hexane. Korean J. Chem. Eng. 2001, 18, 854–861. [Google Scholar]
- Vilaplana, F.; Ribes-Greus, A.; Karlsson, S. Degradation of recycled high-impact polystyrene. Simulation by reprocessing and thermo-oxidation. Polym. Degrad. Stab. 2006, 91, 2163–2170. [Google Scholar] [CrossRef]
- Gewert, B.; Plassmann, M.M.; MacLeod, M. Pathways for degradation of plastic polymers floating in the marine environment. Environ. Sci. Process. Impacts 2015, 17, 1513–1521. [Google Scholar] [PubMed]
- Fotopoulou, K.N.; Karapanagioti, H.K. Degradation of various plastics in the environment. In Hazardous Chemicals Associated with Plastics in the Marine Environment; Springer: Berlin/Heidelberg, Germany, 2019; pp. 71–92. [Google Scholar] [CrossRef]
- Chen, H.; Wan, K.; Zhang, Y.; Wang, Y. Waste to wealth: Chemical recycling and chemical upcycling of waste plastics for a great future. ChemSusChem 2021, 14, 4123–4136. [Google Scholar] [CrossRef]
- Huang, J.; Cheng, X.; Meng, H.; Pan, G.; Wang, S.; Wang, D. Density functional theory study on the catalytic degradation mechanism of polystyrene. AIP Adv. 2020, 10, 085004. [Google Scholar]
- Zhou, J.; Hsu, T.-G.; Wang, J. Mechanochemical degradation and recycling of synthetic polymers. Angew. Chem. 2023, 135, e202300768. [Google Scholar] [CrossRef]
- Olivieri, F.; Caputo, A.; Leonetti, D.; Castaldo, R.; Avolio, R.; Cocca, M.; Errico, M.E.; Iannotta, L.; Avella, M.; Carfagna, C.; et al. Compositional Analysis and Mechanical Recycling of Polymer Fractions Recovered via the Industrial Sorting of Post-Consumer Plastic Waste: A Case Study toward the Implementation of Artificial Intelligence Databases. Polymers 2024, 16, 2898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, C.; Jiang, H. Thermal degradation mechanism of polystyrene by TG-MS. Chem. Ind. Eng. Prog. 2008, 27, 609. [Google Scholar]
- Maafa, I.M. Pyrolysis of polystyrene waste: A review. Polymers 2021, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Awodi, E.; Adewumi, K.C. Exploring the Aesthetic Applications of Expanded Polystyrene: An Interdisciplinary Review. Afr. J. Inter/Multidiscip. Stud. 2024, 6, 1–15. [Google Scholar]
- Zhuo, Y.; He, J.; Li, W.; Deng, J.; Lin, Q. A review on takeaway packaging waste: Types, ecological impact, and disposal route. Environ. Pollut. 2023, 337, 122518. [Google Scholar] [CrossRef]
- Gautam, B.; Tsai, T.-H.; Chen, J.-T. Towards Sustainable Solutions: A Review of Polystyrene Upcycling and Degradation Techniques. Polym. Degrad. Stab. 2024, 225, 110779. [Google Scholar] [CrossRef]
- Drevin, I.; Johansson, B.-L.; Son, E.L. Pyrolysis in biotechnology. Biotechnol. Genet. Eng. Rev. 2001, 18, 3–28. [Google Scholar] [CrossRef]
- Miandad, R.; Nizami, A.S.; Rehan, M.; Barakat, M.A.; Khan, M.I.; Mustafa, A.; Ismail, I.M.I.; Murphy, J.D. Influence of temperature and reaction time on the conversion of polystyrene waste to pyrolysis liquid oil. Waste Manag. 2016, 58, 250–259. [Google Scholar]
- Onwudili, J.A.; Insura, N.; Williams, P.T. Composition of products from the pyrolysis of polyethylene and polystyrene in a closed batch reactor: Effects of temperature and residence time. J. Anal. Appl. Pyrolysis 2009, 86, 293–303. [Google Scholar] [CrossRef]
- Marquez, C.; Martin, C.; Linares, N.; De Vos, D. Catalytic routes towards polystyrene recycling. Mater. Horiz. 2023, 10, 1625–1640. [Google Scholar] [CrossRef] [PubMed]
- Sant, M.O.; Pedrosa, A.M.G.; de Souza, M.J.B. Study of the Conversion of Postconsumer Polystyrene on CeO2/HZSM-5 Type Materials. J. Mater. Sci. Chem. Eng. 2024, 12, 29–41. [Google Scholar]
- Lu, C.; Xiao, H.; Chen, X. Simple pyrolysis of polystyrene into valuable chemicals. e-Polymers 2021, 21, 428–432. [Google Scholar]
- Miandad, R.; Barakat, M.A.; Rehan, M.; Aburiazaiza, A.S.; Ismail, I.M.I.; Nizami, A.S. Plastic waste to liquid oil through catalytic pyrolysis using natural and synthetic zeolite catalysts. Waste Manag. 2017, 69, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Miandad, R.; Rehan, M.; Barakat, M.A.; Aburiazaiza, A.S.; Khan, H.; Ismail, I.M.I.; Dhavamani, J.; Gardy, J.; Hassanpour, A.; Nizami, A.-S. Catalytic pyrolysis of plastic waste: Moving toward pyrolysis based biorefineries. Front. Energy Res. 2019, 7, 437000. [Google Scholar]
- Razzaq, M.; Zeeshan, M.; Qaisar, S.; Iftikhar, H.; Muneer, B. Investigating use of metal-modified HZSM-5 catalyst to upgrade liquid yield in co-pyrolysis of wheat straw and polystyrene. Fuel 2019, 257, 116119. [Google Scholar]
- Li, C.; Yang, Z.; Wu, X.; Shao, S.; Meng, X.; Qin, G. Reactive Molecular Dynamics Simulations of Polystyrene Pyrolysis. Int. J. Mol. Sci. 2023, 24, 16403. [Google Scholar] [CrossRef]
- Abi Bianasari, A.; Khaled, M.S.; Hoang, T.-D.; Reza, M.S.; Bakar, M.S.A.; Azad, A.K. Influence of combined catalysts on the catalytic pyrolysis process of biomass: A systematic literature review. Energy Convers. Manag. 2024, 309, 118437. [Google Scholar]
- Shah, J.; Jan, M.R.; Adnan. Conversion of waste polystyrene through catalytic degradation into valuable products. Korean J. Chem. Eng. 2014, 31, 1389–1398. [Google Scholar]
- Liu, Y.; Qian, J.; Wang, J. Pyrolysis of polystyrene waste in a fluidized-bed reactor to obtain styrene monomer and gasoline fraction. Fuel Process. Technol. 2000, 63, 45–55. [Google Scholar] [CrossRef]
- Huang, J.; He, C.; Pan, G.; Tong, H. A theoretical research on pyrolysis reactions mechanism of coumarone-contained lignin model compound. Comput. Theor. Chem. 2016, 1091, 92–98. [Google Scholar] [CrossRef]
- Huang, J.; He, C.; Li, X.; Pan, G.; Tong, H. Theoretical studies on thermal degradation reaction mechanism of model compound of bisphenol A polycarbonate. Waste Manag. 2018, 71, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; He, C.; Wu, L.; Tong, H. Theoretical studies on thermal decomposition mechanism of arabinofuranose. J. Energy Inst. 2017, 90, 372–381. [Google Scholar] [CrossRef]
- Huang, J.; He, C.; Wu, L.; Tong, H. Thermal degradation reaction mechanism of xylose: A DFT study. Chem. Phys. Lett. 2016, 658, 114–124. [Google Scholar] [CrossRef]
- Huang, J.; Li, X.; Zeng, G.; Cheng, X.; Tong, H.; Wang, D. Thermal decomposition mechanisms of poly (vinyl chloride): A computational study. Waste Manag. 2018, 76, 483–496. [Google Scholar] [CrossRef]
- Younker, J.M.; Beste, A.; Buchanan Iii, A.C. Computational study of bond dissociation enthalpies for lignin model compounds: $β$-5 arylcoumaran. Chem. Phys. Lett. 2012, 545, 100–106. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Chen, X. Unveiling the initial pyrolytic mechanisms of cellulose by DFT study. J. Anal. Appl. Pyrolysis 2015, 113, 621–629. [Google Scholar] [CrossRef]
- Li, R.; Zhang, Z.; Liang, X.; Shen, J.; Wang, J.; Sun, W.; Wang, D.; Jiang, J.; Li, Y. Polystyrene waste thermochemical hydrogenation to ethylbenzene by a N-bridged Co, Ni dual-atom catalyst. J. Am. Chem. Soc. 2023, 145, 16218–16227. [Google Scholar] [CrossRef]
- Xuan, W.; Yan, S.; Dong, Y. Exploration of Pyrolysis Behaviors of Waste Plastics (Polypropylene Plastic/Polyethylene Plastic/Polystyrene Plastic): Macro-Thermal Kinetics and Micro-Pyrolysis Mechanism. Processes 2023, 11, 2764. [Google Scholar] [CrossRef]
- da Mata Costa, L.P.; Brandão, A.L.T.; Pinto, J.C. Modeling of polystyrene degradation using kinetic Monte Carlo. J. Anal. Appl. Pyrolysis 2022, 167, 105683. [Google Scholar]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuß, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Giroday, T.; Montero-Campillo, M.M.; Mora-Diez, N. Thermodynamic stability of PFOS: M06-2X and B3LYP compari-son. Comput. Theor. Chem. 2014, 1046, 81–92. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E. Revisión, B. 01, Gaussiano.Inc., Wallingford CT2018. Available online: https://www.researchgate.net/publication/220020584_Gaussian_Gaussian_Inc_Wallingford_CT (accessed on 20 January 2025).
- Balakrishnan, R.K.; Guria, C. Thermal degradation of polystyrene in the presence of hydrogen by catalyst in solution. Polym. Degrad. Stab. 2007, 92, 1583–1591. [Google Scholar]
- Ntwampe, I.O.; Jewell, L.L.; Hildebrandt, D.; Glasser, D. The effect of mixing on the treatment of paint wastewater with Fe3+ and Al3+ salts. J. Environ. Chem. Ecotoxicol. 2013, 5, 7–16. [Google Scholar]
- Aramendía, M.A.; Borau, V.; Jiménez, C.; Marinas, J.M.; Romero, F.J. Vapour-phase reaction of acetophenone with methanol or dimethyl carbonate on magnesium oxide and magnesium phosphates. J. Catal. 1999, 183, 119–127. [Google Scholar]
- Razouk, R.I.; Mikhail, R.S. Surface properties of magnesium oxide. J. Phys. Chem. 1957, 61, 886–891. [Google Scholar]
- Feliu, A.M. Catalizadores Bifuncionales Para Aumentar el Indice de Cetano de Combustibles Diésel Mediante Combinación de Hidrogenación y Apertura del Anillo Nafténico. Master’s Thesis, Universitat Politècnica de València, València, Spain, 2003. [Google Scholar]
- Lopes, M.; Dussan, K.; Leahy, J.J. Enhancing the conversion of D-xylose into furfural at low temperatures using chloride salts as co-catalysts: Catalytic combination of AlCl3 and formic acid. Chem. Eng. J. 2017, 323, 278–286. [Google Scholar]
- Hernández-Fernández, J.; Cano, H.; Aldas, M. Impact of Traces of Hydrogen Sulfide on the Efficiency of Ziegler–Natta Catalyst on the Final Properties of Polypropylene. Polymers 2022, 14, 3910. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; Guerra, Y.; Espinosa, E. Development and Application of a Principal Component Analysis Model to Quantify the Green Ethylene Content in Virgin Impact Copolymer Resins During Their Synthesis on an Industrial Scale. J. Polym. Environ. 2022, 30, 4800–4808. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; Lopez-Martinez, J.; Barceló, D. Development and validation of a methodology for quanti-fying partsper-billion levels of arsine and phosphine in nitrogen, hydrogen, and liquefied petroleum gas using a variable pressure sampler coupled to gas chromatography-mass spectrometry. J. Chromatogr. A 2021, 1637, 461833. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Fernández, J.; Castro-Suarez, J.R.; Toloza, C.A.T. Iron Oxide Powder as Responsible for the Generation of Industrial PolypropyleneWaste and as a Co-Catalyst for the Pyrolysis of Non-Additive Resins. Int. J. Mol. Sci. 2022, 23, 11708. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Fernández, J.; Vivas-Reyes, R.; Toloza, C.A.T. Experimental Study of the Impact of Trace Amounts of Acetylene and Methylacetylene on the Synthesis, Mechanical and Thermal Properties of Polypropylene. Int. J. Mol. Sci. 2022, 23, 12148. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; González-Cuello, R.; Ortega-Toro, R. Parts per Million of Propanol and Arsine as Respon-sible for the Poisoning of the Propylene Polymerization Reaction. Polymers 2023, 15, 3619. [Google Scholar] [CrossRef]
- Joaquin, H.-F.; Juan, L. Quantification of poisons for Ziegler Natta catalysts and effects on the production of poly-propylene by gas chromatographic with simultaneous detection: Pulsed discharge helium ionization, mass spec-trometry and flame ionization. J. Chromatogr. A 2019, 1614, 460736. [Google Scholar] [CrossRef]
- Hernández-Fernández, J.; López-Martínez, J. Experimental study of the auto-catalytic effect of triethylaluminum and TiCl4 residuals at the onset of non-additive polypropylene degradation and their impact on thermo-oxidative deg-radation and pyrolysis. J. Anal. Appl. Pyrolysis 2021, 155, 105052. [Google Scholar] [CrossRef]
- Hernández-Fernández, J. Quantification of oxygenates, sulphides, thiols and permanent gases in propylene. A multiple linear regression model to predict the loss of efficiency in polypropylene production on an industrial scale. J. Chromatogr. A 2020, 1628, 461478. [Google Scholar] [CrossRef]
- Joaquin, H.-F.; Juan, L.-M. Autocatalytic influence of different levels of arsine on the thermal stability and pyrolysis of polypropylene. J. Anal. Appl. Pyrolysis 2022, 161, 105385. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
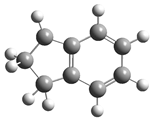
| Optimized Structure | Number | Optimized Structure | Number |
|---|---|---|---|
 | 1 | 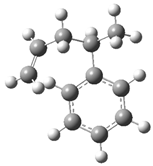 | 17 |
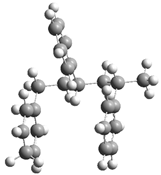 | 2 | 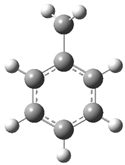 | 18 |
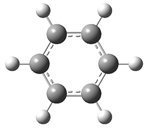 | 3 | 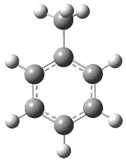 | 19 |
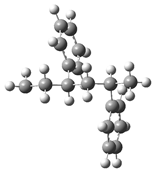 | 4 | 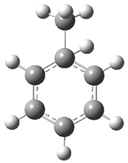 | 20 |
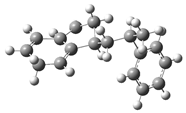 | 5 | 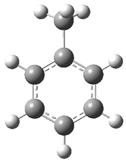 | 21 |
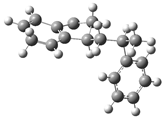 | 6 |  | 22 |
 | 7 | 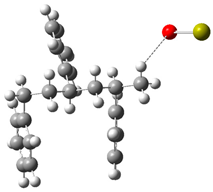 | 23 |
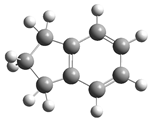 | 8 | 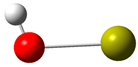 | 24 |
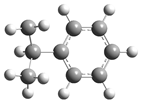 | 9 | 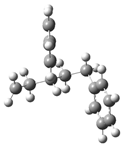 | 25 |
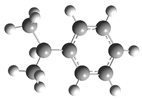 | 10 | 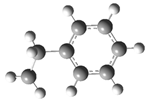 | 26 |
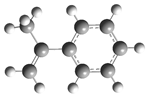 | 11 | 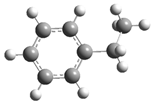 | 27 |
 | 12 |  | 28 |
 | 13 |  | 29 |
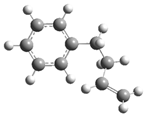 | 14 | 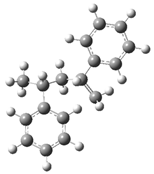 | 30 |
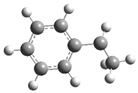 | 15 | 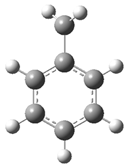 | 31 |
 | 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández Fernández, J.A.; Prieto Palomo, J.A.; Ortega-Toro, R. Application of Computational Studies Using Density Functional Theory (DFT) to Evaluate the Catalytic Degradation of Polystyrene. Polymers 2025, 17, 923. https://doi.org/10.3390/polym17070923
Hernández Fernández JA, Prieto Palomo JA, Ortega-Toro R. Application of Computational Studies Using Density Functional Theory (DFT) to Evaluate the Catalytic Degradation of Polystyrene. Polymers. 2025; 17(7):923. https://doi.org/10.3390/polym17070923
Chicago/Turabian StyleHernández Fernández, Joaquín Alejandro, Jose Alfonso Prieto Palomo, and Rodrigo Ortega-Toro. 2025. "Application of Computational Studies Using Density Functional Theory (DFT) to Evaluate the Catalytic Degradation of Polystyrene" Polymers 17, no. 7: 923. https://doi.org/10.3390/polym17070923
APA StyleHernández Fernández, J. A., Prieto Palomo, J. A., & Ortega-Toro, R. (2025). Application of Computational Studies Using Density Functional Theory (DFT) to Evaluate the Catalytic Degradation of Polystyrene. Polymers, 17(7), 923. https://doi.org/10.3390/polym17070923