
Effect of Poly (Caprolactone) Introduction Site on the Network Structure and Properties of Glycidyl Azide Polymer Adhesive


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Synthesis of GAP/PCL Block Copolymer (PCL-b-GAP-b-PCL)
2.3. Synthesis of GAP/PCL Graft Copolymer (GAP-g-PCL)
2.4. Preparation of GAP and GAP/PCL Elastomer
2.5. Characterizations
3. Results and Discussion
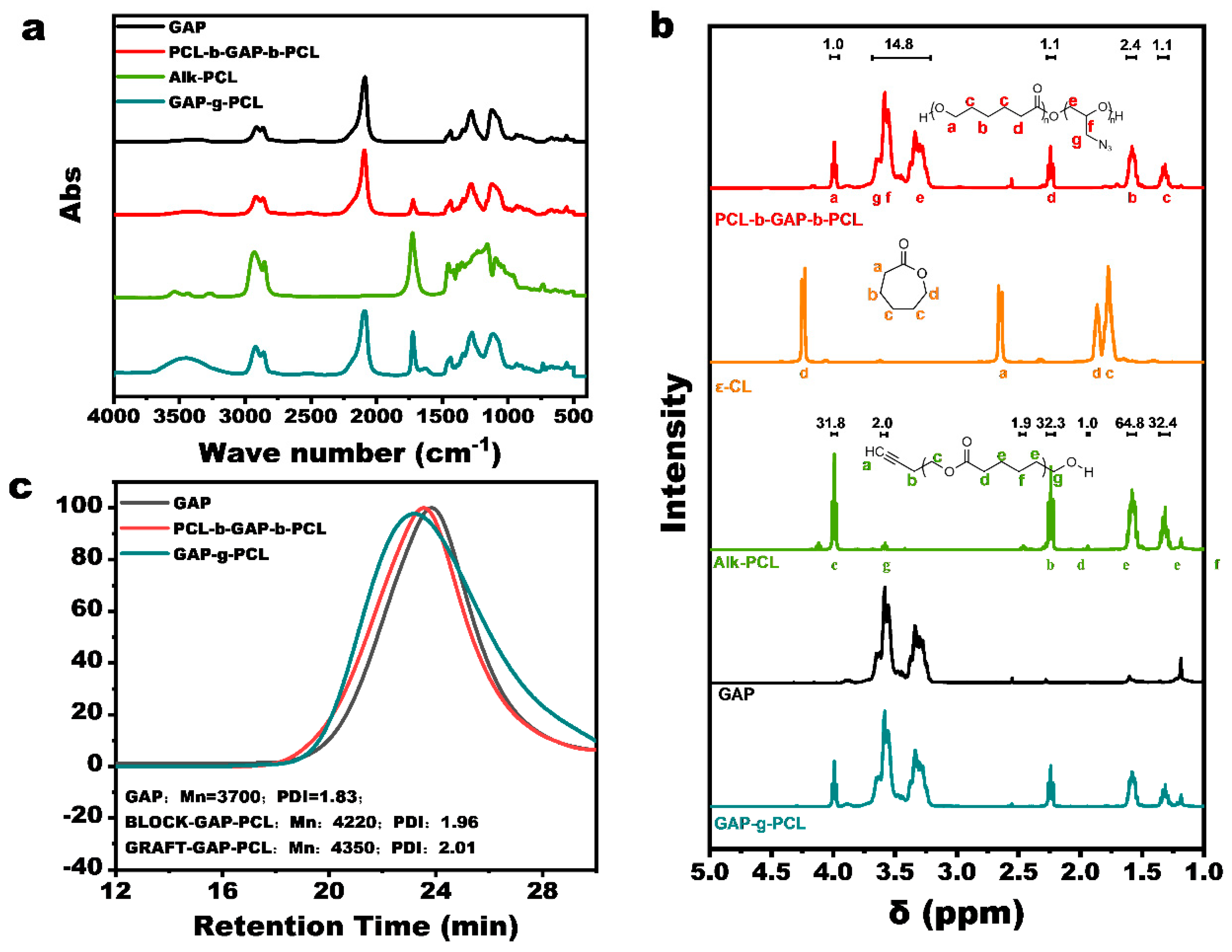
3.1. Synthesis and Characterization of GAP/PCL Prepolymers
3.1.1. Synthesis and Characterization of PCL-b-GAP-b-PCL
3.1.2. Synthesis and Characterization of GAP-g-PCL
3.2. Characterization of GAP/PCL Prepolymer Properties
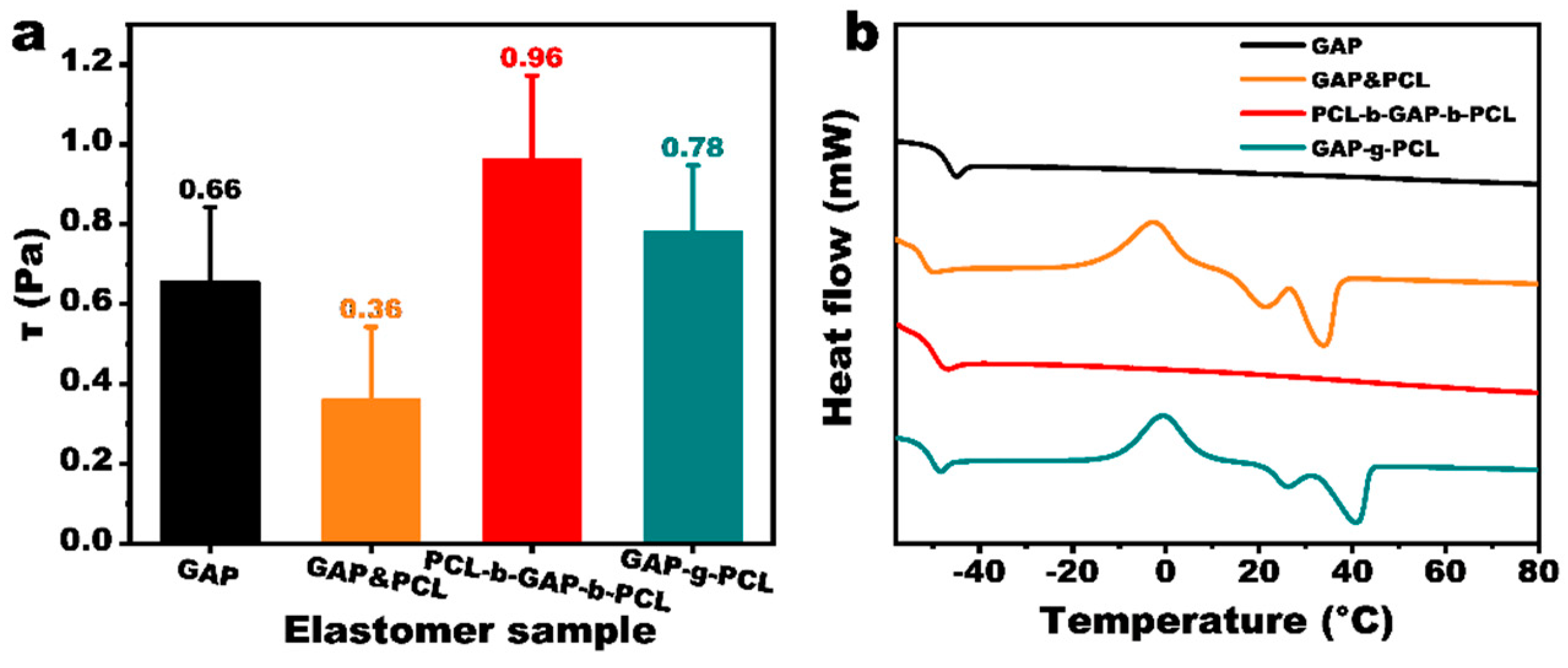
3.3. Network Structure of GAP/PCL Elastomers
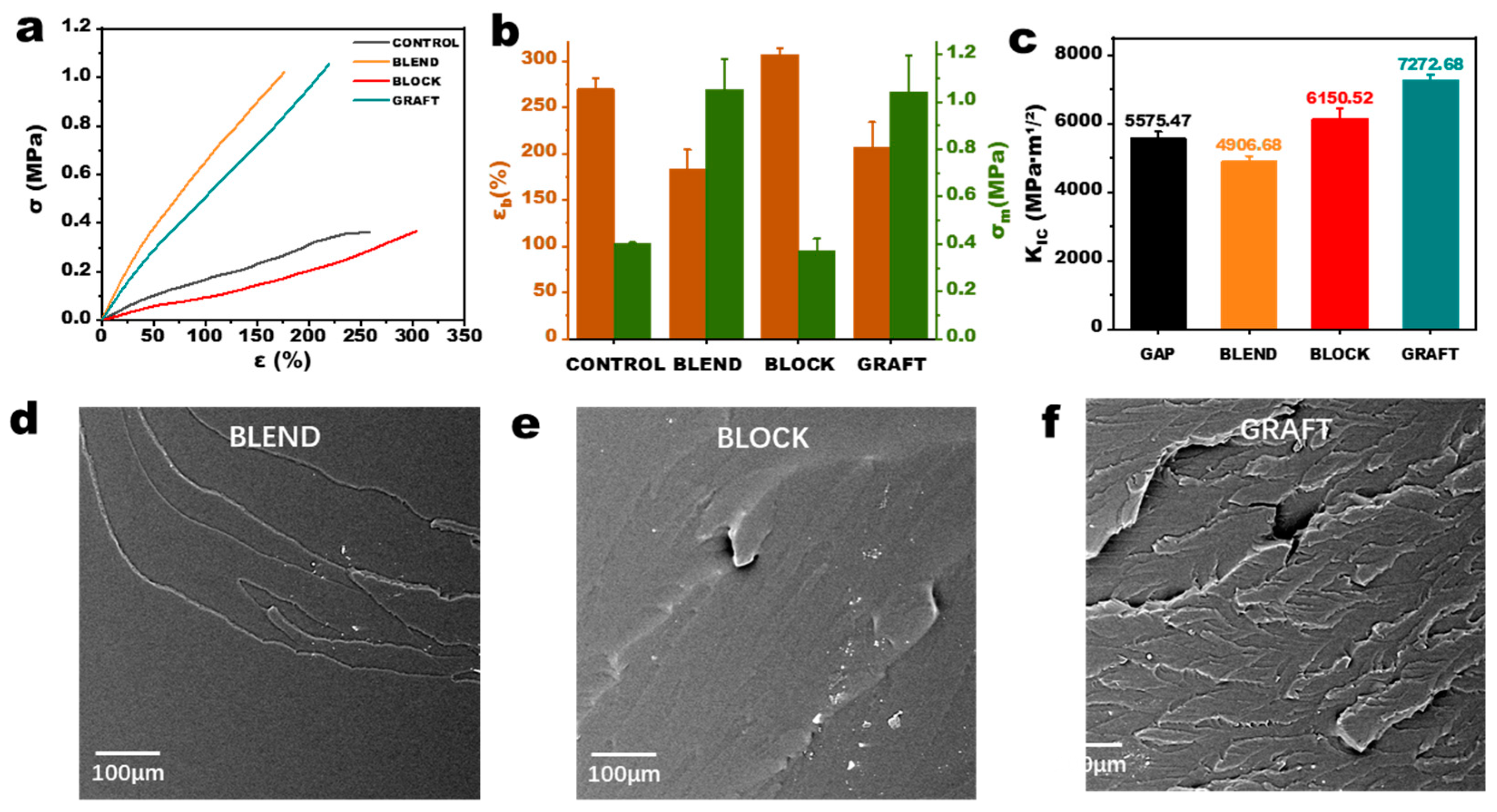
3.4. Static Mechanical Properties of GAP/PCL Elastomers
3.5. Dynamic Mechanical Properties of GAP/PCL Elastomers
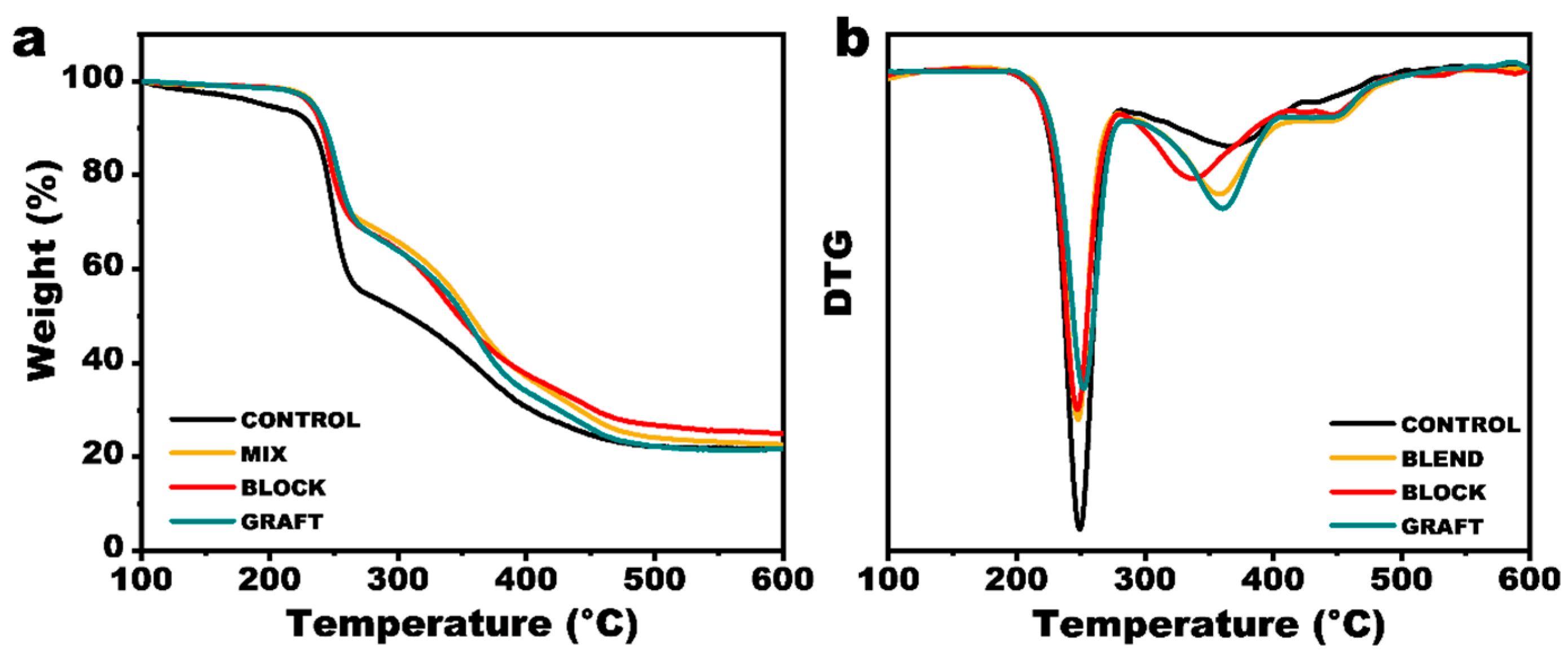
3.6. Thermal Stability of GAP/PCL Elastomers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, Z.; Zhang, J.; Xu, S.; Li, H.; Zhou, H.; Zheng, J.; Pang, A.; Yang, Y. Synthesis of two novel neutral polymeric bonding agents to enhance the mechanical properties of composite solid propellants. RSC Adv. 2022, 12, 19946–19952. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, Z.; Sun, P.; Dong, J.; Li, Q.; Xu, B.; Chen, F.; Liao, X. Study on the thermal stability of NG/NC/RDX triple-base gun propellants modified by oligomer GAP (Mn = 330). Colloids Surf. A Physicochem. Eng. Asp. 2024, 689, 133731. [Google Scholar] [CrossRef]
- Touidjine, S.; Boulkadid, M.K.; Trache, D.; Akbi, H.; Guettiche, D.; Belkhiri, S.; Nourine, M. Review on energetic copolymer binders for propulsion applications: Synthesis and properties. J. Polym. Sci. 2023, 61, 2254–2275. [Google Scholar] [CrossRef]
- Cheng, T. Review of novel energetic polymers and binders—High energy propellant ingredients for the new space race. Des. Monomers Polym. 2019, 22, 54–65. [Google Scholar] [CrossRef]
- Jarosz, T.; Stolarczyk, A.; Wawrzkiewicz-Jalowiecka, A.; Pawlus, K.; Miszczyszyn, K. Glycidyl Azide Polymer and its Derivatives-Versatile Binders for Explosives and Pyrotechnics: Tutorial Review of Recent Progress. Molecules 2019, 24, 4475. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Li, J.; Zhang, X.; Liu, W.; Liu, S.; Zheng, M.; Yang, W.; Luo, Y. Novel azide polymer/NC/RDX composite microspheres: Low sensitivity and excellent thermal stability. J. Energetic Mater. 2023, 1–18. [Google Scholar] [CrossRef]
- Jin, P.; Li, J.; Zhang, X.; Liu, S.; Yang, W.; Liu, W.; Luo, Y. Preparation and properties of different azide polymer-modified nitrocellulose spherical powder. J. Therm. Anal. Calorim. 2023, 148, 9661–9671. [Google Scholar] [CrossRef]
- Fu, J.; Ren, H.; Liu, X.; Sun, J.; Wu, G. Molecular revelation of the thermal decomposition mechanism of glycidyl azide polymer in nitrate esters matrix. Combust. Flame 2024, 268, 113648. [Google Scholar] [CrossRef]
- Sahu, S.K.; Panda, S.P.; Sadafule, D.S.; Kumbhar, C.G.; Kulkarni, S.G.; Thakur, J.V. Thermal and photodegradation of glycidyl azide polymers. Polym. Degrad. Stab. 1998, 68, 495–500. [Google Scholar] [CrossRef]
- He, L.; Zhou, J.; Wang, Y.; Ma, Z.; Chen, C. Mechanical and Thermal Properties of Polyether Polytriazole Elastomers Formed by Click-Chemical Reaction Curing Glycidyl Azide Polymer. Molecules 2020, 25, 1988. [Google Scholar] [CrossRef]
- Xu, M.; Lu, X.; Liu, N.; Zhang, Q.; Mo, H.; Ge, Z. Fluoropolymer/Glycidyl Azide Polymer (GAP) Block Copolyurethane as New Energetic Binders: Synthesis, Mechanical Properties, and Thermal Performance. Polymers 2021, 13, 2706. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, W.; Jin, P.; Ding, S.; Liu, S.; Zhao, S.; Yang, W.; Liu, W.; Luo, Y. Effect of azide polyether pyrolysis property on combustion and heat release of boron-based fuel-rich propellant. Combust. Flame 2022, 245, 112269. [Google Scholar] [CrossRef]
- Gaur, B.; Lochab, B.; Choudhary, V.; Varma, I.K. Azido Polymers—Energetic Binders for Solid Rocket Propellants. J. Macromol. Sci. Part C Polym. Rev. 2003, 43, 505–545. [Google Scholar] [CrossRef]
- Lu, Y.-Y.; Shu, Y.-J.; Liu, N.; Shu, Y.; Wang, K.; Wu, Z.-K.; Wang, X.-C.; Ding, X.-Y. Theoretical simulations on the glass transition temperatures and mechanical properties of modified glycidyl azide polymer. Comput. Mater. Sci. 2017, 139, 132–139. [Google Scholar] [CrossRef]
- Xu, M.; Ge, Z.; Lu, X.; Mo, H.; Ji, Y.; Hu, H. Fluorinated glycidyl azide polymers as potential energetic binders. RSC Adv. 2017, 7, 47271–47278. [Google Scholar] [CrossRef]
- Reshmi, S.K.; Vijayalakshmi, K.P.; Thomas, D.; Arunan, E.; Reghunadhan, C.P. Glycidyl Azide Polymer Crosslinked Through Triazoles by Click Chemistry: Curing, Mechanical and Thermal Properties. Propell. Explos. Pyrotech. 2013, 38, 525–532. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Q.; Peng, R.; Jin, B. Interpenetrating polymer networks of polyurethane and polytriazole: Effect of the composition and number average molecular weight of glycidyl azide polymer on the mechanical properties and morphology. Polym. Adv. Technol. 2024, 35, e6409. [Google Scholar] [CrossRef]
- Bayat, Y.; Chizari, M. Designing a Highly Energetic PCL-GAP-PCL-based PU Elastomer; Investigation of the Effect of Plasticizers on Its Properties. Cent. Eur. J. Energetic Mater. 2019, 16, 33–48. [Google Scholar] [CrossRef]
- Kim, H.; Jang, Y.; Noh, S.; Jeong, J.; Kim, D.; Kang, B.; Kang, T.; Choi, H.; Rhee, H. Ecofriendly synthesis and characterization of carboxylated GAP copolymers. RSC Adv. 2018, 8, 20032–20038. [Google Scholar] [CrossRef]
- Deng, J.; Li, G.; Xia, M.; Lan, Y.; Luo, Y. Improvement of mechanical characteristics of glycidyl azide polymer binder system by addition of flexible polyether. J. Appl. Polym. Sci. 2016, 133, 43840. [Google Scholar] [CrossRef]
- Ma, S.; Du, W.; Luo, Y. Simulation of GAP/HTPB phase behaviors in plasticizers and its application in composite solid propellant. e-Polymers 2018, 18, 529–540. [Google Scholar] [CrossRef]
- Sun, B. Characterization of the Plasticized GAP/PEG and GAP/PCL Block Copolyurethane Binder Matrices and its Propellants. Propell. Explos. Pyrotech. 2008, 33, 131–138. [Google Scholar] [CrossRef]
- Min, B.S.; Ko, S.W. Characterization of segmented block copolyurethane network based on glycidyl azide polymer and polycaprolactone. Macromol. Res. 2007, 15, 225–233. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Ma, S.; Luo, Y. Compatibility, mechanical and thermal properties of GAP/P(EO-co-THF) blends obtained upon a urethane-curing reaction. Polym. Bull. 2017, 74, 4607–4618. [Google Scholar] [CrossRef]
- Yang, Z.; Xin-Ping, L.; Qing-Xuan, Z. Simulation study of the morphologies of energetic block copolymers based on glycidyl azide polymer. J. Appl. Polym. Sci. 2012, 129, 480–486. [Google Scholar] [CrossRef]
- Hou, S.; Lai, Z.; Wei, M. Plate gap effect on vicosity and rheological model of shear thickening fluid. Korea-Aust. Rheol. J. 2023, 35, 11–18. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Song, X.; An, C. Rheological Behavior of Concentrated GAP/ AP Propellant Suspensions Plasticized by Diglycerol Tetranitrate. Propell. Explos. Pyrotech. 2022, 47, e202200049. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Song, X.; An, C.; Li, F. Rheological Impact of Particle Size Gradation on GAP Propellant Slurries. ACS Omega 2022, 7, 38536–38542. [Google Scholar] [CrossRef]
- Jin, L.; Qian, J.; Fang, Q.Z.; Hu, Q.W.; Yu, M.H. Study on mechanical properties of solid propellant NEPE. Propell. Explos. Pyrot. 2023, 48, e202200338. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, C.; Wang, Z.; Zhu, F.; Yang, D.; Liu, C.; Bai, C.; Li, G.; Luo, Y. Effect of Poly (Caprolactone) Introduction Site on the Network Structure and Properties of Glycidyl Azide Polymer Adhesive. Polymers 2025, 17, 661. https://doi.org/10.3390/polym17050661
Tu C, Wang Z, Zhu F, Yang D, Liu C, Bai C, Li G, Luo Y. Effect of Poly (Caprolactone) Introduction Site on the Network Structure and Properties of Glycidyl Azide Polymer Adhesive. Polymers. 2025; 17(5):661. https://doi.org/10.3390/polym17050661
Chicago/Turabian StyleTu, Chengzhao, Zhengyuan Wang, Fengdan Zhu, Dengsheng Yang, Chang Liu, Chaofei Bai, Guoping Li, and Yunjun Luo. 2025. "Effect of Poly (Caprolactone) Introduction Site on the Network Structure and Properties of Glycidyl Azide Polymer Adhesive" Polymers 17, no. 5: 661. https://doi.org/10.3390/polym17050661
APA StyleTu, C., Wang, Z., Zhu, F., Yang, D., Liu, C., Bai, C., Li, G., & Luo, Y. (2025). Effect of Poly (Caprolactone) Introduction Site on the Network Structure and Properties of Glycidyl Azide Polymer Adhesive. Polymers, 17(5), 661. https://doi.org/10.3390/polym17050661