
Kinetics of Hydroxyl Growth on Natural Rubber Depolymerization with H2O2/Fenton Using Infrared Spectroscopy

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Synthesis of HTNR
2.2.2. Analysis
2.2.3. Kinetics Model
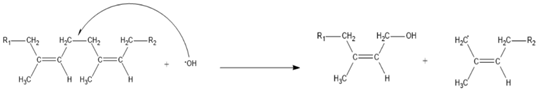
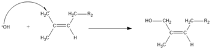


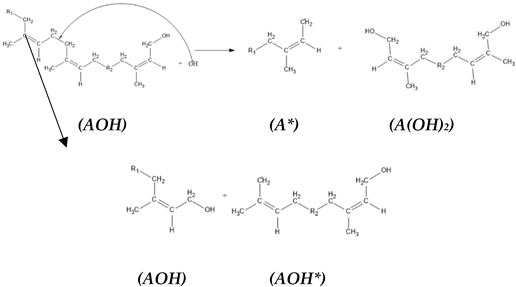
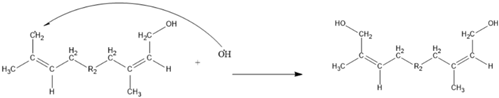
3. Results and Discussion
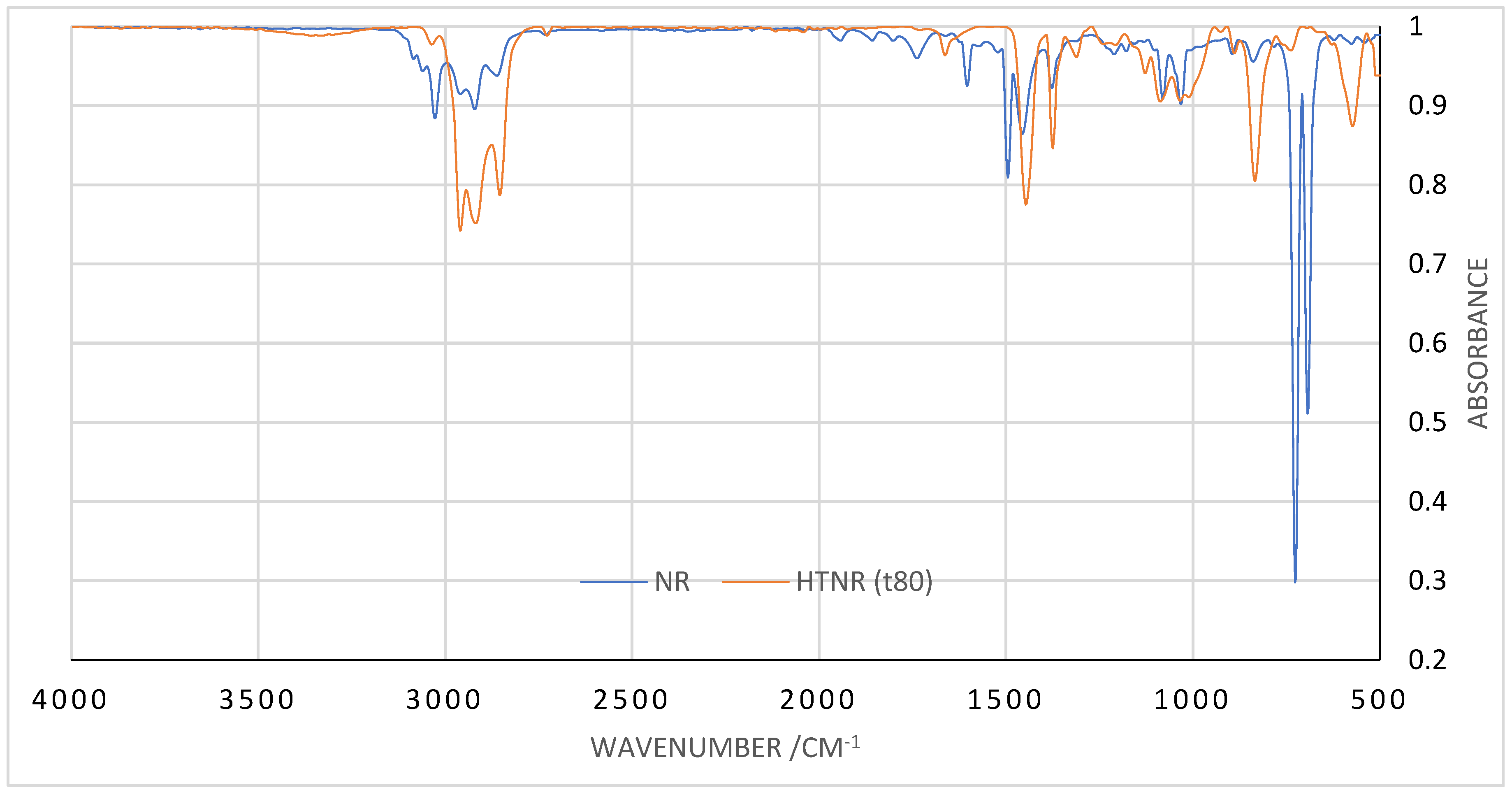
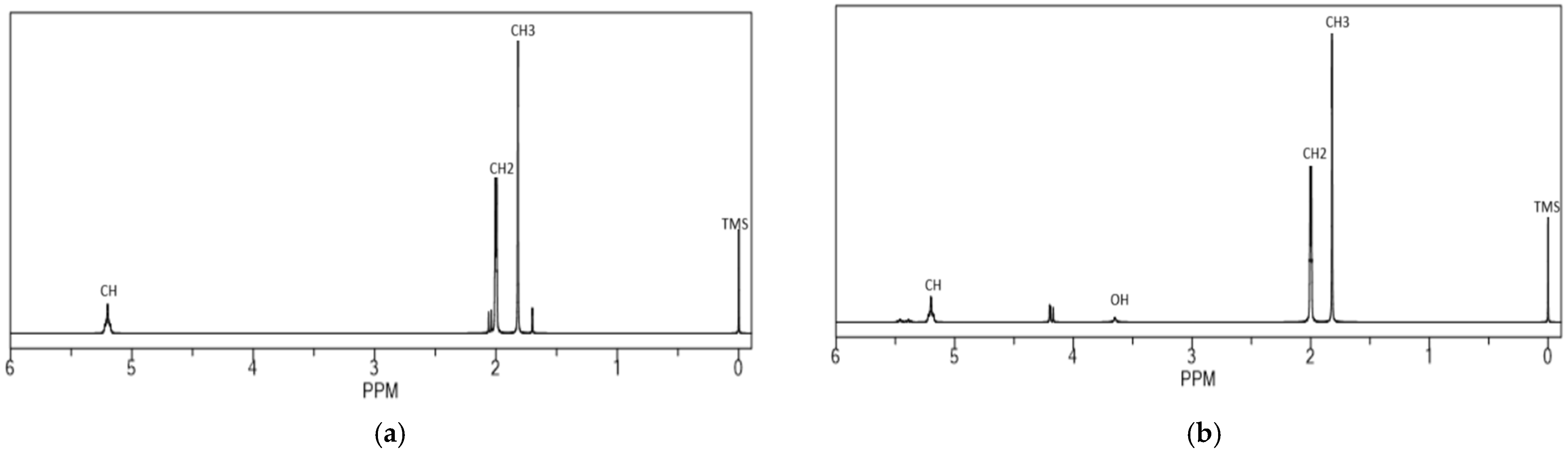
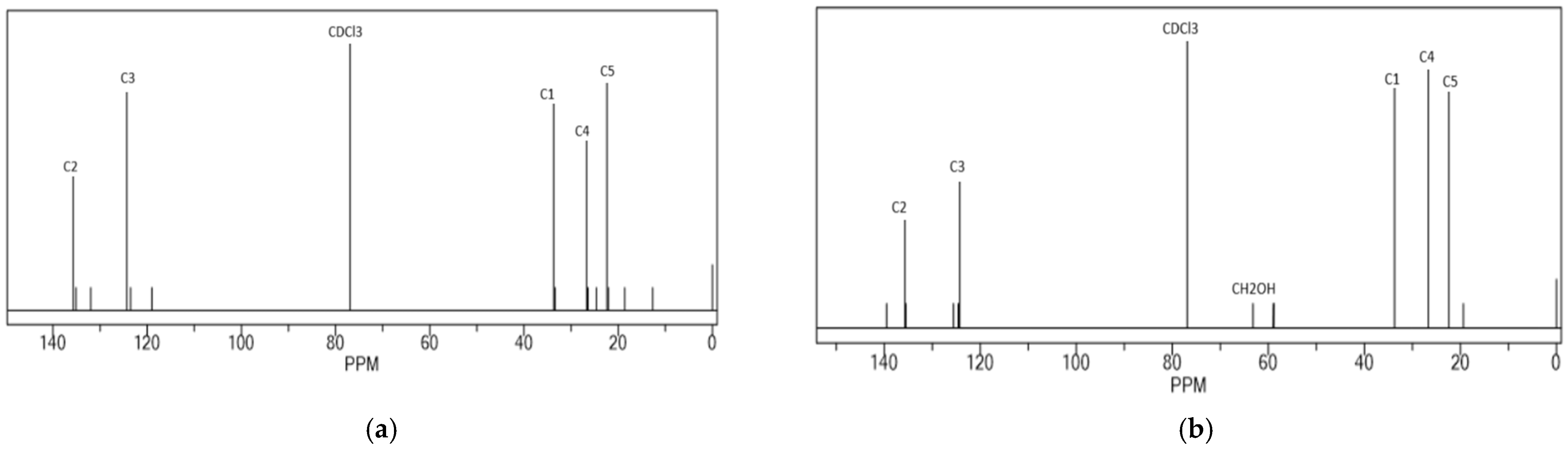
3.1. HTNR Characterization
3.2. The Role of Infrared Absorption in Depolymerization Analysis
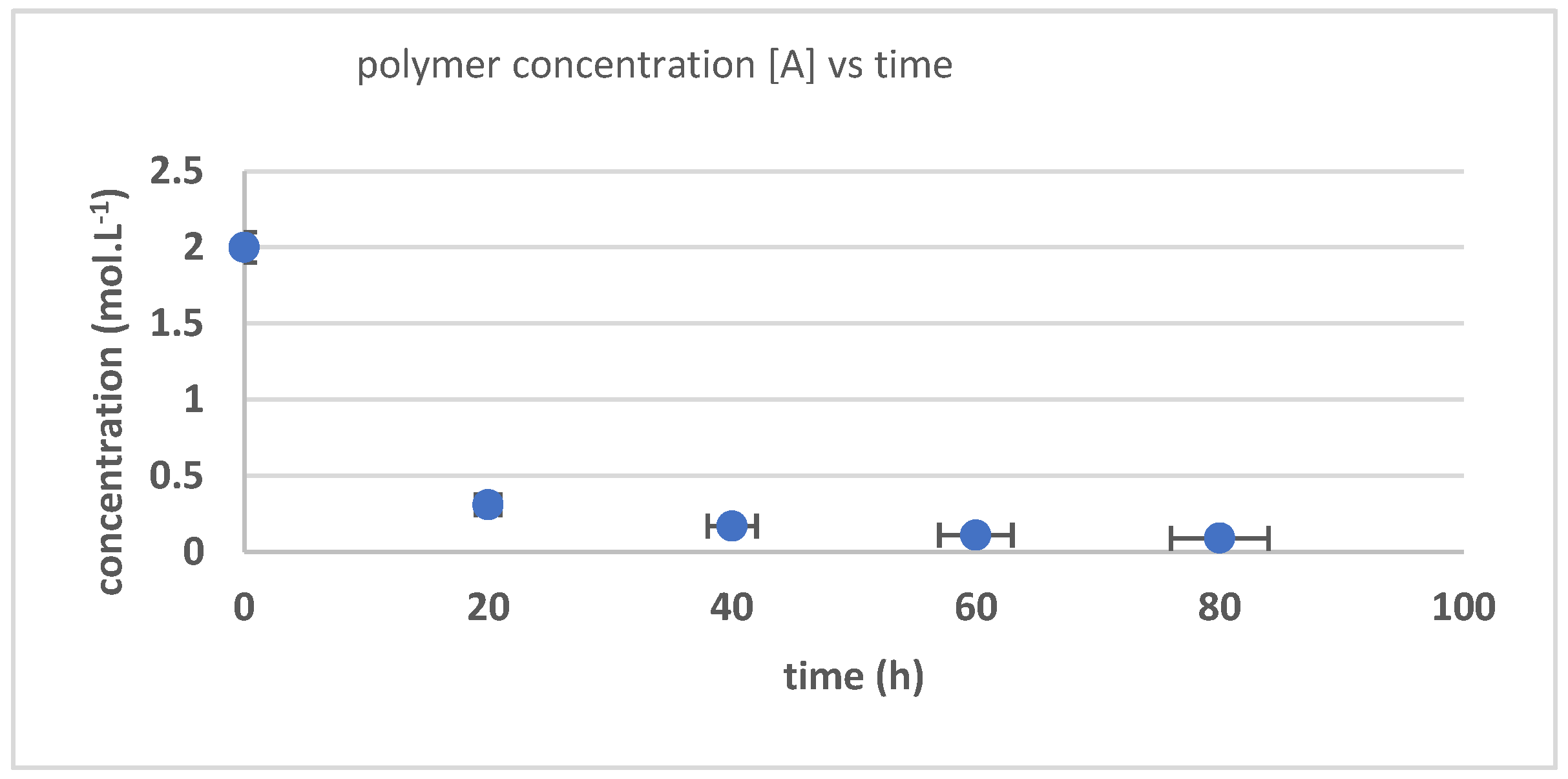
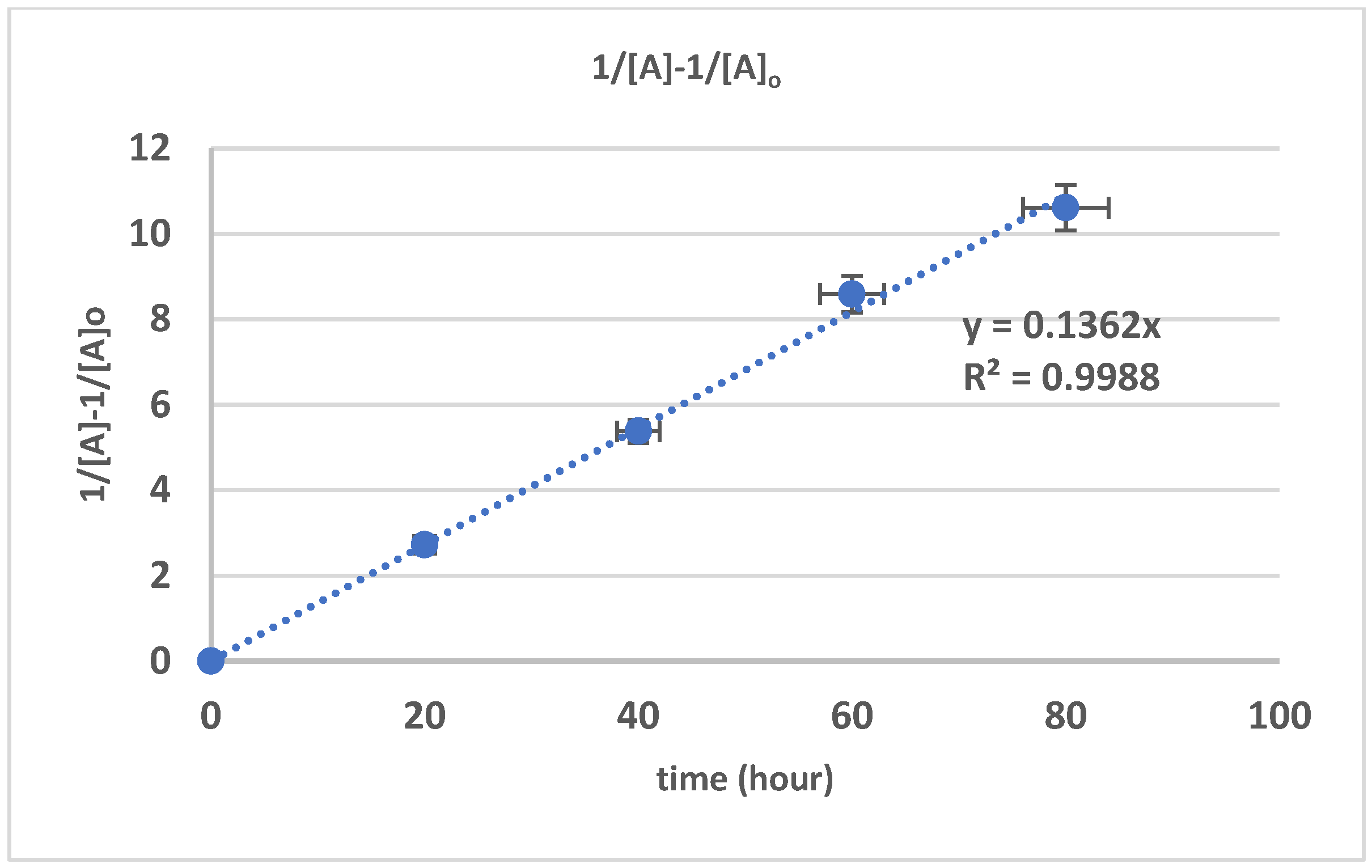
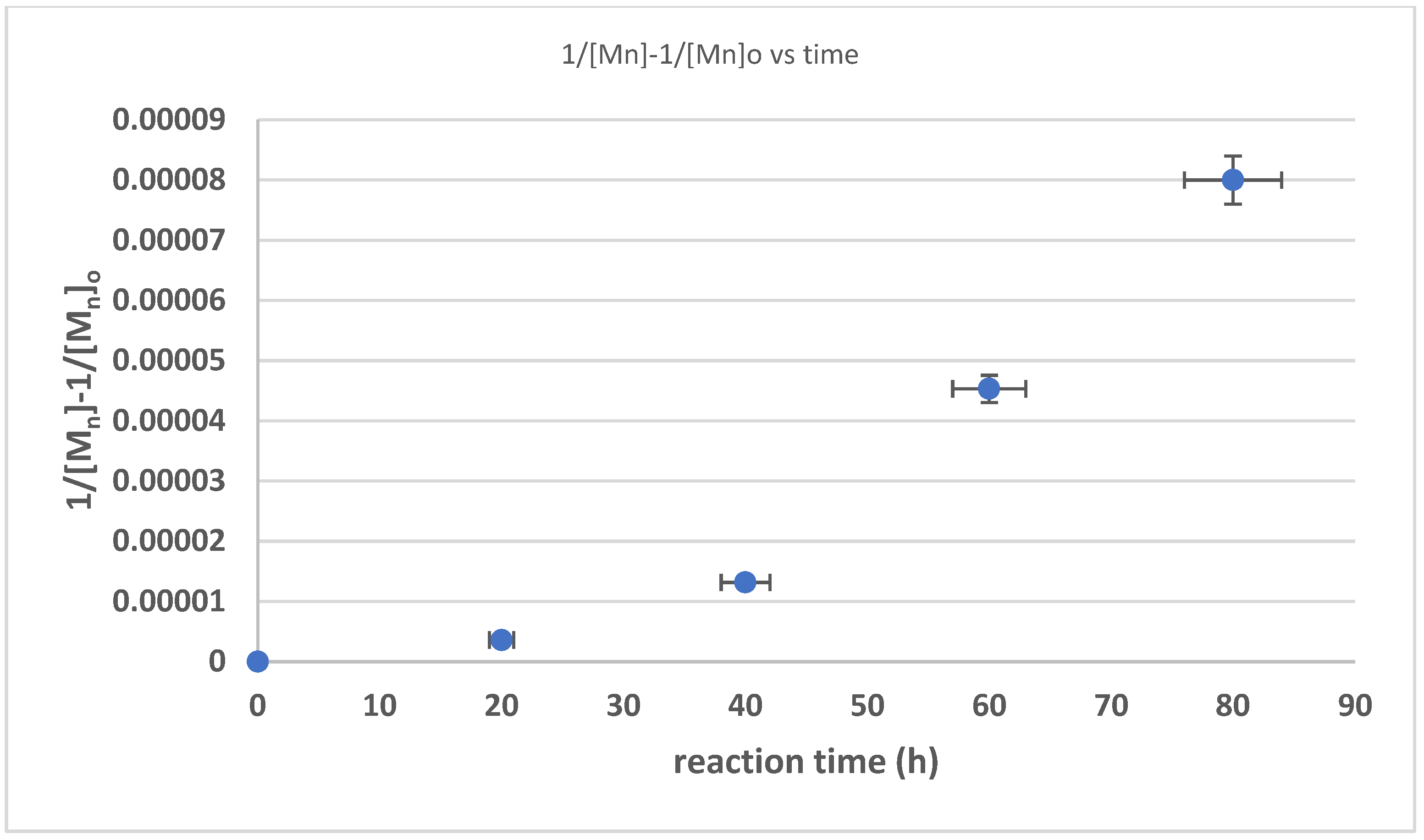
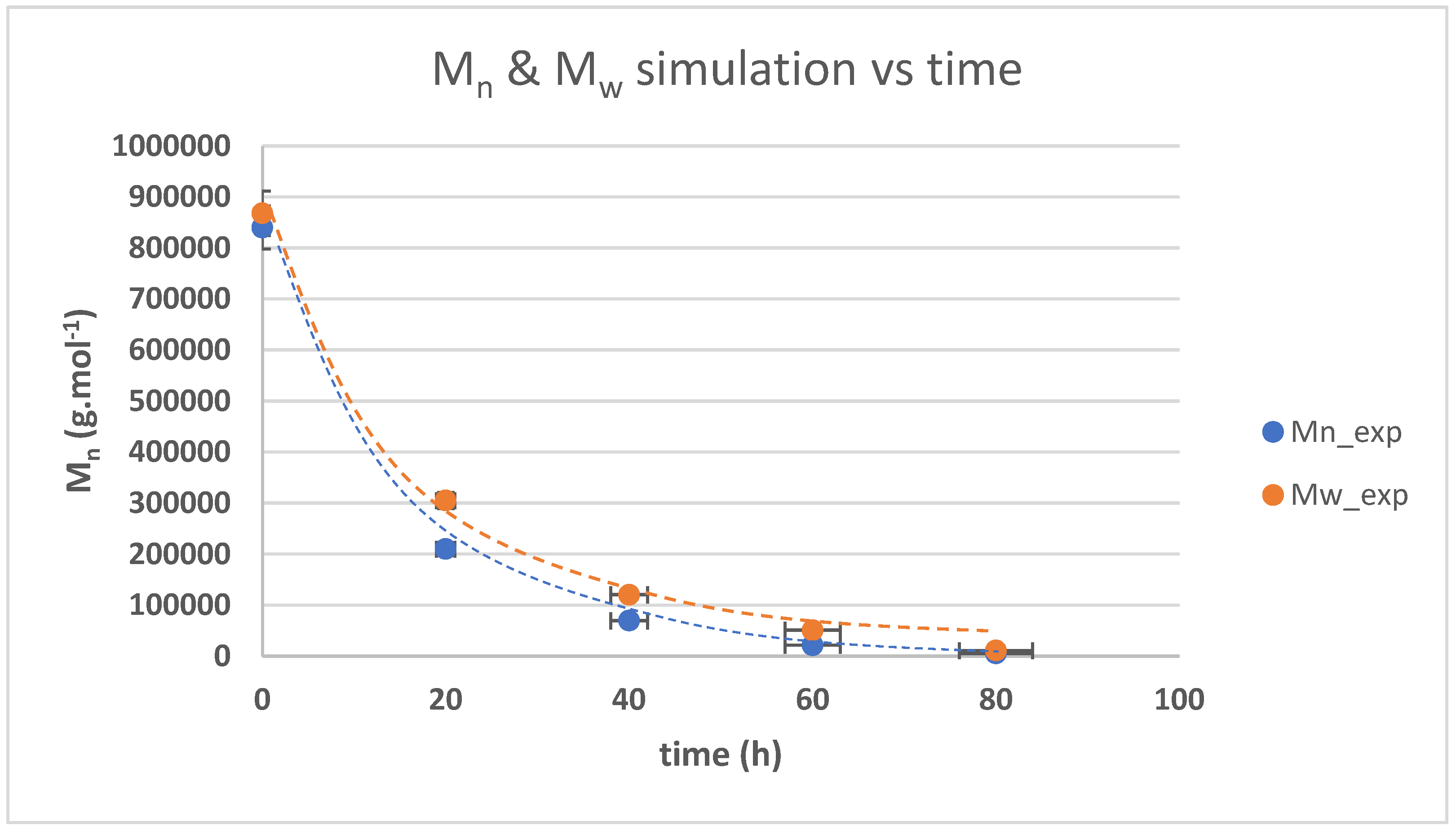
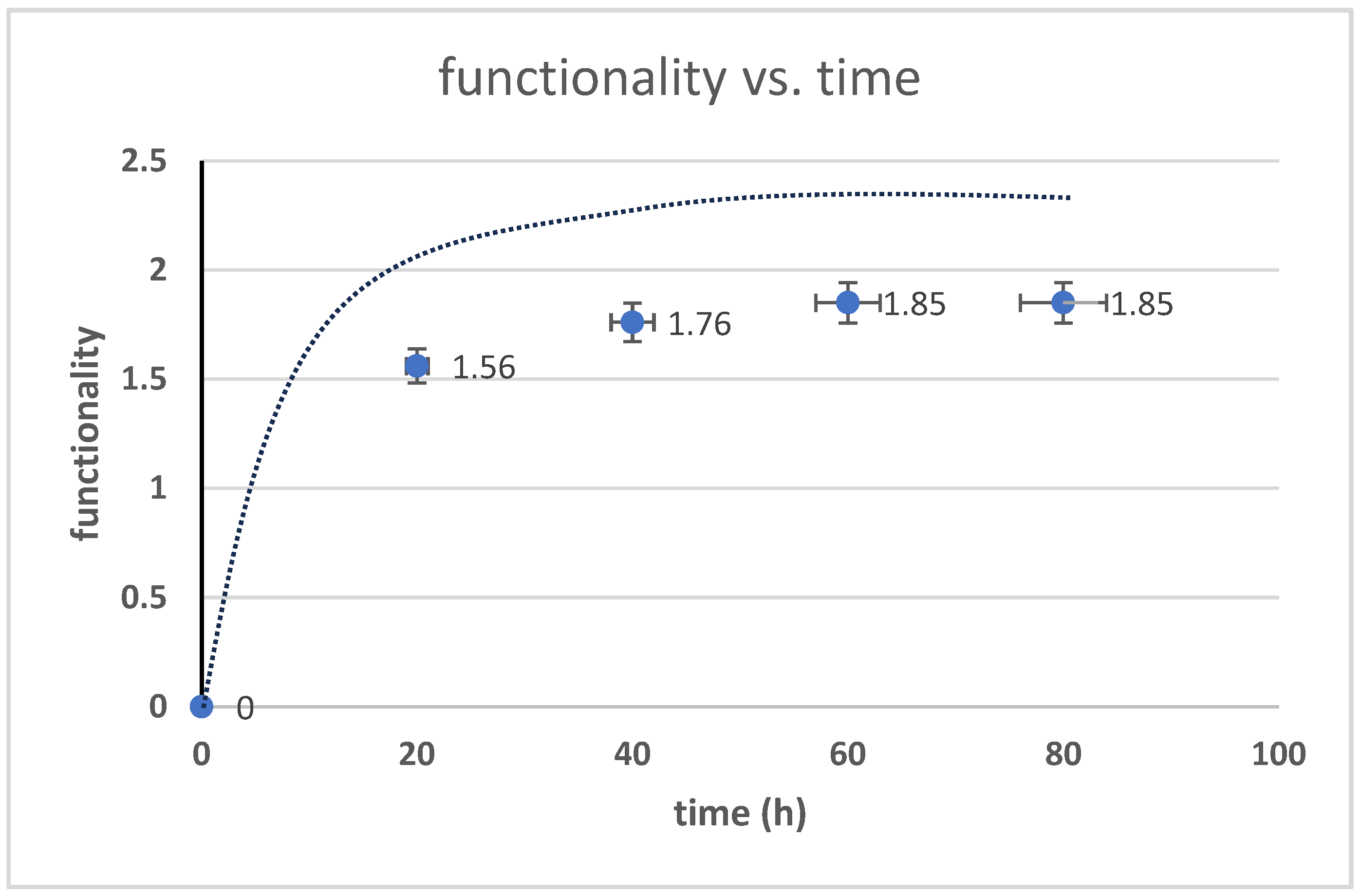
3.3. The Kinetics of HTNR Formation Reaction
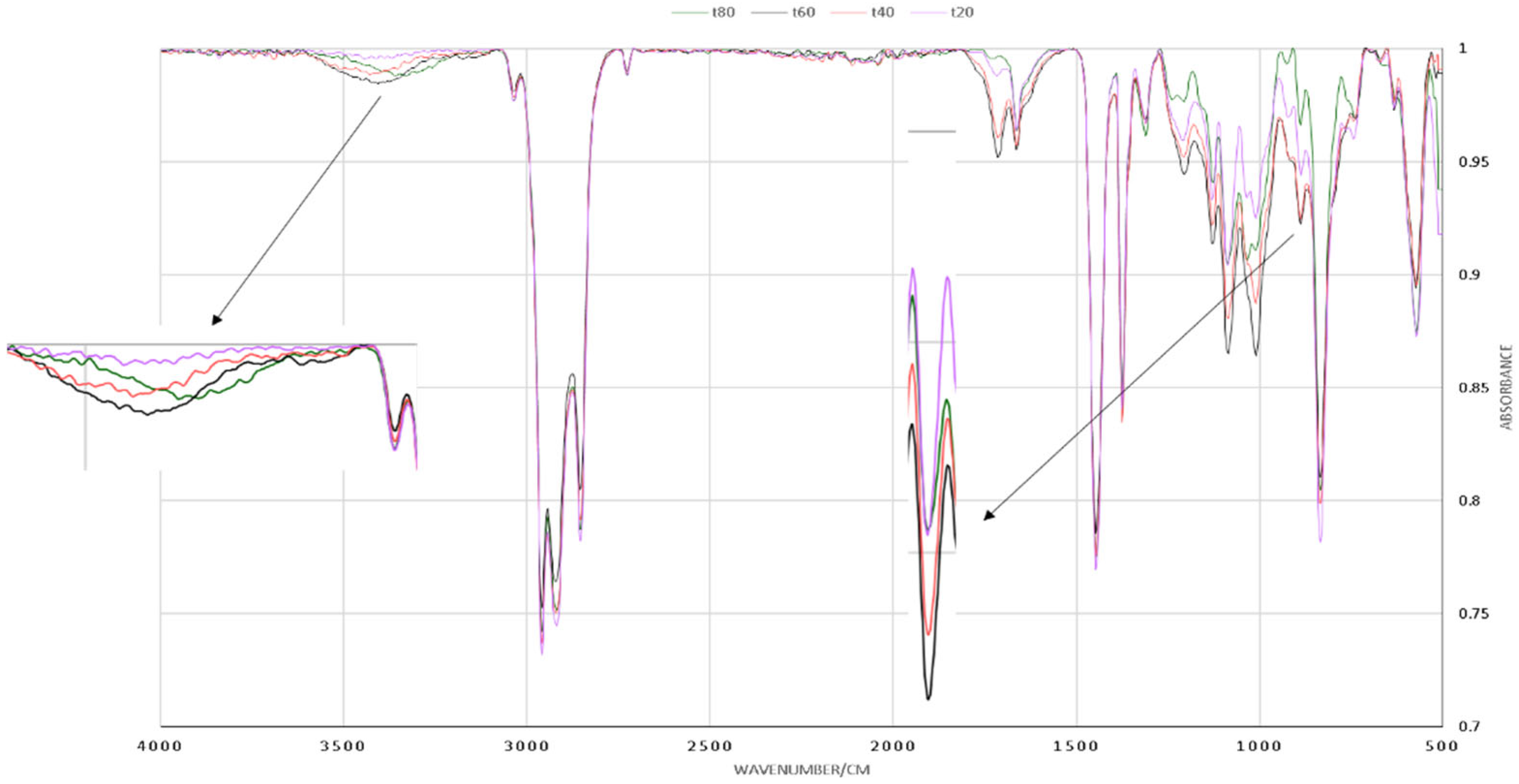
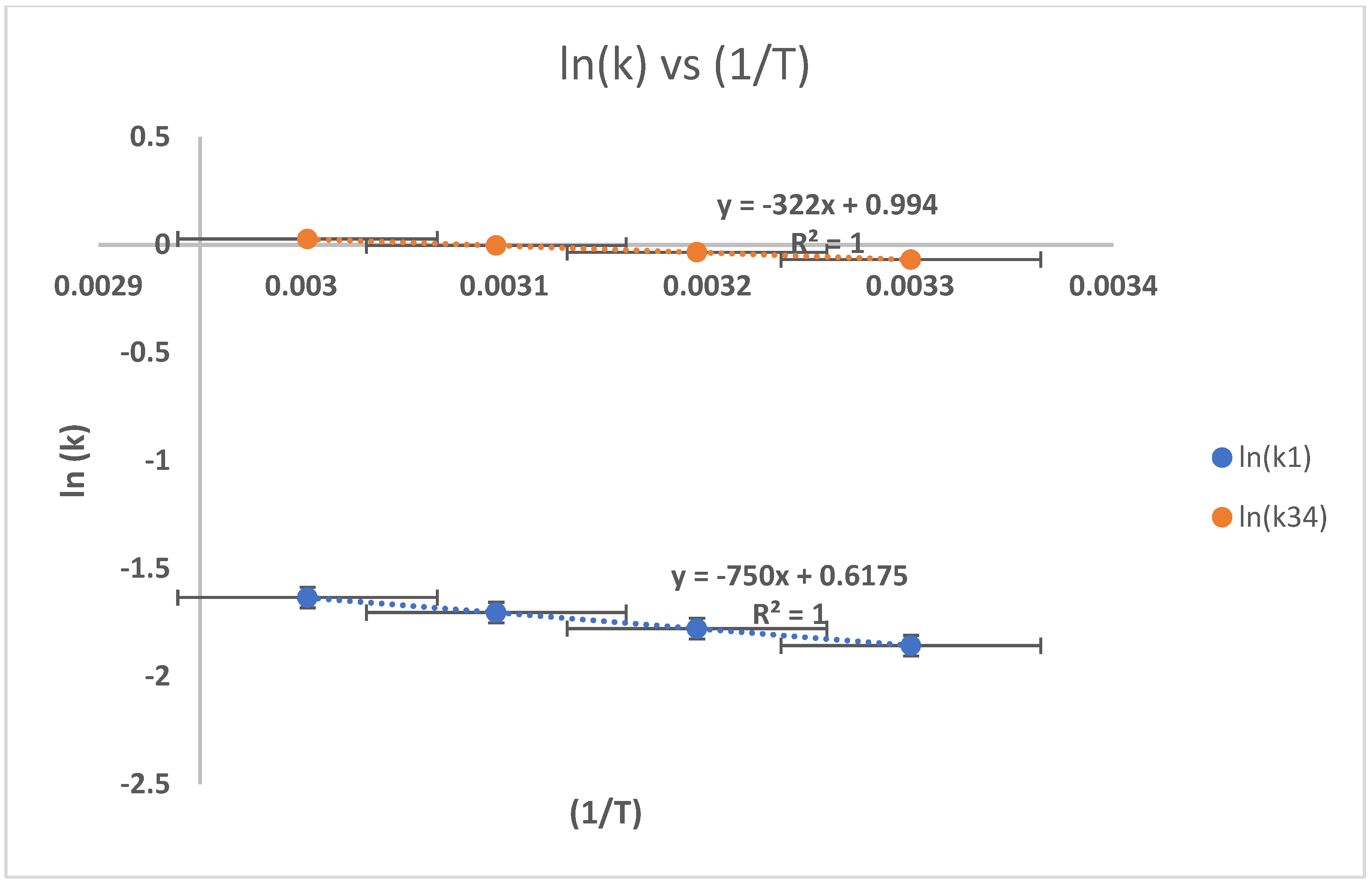
3.4. The Influence of Reaction Temperature
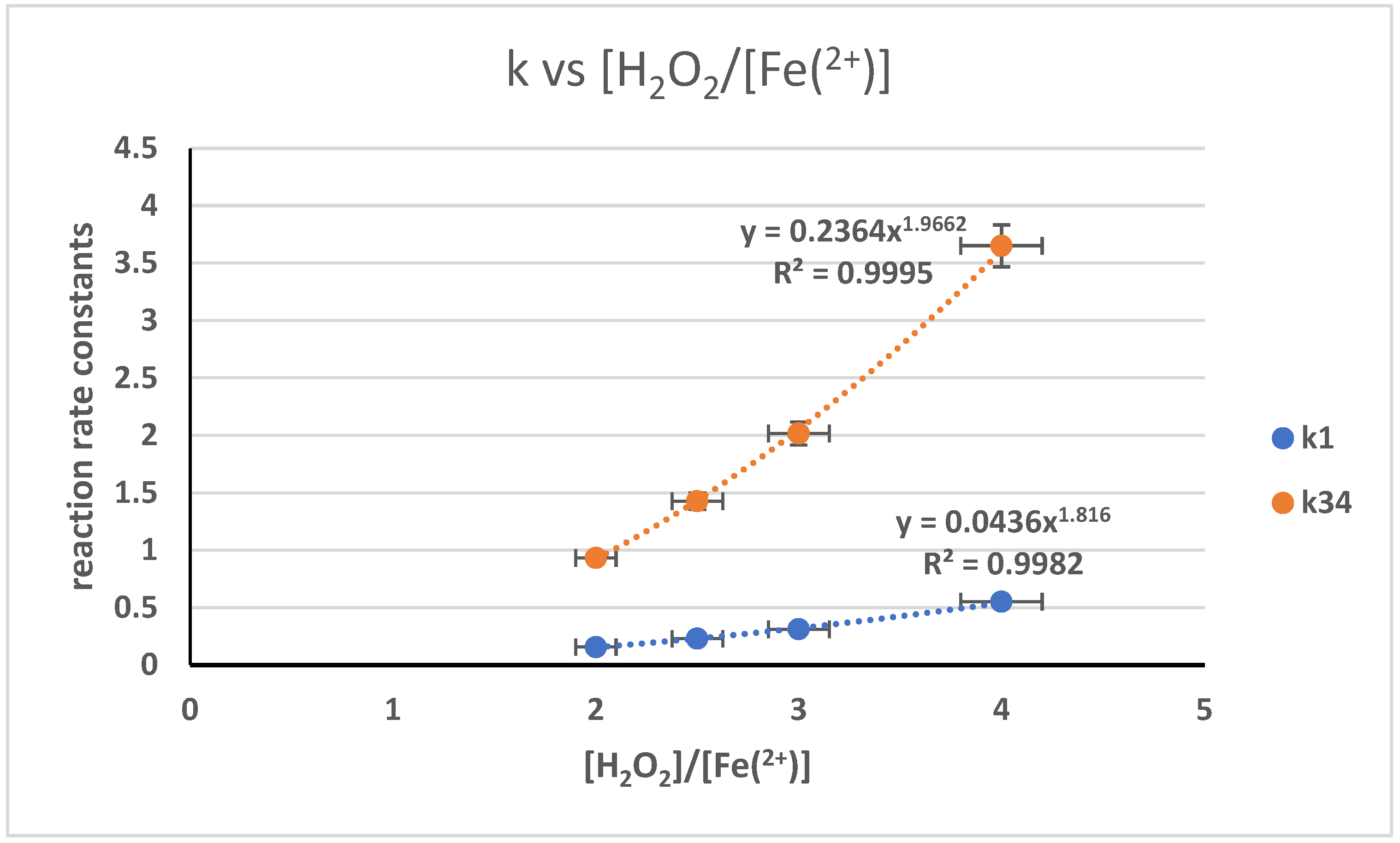
3.5. The Influence of Reactant and Catalyst Ratio
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Nor, H.M.; Ebdon, J.R. Telechelic Liquid Natural Rubber: A Review. Prog. Polym. Sci. 1998, 23, 143–177. [Google Scholar] [CrossRef]
- Saleh, A.B.; Ishak, Z.A.M.; Mahmood, W.A.K.; Hashim, A.S. On the Stability of Natural Rubber Latex Depolymerization by Hydrogen Peroxide and Subsequent Epoxidation by Peracetic Acid; Universiti Sains Malaysia: Penang, Malaysia, 2004. [Google Scholar]
- Cifriadi, A.; Chalid, M.; Puspitasari, S. Characterization of Hydrogenated Natural Rubber Synthesized by Diimide Transfer Hydrogenation. Int. J. Technol. 2017, 8, 448. [Google Scholar] [CrossRef]
- Ly, P.H. Reinforcement of Natural Rubber from Hydroxyl-Terminated Liquid Natural Rubber Grafted Carbon Black. I. Grafting of Acyl Chloride Capped Liquid Natural Rubber onto Carbon Black. J. Macromol. Sci. Part A 1996, 33, 1931–1937. [Google Scholar] [CrossRef]
- Sukhawipat, N.; Saetung, N.; Pasetto, P.; Pilard, J.-F.; Bistac, S.; Saetung, A. A Novel High Adhesion Cationic Waterborne Polyurethane for Green Coating Applications. Prog. Org. Coat. 2020, 148, 105854. [Google Scholar] [CrossRef]
- Gopakumar, S.; Nair, M.R.G. Natural Rubber–Polyurethane Block Copolymers: Nonlinear Structural Variations with NCO/OH Ratio. Polym. Eng. Sci. 2006, 46, 1812–1821. [Google Scholar] [CrossRef]
- Saetung, A.; Kaenhin, L.; Klinpituksa, P.; Rungvichaniwat, A.; Tulyapitak, T.; Munleh, S.; Campistron, I.; Pilard, J. Synthesis, Characteristic, and Properties of Waterborne Polyurethane Based on Natural Rubber. J. Appl. Polym. Sci. 2012, 124, 2742–2752. [Google Scholar] [CrossRef]
- Ha, N.T.; Shiobara, K.; Yamamoto, Y.; Fukuhara, L.; Nghia, P.T.; Kawahara, S. Preparation and Characterization of Hydrogenated Natural Rubber with Hydroxyl Groups. Polym. Adv. Technol. 2015, 26, 1504–1511. [Google Scholar] [CrossRef]
- Tsupphayakorn-aek, P.; Suwan, A.; Tulyapitak, T.; Saetung, N.; Saetung, A. A Novel UV-Curable Waterborne Polyurethane-Acrylate Coating Based on Green Polyol from Hydroxyl Telechelic Natural Rubber. Prog. Org. Coat. 2022, 163, 106585. [Google Scholar] [CrossRef]
- Panwiriyarat, W.; Tanrattanakul, V.; Pilard, J.-F.; Pasetto, P.; Khaokong, C. Preparation and Properties of Bio-Based Polyurethane Containing Polycaprolactone and Natural Rubber. J. Polym. Environ. 2013, 21, 807–815. [Google Scholar] [CrossRef]
- Zainudin, Z.; Baharulrazi, N.; Che Man, S. Natural Rubber Derivatives for Adhesives Applications: A Review. Chem. Eng. Trans. 2020, 83, 493–498. [Google Scholar] [CrossRef]
- Kandil, H.; Abo-Salem, H.M. A Novel Thiourea Based Interfacial Modifier for Silica-filled Natural Rubber Composites. J. Appl. Polym. Sci. 2023, 140, e53550. [Google Scholar] [CrossRef]
- Katueangngan, K.; Tulyapitak, T.; Saetung, A.; Soontaranon, S.; Nithi-uthai, N. Renewable Interfacial Modifier for Silica Filled Natural Rubber Compound. Procedia Chem. 2016, 19, 447–454. [Google Scholar] [CrossRef]
- Srisuwan, S.; Ruksakulpiwat, Y.; Chumsamrong, P. Physical Properties of Poly (Lactic Acid)/Hydroxyl Terminated Natural Rubber Blends. Macromol. Symp. 2015, 354, 118–124. [Google Scholar] [CrossRef]
- Triampanichkul, N.; Boochathum, P. Hydroxylated Natural Rubber Effect on Crystallinity and Mechanical Properties of PLA. Key Eng. Mater. 2019, 798, 310–315. [Google Scholar] [CrossRef]
- Baharulrazi, N.; Mohd Nor, H.; Wan Ali, W.K. Hydroxyl Terminated Natural Rubber (HTNR) as a Binder in Solid Rocket Propellant. Appl. Mech. Mater. 2014, 695, 174–178. [Google Scholar] [CrossRef]
- Gupta, S.K.; Kurup, M.R.; Devadoss, E.; Muthiah, R.M.; Thomas, S. Development and Evaluation of a Novel Binder Based on Natural Rubber and High-Energy Polyurethane/Composite Propellants. J. Appl. Polym. Sci. 1985, 30, 1095–1112. [Google Scholar] [CrossRef]
- Krishnamurthy, V.N.; Thomas, S. ISRO Polyol-The Versatile Binder for Composite Solid Propellants for Launch Vehicles and Missiles. Def. Sci. J. 1987, 37, 29–37. [Google Scholar] [CrossRef]
- Onn, M.; Md Nor, H.; Wan Ali, W.K. Development of Solid Rocket Propellant Based on Isophorone Diisocyanate–Hydroxyl Terminated Natural Rubber Binder. J. Teknol. 2014, 69, 53–58. [Google Scholar] [CrossRef]
- Thomas, S.; Varghese, T.L.; Gupta, S.K.; Ram, T.S.; Krishnamurthy, V.N. Natural Rubber Based Fuel Rich Propellant for Ramjet Rocket. Def. Sci. J. 1992, 42, 141. [Google Scholar] [CrossRef]
- Sillapasuwan, A.; Saekhow, P.; Rojruthai, P.; Sakdapipanich, J. The Preparation of Hydroxyl-Terminated Deproteinized Natural Rubber Latex by Photochemical Reaction Utilizing Nanometric TiO2 Depositing on Quartz Substrate as a Photocatalyst. Polymers 2022, 14, 2877. [Google Scholar] [CrossRef]
- Banan, A.; Mehdipour, H. Controlled Degradation and Functionalization of Natural Rubber by Ozonolysis in Organic Solvent. J. Polym. Res. 2021, 28, 333. [Google Scholar] [CrossRef]
- Ibrahim, S.; Daik, R.; Abdullah, I. Functionalization of Liquid Natural Rubber via Oxidative Degradation of Natural Rubber. Polymers 2014, 6, 2928–2941. [Google Scholar] [CrossRef]
- Ravindran, T.; Nayar, M.R.G.; Francis, J.D. A Novel Method for the Preparation of Hydroxyl Terminated Liquid Natural Rubber. Die Makromol. Chem. Rapid Commun. 1986, 7, 159–163. [Google Scholar] [CrossRef]
- Ravindran, T.; Nayar, M.R.G.; Francis, D.J. Production of Hydroxyl-terminated Liquid Natural Rubber—Mechanism of Photochemical Depolymerization and Hydroxylation. J. Appl. Polym. Sci. 1988, 35, 1227–1239. [Google Scholar] [CrossRef]
- Pham, H.L.; Do, B.T.; Pham, T.S.; Le, D.G. Synthesis and Characterisation of Hydroxyl-Terminated Liquid Natural Rubber by Photo-Fenton Reaction. ASEAN J. Sci. Technol. Dev. 2017, 30, 29–36. [Google Scholar] [CrossRef]
- Giang, L.D.; Thao, D.L.M.; Huong, H.T.; Thu Hiep, L.T. Synthesis of Hydroxyl Terminated Liquid Natural Rubber by Oxidative Depolymerization of Deproteinized Natural Rubber. Vietnam J. Sci. Technol. 2016, 54, 340. [Google Scholar] [CrossRef]
- Nijpanich, S.; Nimpaiboon, A.; Rojruthai, P.; Sakdapipanich, J. Hydroxyl-Terminated Saponified Natural Rubber Based on the H2O2/P25-TiO2 Powder/UVC-Irradiation System. Polymers 2021, 13, 1319. [Google Scholar] [CrossRef]
- Baharulrazi, N.; Man, S.H.C.; Mohamad, Z.; Nor, H.M.; Ali, W.K.W. Effect of Various Alcohols and Reaction Time on the Properties of Hydroxyl Terminated Natural Rubber Synthesized via Oxidative Degradation. Chem. Eng. Trans. 2017, 56, 583–588. [Google Scholar]
- Aina Shafinaz, M.B.K.; Baharulrazi, N.; Che Man, S.H. Preparation of Reinforced Hydroxyl Terminated Liquid Epoxidized Natural Rubber Nanocomposite by Grafting of Graphene Oxide. Mater. Today. Proc. 2019, 17, 761–767. [Google Scholar] [CrossRef]
- Wisetkhamsai, K.; Patthaveekongka, W.; Arayapranee, W. Study on Degradation of Natural Rubber Latex Using Hydrogen Peroxide and Sodium Nitrite in the Presence of Formic Acid. Polymers 2023, 15, 1031. [Google Scholar] [CrossRef]
- Ibrahim, S.; Othman, N.; Sreekantan, S.; Tan, K.S.; Mohd Nor, Z.; Ismail, H. Preparation and Characterization of Low-Molecular-Weight Natural Rubber Latex via Photodegradation Catalyzed by Nano TiO2. Polymers 2018, 10, 1216. [Google Scholar] [CrossRef] [PubMed]
- Saetung, N.; Campistron, I.; Pascual, S.; Soutif, J.-C.; Pilard, J.-F.; Fontaine, L. Synthesis of Natural Rubber-Based Telechelic Cis-1,4-Polyisoprenes and Their Use to Prepare Block Copolymers via RAFT Polymerization. Eur. Polym. J. 2011, 47, 1151–1159. [Google Scholar] [CrossRef]
- Azhar, N.H.A.; Rasid, H.M.; Yusoff, S.F. Epoxidation and Hydroxylation of Liquid Natural Rubber. Sains Malays. 2017, 46, 485–491. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical Thermodynamics of Random Networks. Proc. R. Soc. Lond. A Math. Phys. Sci. 1976, 351, 351–380. [Google Scholar] [CrossRef]
- Wibowo, H.B.; Dharmawan, W.C.; Wibowo, R.S.M. Bulk Polymerization Kinetics of Hydroxy Terminated Polybutadiene and Toluene Diisocyanate with Infrared Spectroscopy. Indones. J. Chem. 2018, 18, 552. [Google Scholar] [CrossRef]
- Wibowo, H.; Sitompul, H.; Budi, R.; Hartaya, K.; Abdillah, L.; Ardianingsih, R.; Wibowo, R. Hexogen Coating Kinetics with Polyurethane-Based Hydroxyl-Terminated Polybutadiene (HTPB) Using Infrared Spectroscopy. Polymers 2022, 14, 1184. [Google Scholar] [CrossRef]
- Nogueira, R.F.P.; Trovó, A.G.; Silva, M.R.A.D.; Villa, R.D.; Oliveira, M.C.D. Fundamentos e Aplicações Ambientais Dos Processos Fenton e Foto-Fenton. Quim. Nova 2007, 30, 400–408. [Google Scholar] [CrossRef]
- dos Santos, K.J.L.; dos Santos, G.E.D.S.; de Sá, Í.M.G.L.; Ide, A.H.; da Silva Duarte, J.L.; de Carvalho, S.H.V.; Soletti, J.I.; Meili, L. Wodyetia Bifurcata Biochar for Methylene Blue Removal from Aqueous Matrix. Bioresour. Technol. 2019, 293, 122093. [Google Scholar] [CrossRef]
- Bahruddin; Fadhillah, I.; Septian; Wiranata, A.; Zahrina, I. Molecular Weight of Liquid Natural Rubber (LNR) Product from the Chemical Depolymerization Process of High Molecular Weight Narutal Rubber Latex. J. Phys. Conf. Ser. 2020, 1655, 012091. [Google Scholar] [CrossRef]
- Krause, A.; Lange, A.; Ezrin, M.; Ruby, K. Plastics Analysis Guide: Chemical and Instrumental Methods; Hauser Publishers: Munich, Germany, 1983; ISBN 9783446135871. [Google Scholar]
- Hadjiivanov, K. Identification and Characterization of Surface Hydroxyl Groups by Infrared Spectroscopy. In Advances in Catalysis; Academic Press: Cambridge, MA, USA, 2014; pp. 99–318. [Google Scholar]
- Chalmers, J.M. Infrared Spectroscopy in Analysis of Polymers and Rubbers. In Encyclopedia of Analytical Chemistry; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Remizov, A.B.; Kamalova, D.I.; Skochilov, R.A.; Suvorova, I.A.; Batyrshin, N.N.; Kharlampidi, K.E. FT-IR Study of Self-Association of Some Hydroperoxides. J. Mol. Struct. 2004, 700, 73–79. [Google Scholar] [CrossRef]
- Rolere, S.; Liengprayoon, S.; Vaysse, L.; Sainte-Beuve, J.; Bonfils, F. Investigating Natural Rubber Composition with Fourier Transform Infrared (FT-IR) Spectroscopy: A Rapid and Non-Destructive Method to Determine Both Protein and Lipid Contents Simultaneously. Polym. Test. 2015, 43, 83–93. [Google Scholar] [CrossRef]
- Merriman, S.; Chandra, D.; Borowczak, M.; Dhinojwala, A.; Benko, D. Simultaneous Determination of Additive Concentration in Rubber Using ATR-FTIR Spectroscopy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2022, 281, 121614. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, H.B. KAJIAN PROGRAM PENINGKATAN KINERJA PROPELAN KOMPOSIT BERBASIS AP/HTPB/AL. J. Teknol. Dirgant. 2019, 16, 123–138. [Google Scholar] [CrossRef]
- Grishchenko, V.K.; Boiko, V.P.; Svistova, E.I.; Yatsimirskaya, T.S.; Valuev, V.I.; Dmitrieva, T.S. Hydrogen-peroxide-initiated Polymerization of Isoprene in Alcohol Solutions. J. Appl. Polym. Sci. 1992, 46, 2081–2087. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | Mn_exp/g/mol | Mw_exp/g/mol | 1/[Mn} − 1/[Mn]0 | Dispersity |
|---|---|---|---|---|
| 0 | 840,000 | 868,000 | 0 | 1.033333 |
| 20 | 210,000 | 305,100 | 3.57 × 10−6 | 1.452857 |
| 40 | 69,700 | 120,500 | 1.32 × 10−5 | 1.728838 |
| 60 | 21,500 | 51,000 | 4.53 × 10−5 | 2.372093 |
| 80 | 5300 | 11,100 | 8.00 × 10−5 | 2.09434 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wibowo, H.B.; Sutrisno, S.; Sitompul, H.R.D.; Ardianingsih, R.; Abdilah, L.H.; Hartaya, K.; Witjakso, A. Kinetics of Hydroxyl Growth on Natural Rubber Depolymerization with H2O2/Fenton Using Infrared Spectroscopy. Polymers 2025, 17, 1847. https://doi.org/10.3390/polym17131847
Wibowo HB, Sutrisno S, Sitompul HRD, Ardianingsih R, Abdilah LH, Hartaya K, Witjakso A. Kinetics of Hydroxyl Growth on Natural Rubber Depolymerization with H2O2/Fenton Using Infrared Spectroscopy. Polymers. 2025; 17(13):1847. https://doi.org/10.3390/polym17131847
Chicago/Turabian StyleWibowo, Heri Budi, Sutrisno Sutrisno, Hamonangan Rekso Diputro Sitompul, Retno Ardianingsih, Luthfia Hajar Abdilah, Kendra Hartaya, and Ario Witjakso. 2025. "Kinetics of Hydroxyl Growth on Natural Rubber Depolymerization with H2O2/Fenton Using Infrared Spectroscopy" Polymers 17, no. 13: 1847. https://doi.org/10.3390/polym17131847
APA StyleWibowo, H. B., Sutrisno, S., Sitompul, H. R. D., Ardianingsih, R., Abdilah, L. H., Hartaya, K., & Witjakso, A. (2025). Kinetics of Hydroxyl Growth on Natural Rubber Depolymerization with H2O2/Fenton Using Infrared Spectroscopy. Polymers, 17(13), 1847. https://doi.org/10.3390/polym17131847