
Processing of Thermotropic Fully Aromatic Polyesters by Powder Molding Accompanied by Solid-State Post-Polymerization

 , , , ,
, , , ,  and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
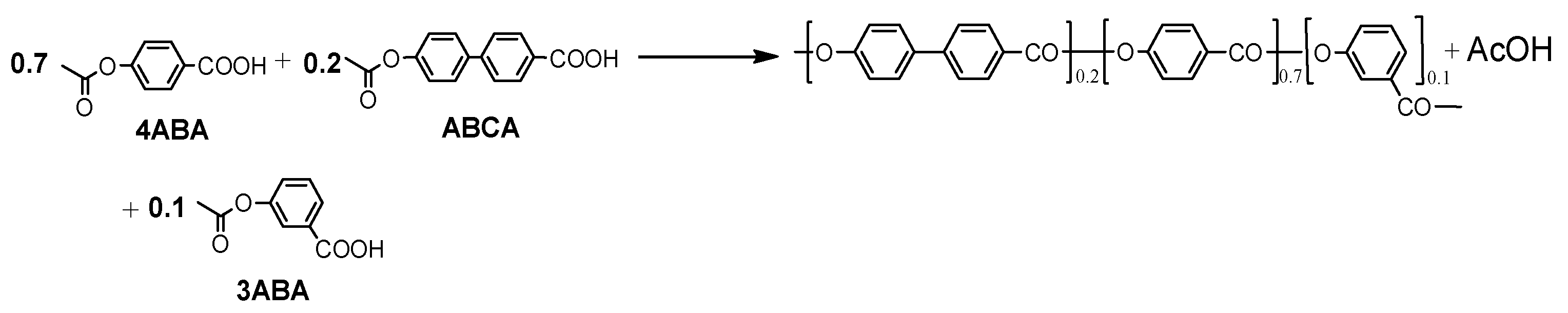
2.2. Prepolymer Synthesis
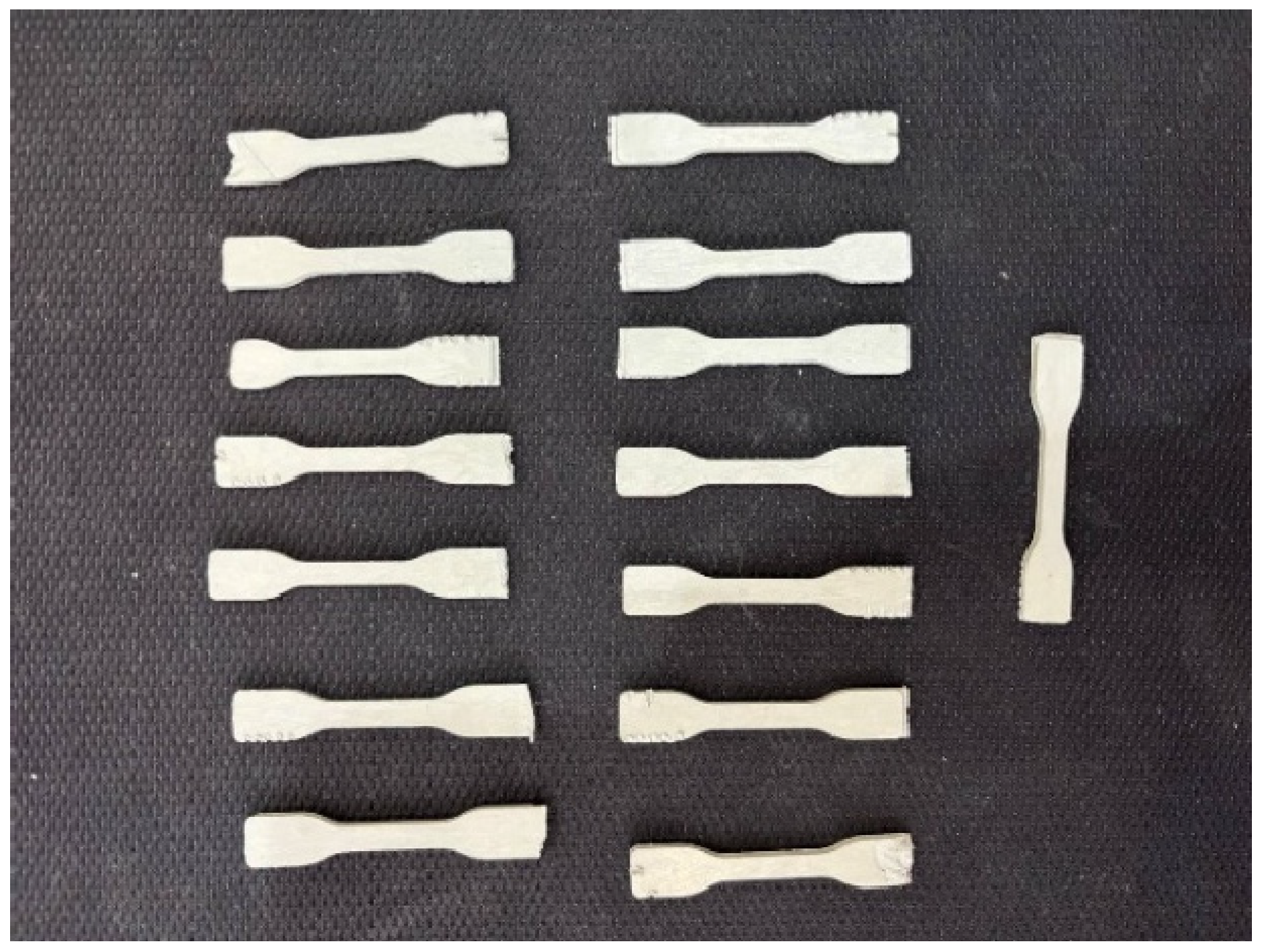
2.3. Sample Preparation
2.4. Polymer Compounding
2.5. Solid-State Polymerization
2.6. Polymer Characterization
3. Results and Discussion
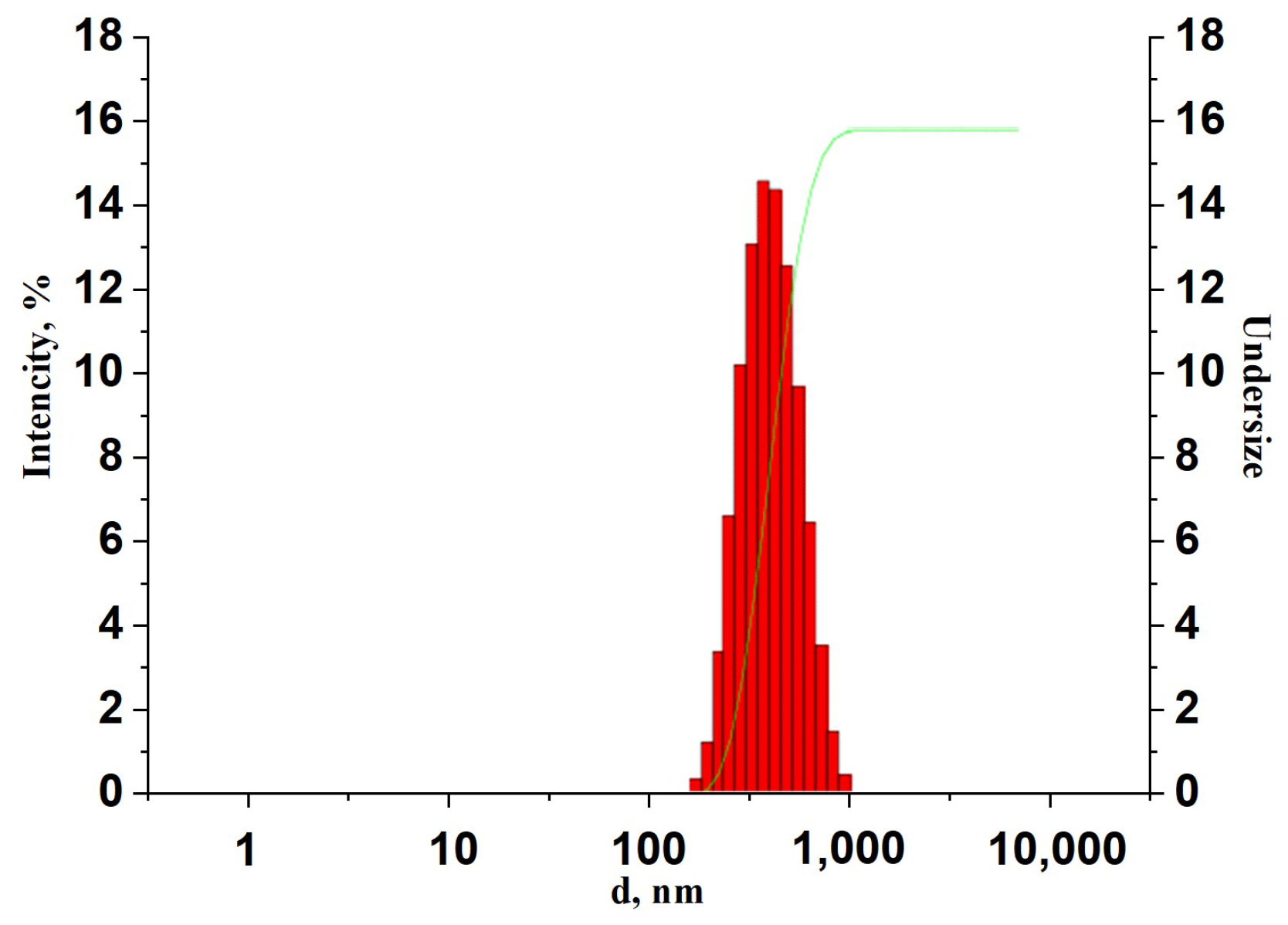
3.1. Synthesis and Sample Preparation
3.2. FTIR Spectroscopy
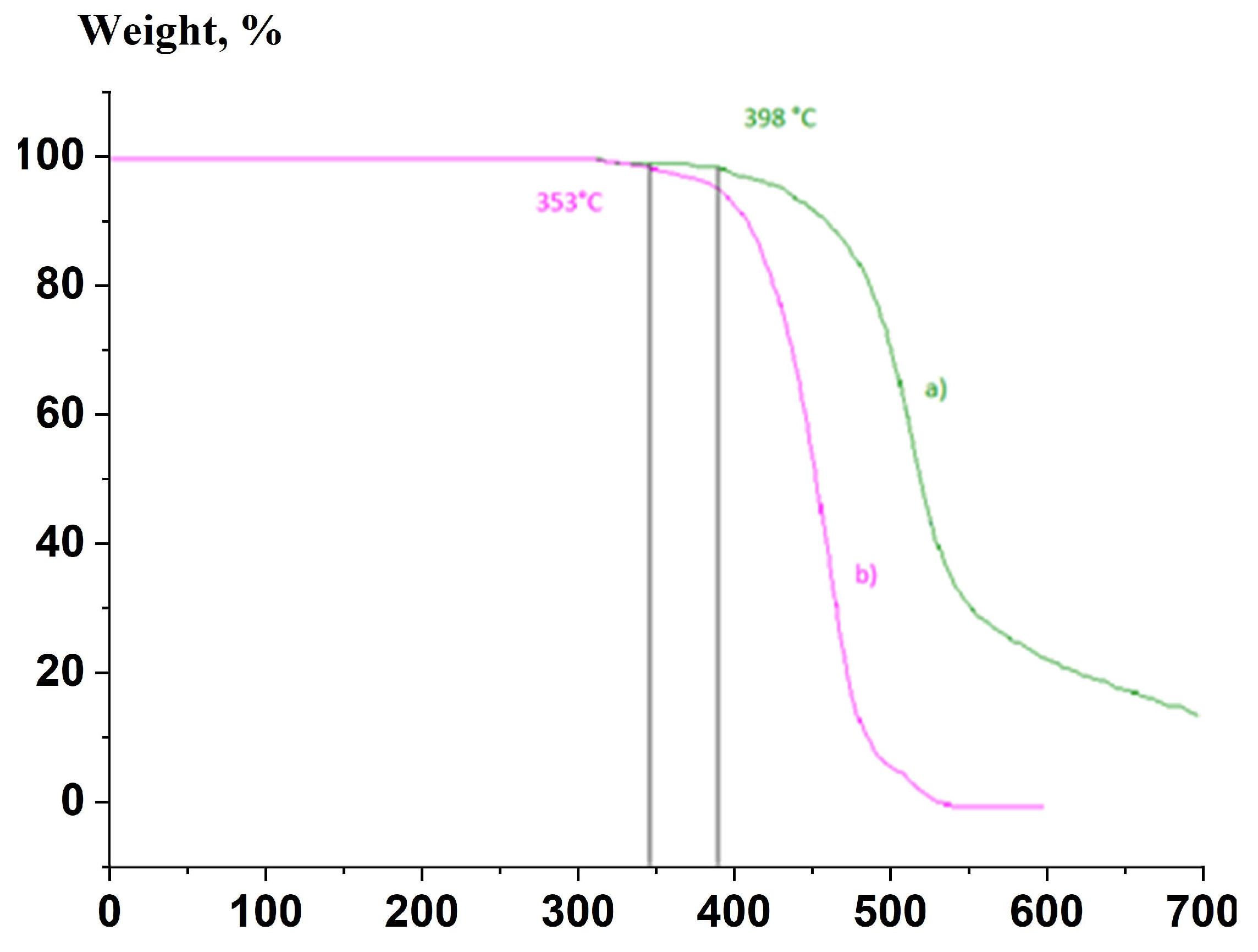
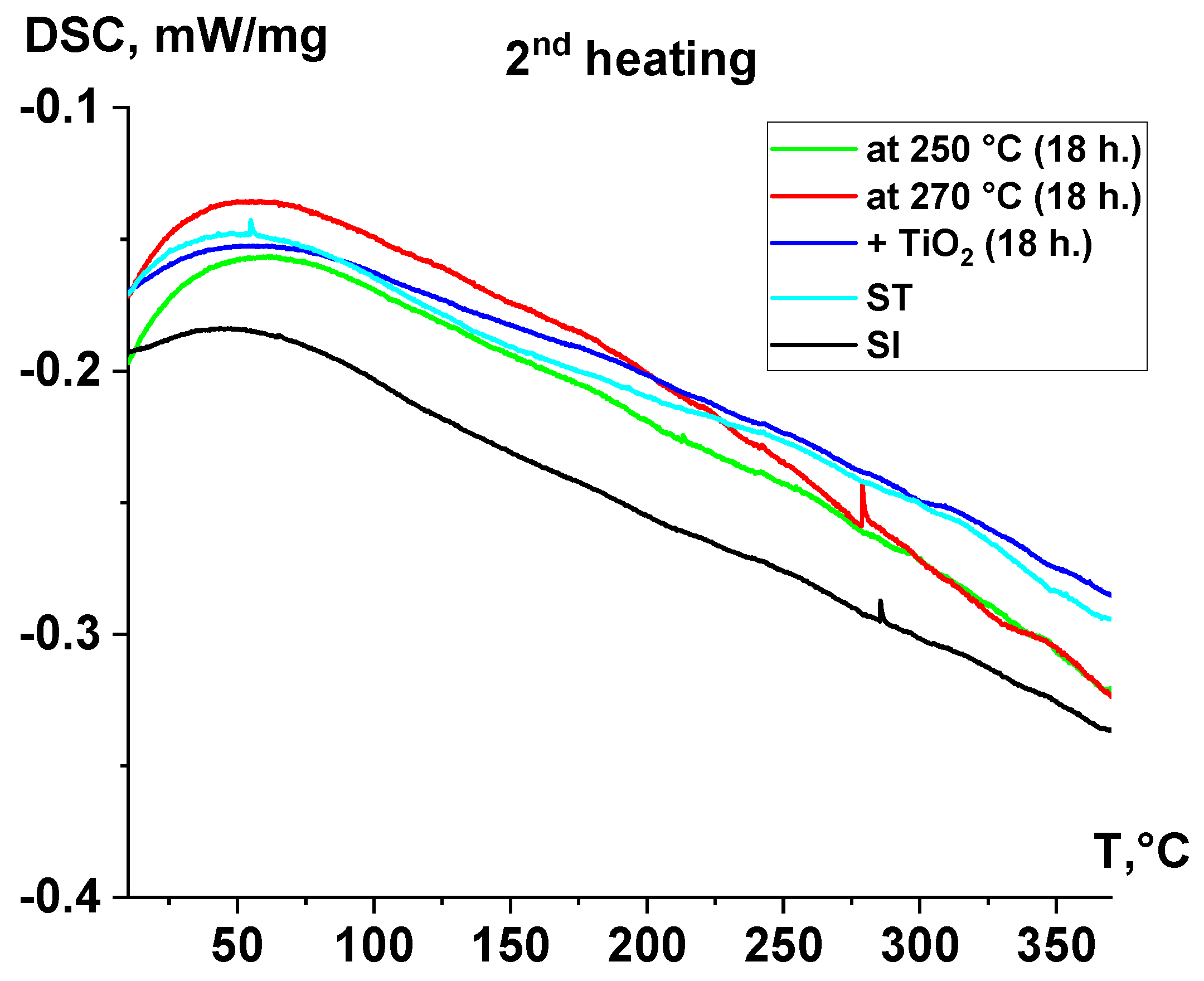
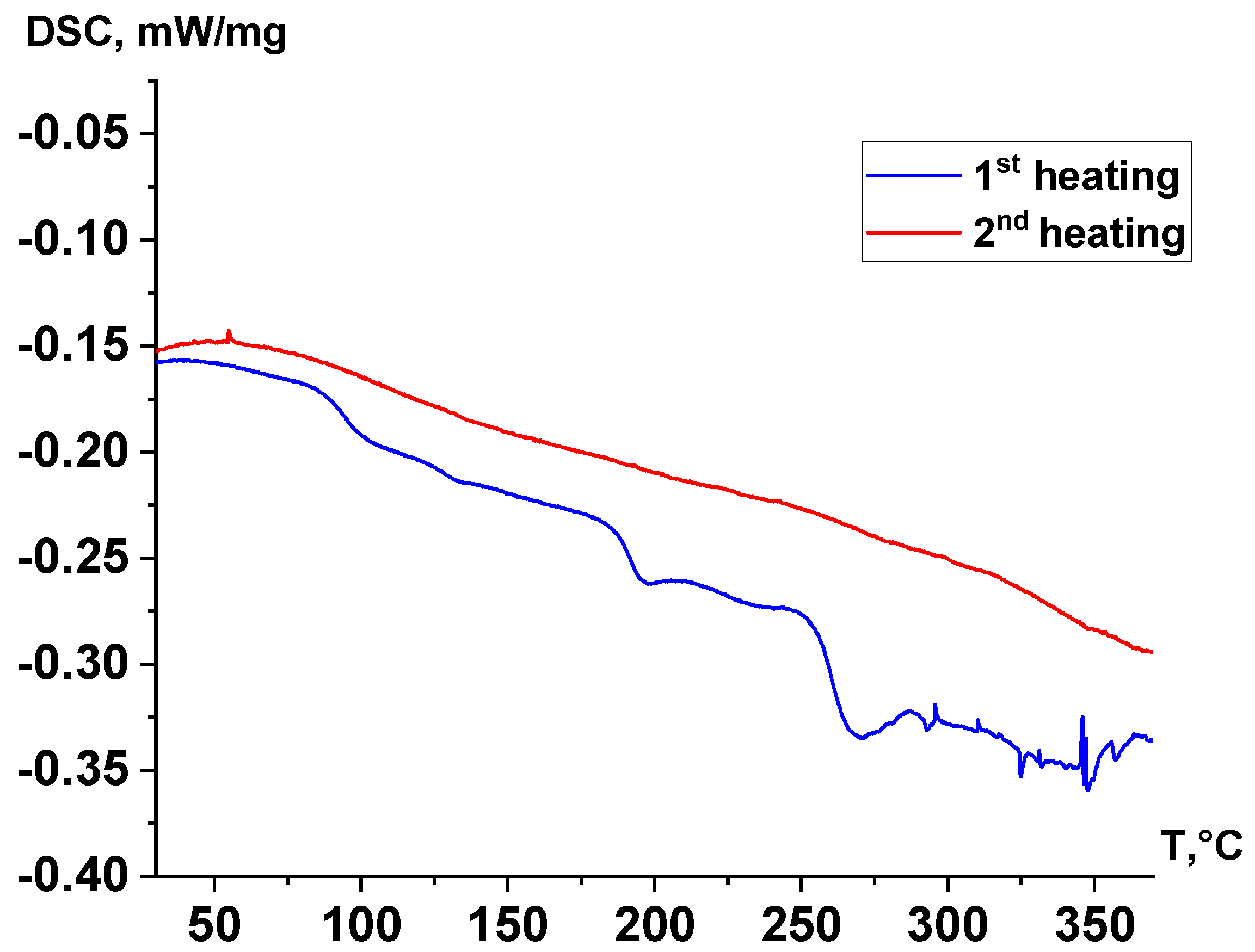
3.3. Thermal Characterization
3.4. Optical Spectroscopy
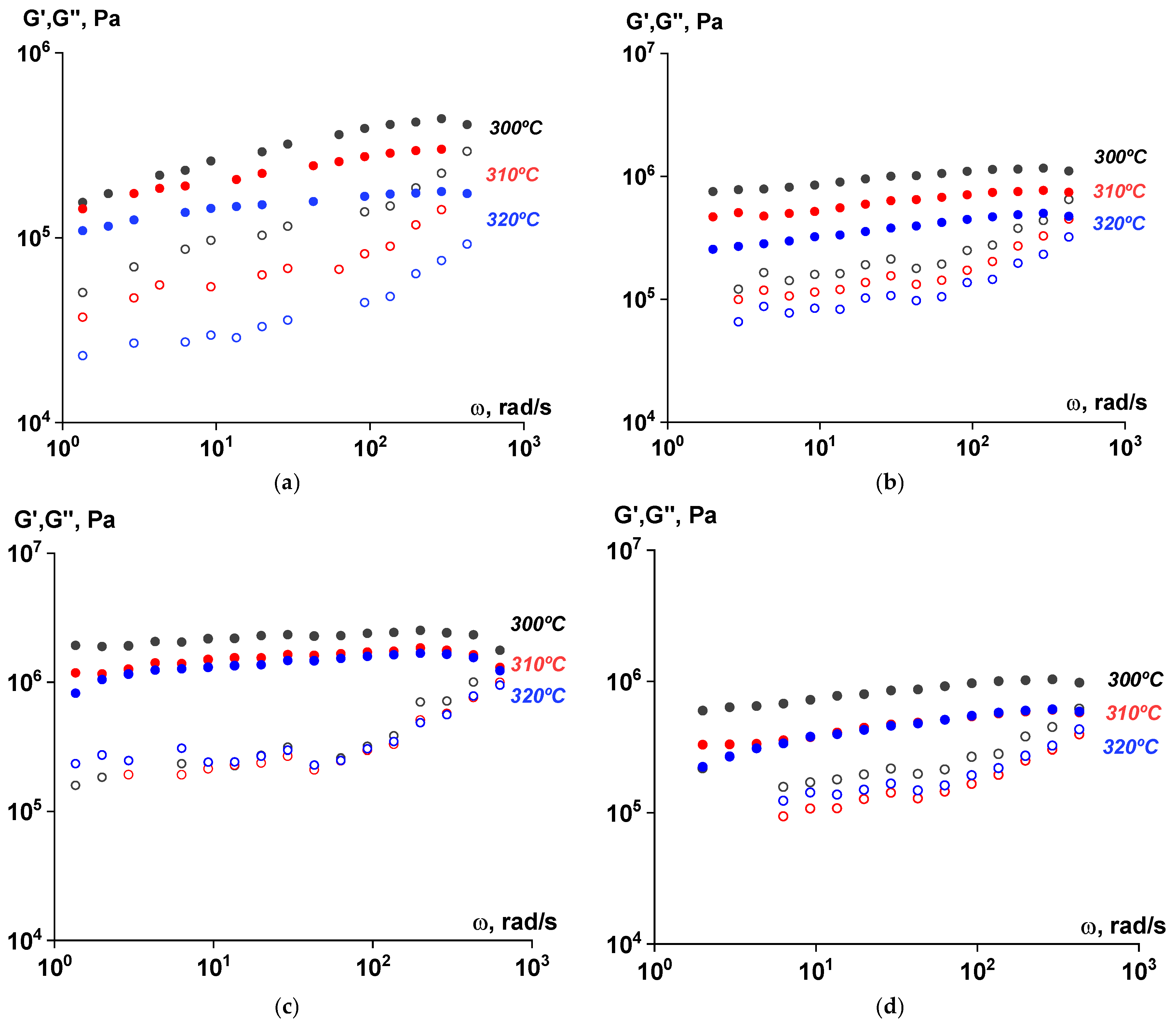
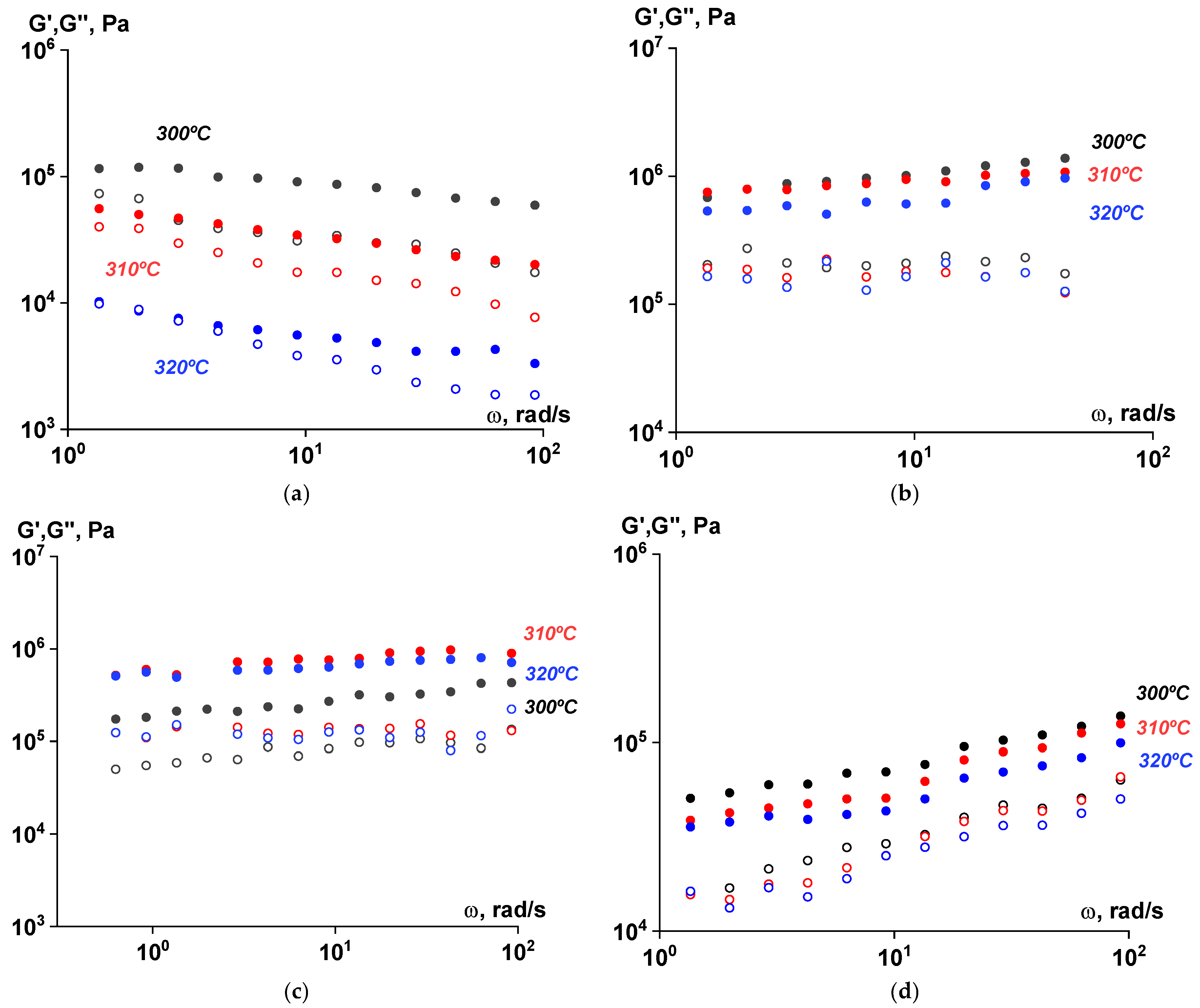
3.5. Rheological Properties
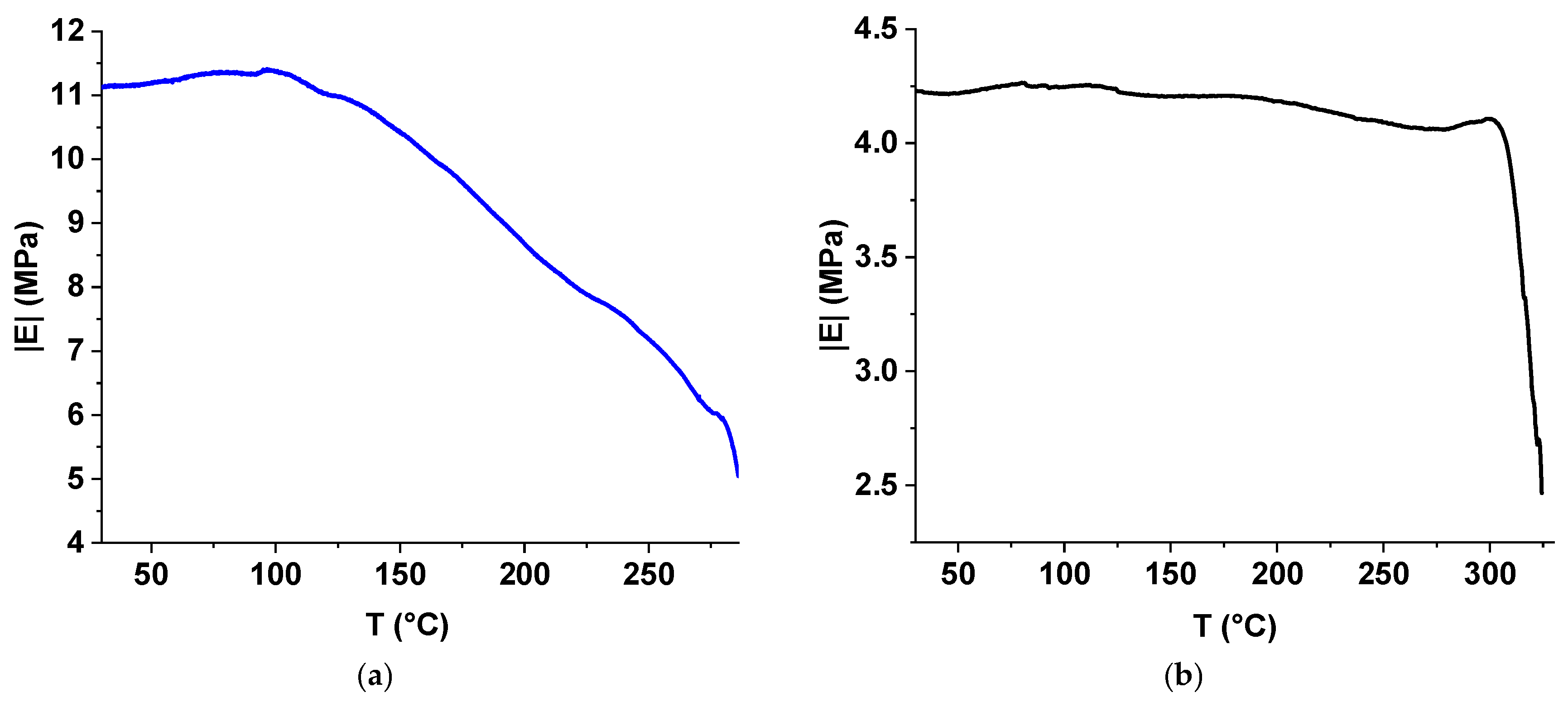
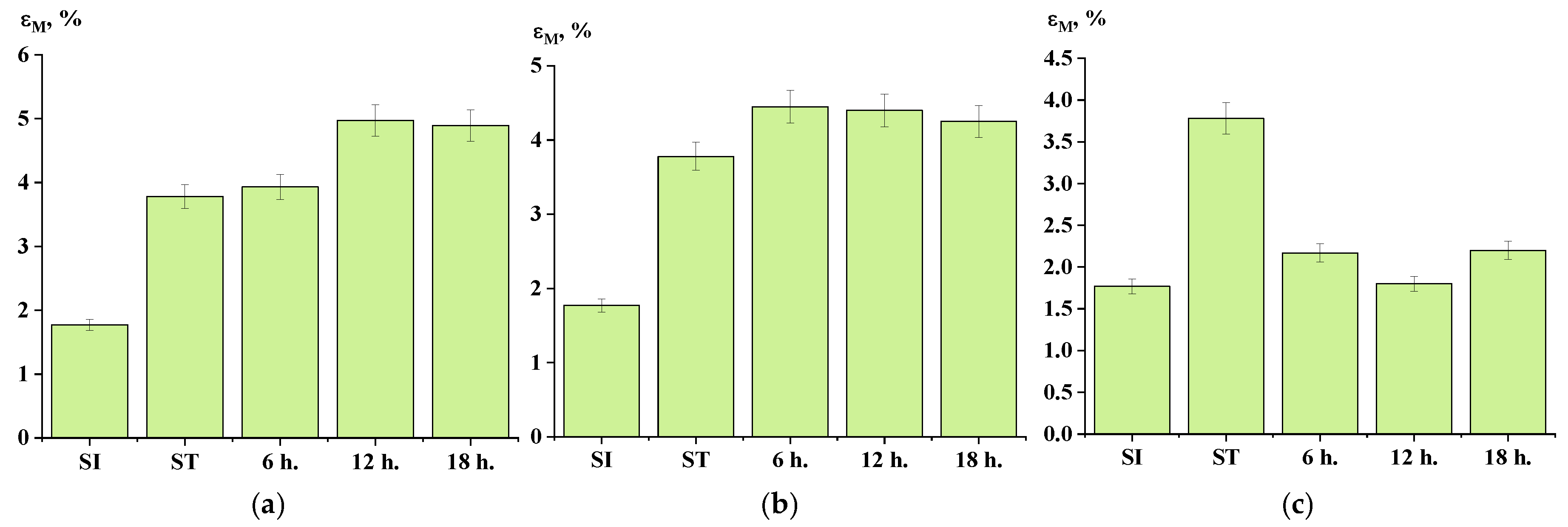
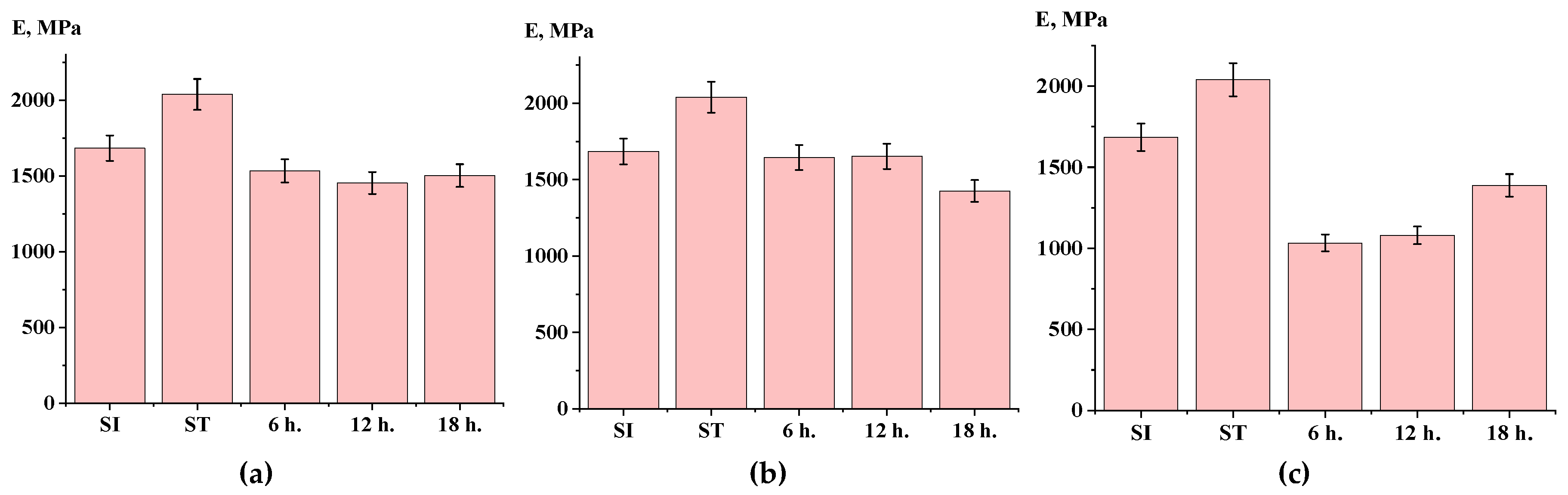
3.6. Mechanical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mikhaylov, P.A.; Zuev, K.V.; Golubev, Y.V.; Kulichikhin, V.G. Fully Aromatic Thermotropic Copolyesters Based on Vanillic, Hydroxybenzoic, and Hydroxybiphenylcarboxylic Acids. Polymers 2024, 16, 1501. [Google Scholar] [CrossRef]
- Mikhaylov, P.A.; Kalita, A.G.; Kulichikhin, V.G. Synthesis of New Thermotropic Fully Aromatic Copolyesters from Hydroxybenzoic and Hydroxybiphenylcarboxylic Acids. Polym. Sci. Ser. B 2022, 64, 393–401. [Google Scholar] [CrossRef]
- Kulichikhin, V.G.; Platé, N.A. Blend Composites Based on Liquid Crystal Thermoplast. Review. Polym. Sci. USSR 1991, 33, 1–37. [Google Scholar] [CrossRef]
- Economy, J. Aromatic Polyesters of p-Hydroxybenzoic Acid. Mol. Cryst. Liq. Cryst. Inc. Nonlinear Opt. 1989, 169, 1–22. [Google Scholar] [CrossRef]
- Jin, J.I.; Kang, C.S. Thermotropic Main Chain Polyesters. Prog. Polym. Sci. 1997, 22, 937–973. [Google Scholar] [CrossRef]
- Han, H.S.; Bhowmik, P.K. Wholly Aromatic Liquid-Crystalline Polyesters. Prog. Polym. Sci. 1997, 22, 1431–1502. [Google Scholar] [CrossRef]
- Jackson, W.J. Liquid Crystal Polymers. XI. Liquid Crystal Aromatic Polyesters: Early History and Future Trends. Mol. Cryst. Liq. Cryst. Inc. Nonlinear Opt. 1989, 169, 23–49. [Google Scholar] [CrossRef]
- Sumitoto Chemical Company. Ultra-High Heat Resistant Engineering Plastics. In Sumikasuper LCP Technical Note; Sumitoto Chemical Company: Tokyo, Japan, 2022. [Google Scholar]
- Watanabe, Y.; Fukushima, K.; Kato, T. Degradation of a Wholly Aromatic Main-Chain Thermotropic Liquid-Crystalline Polymer Mediated by Superbases. JACS Au 2024, 4, 2944–2956. [Google Scholar] [CrossRef]
- Mikhailov, P.A.; Zuev, K.V.; Kulichikhin, V.G. Synthesis and Characterization of Novel Wholly Aromatic Copolyesters Based on 4′-Hydroxybiphenyl-3-Carboxylic and 3-Hydroxybenzoic Acids. Polymers 2023, 15, 2133. [Google Scholar] [CrossRef]
- Blackwell, J.; Gutierrez, G. The Structure of Liquid Crystalline Copolyester Fibers Prepared from P-Hydroxybenzoic Acid, 2,6-Dihydroxy Naphthalene, and Terephthalic Acid. Polymer 1982, 23, 671–675. [Google Scholar] [CrossRef]
- Gutierrez, G.A.; Chivers, R.A.; Blackwell, J.; Stamatoff, J.B.; Yoon, H. The Structure of Liquid Crystalline Aromatic Copolyesters Prepared from 4-Hydroxybenzoic Acid and 2-Hydroxy-6-Naphthoic Acid. Polymer 1983, 24, 937–942. [Google Scholar] [CrossRef]
- Chvalun, S.N.; Ishaq, M.; Blackwell, J.; Kricheldorf, H.R. The Structure of Wholly Aromatic Polyesters with Bulky Side Chains: Poly(p-Phenylene Phenylthioterephthalate). J. Macromol. Sci. Part B 1999, 38, 93–106. [Google Scholar] [CrossRef]
- Muhlebach, A.; Johnson, R.D.; Lyerla, J.; Economy, J. Diad Sequence Distribution in Copolyesters of 4-Hydroxybenzoic Acid and 6-Hydroxy-2-Naphthoic Acid. Macromolecules 1988, 21, 3115–3117. [Google Scholar] [CrossRef]
- Calundann, G.W. Polyester of 6-Hydroxy-2-Naphthoic Acid and Para-Hydroxy Benzoic Acid Capable of Readily Undergoing Melt Processing. U.S. Patent 4,161,470, 17 July 1979. [Google Scholar]
- Volksen, W.; Lyerla, J.R.; Economy, J.; Dawson, B. Liquid-Crystalline Copolyesters Based on Poly(Para-Oxybenzoate) and Poly(Para,Para-Biphenylene Terephthalate). J. Polym. Sci. Part A-Polym. Chem. 1983, 21, 2249–2259. [Google Scholar] [CrossRef]
- Cottis, S.G.; Economy, J.; Wohrer, L.C. Process for Spinning High Modulus Oxybenzoyl Copolyester Fibers. U.S. Patent 3,975,487, 17 August 1976. [Google Scholar]
- Heifferon, K.V.; Spiering, G.A.; Talley, S.J.; Hegde, M.; Moore, R.B.; Turner, S.R.; Long, T.E. Synthesis and Characterization of a Nematic Fully Aromatic Polyester Based on Biphenyl 3,4′-Dicarboxylic Acid. Polym. Chem. 2019, 10, 4287–4296. [Google Scholar] [CrossRef]
- Kitai, T.; Nakayama, T.; Murouchi, S. Fully-Aromatic Thermotropic Liquid Crystal Polyester Resin Composition, Molded Object, and Led Reflector. U.S. Patent 8,715,526, 6 May 2014. [Google Scholar]
- Akiyama, N.; Matsuura, H. Wholly Aromatic Liquid Crystalline Polyester Resin. U.S. Patent 2019/00300700, 3 October 2019. [Google Scholar]
- Lee, G.J.; Burdett, B.A.; Brewbaker, L.; Marshall, B. Melt Processable Thermotropic Aromatic Copolyesters and Process for Preparing Same. European Patent EP0394813, 18 April 1990. [Google Scholar]
- Shintaro, S.; Iwase, H.; Harada, S. Method for Producing Liquid-Crystalline Polyester Resin Composition. U.S. Patent 9,283,707, 15 March 2016. [Google Scholar]
- Kricheldorf, H.R.; Struve, O.; Schwarz, G. Whiskers: 13. Polyesters Based on 4,4′-Biphenyldiol and Biphenyl-4,4′-Dicarboxylic Acid. Polymer 1996, 37, 4311–4320. [Google Scholar] [CrossRef]
- ISO 527-2:2012; Plastics—Determination of Tensile Properties Part 2: Test Conditions for Moulding and Extrusion Plastics. ISO: Geneva, Switzerland, 2012.
- ISO 1133-1:2022; Plastics—Determination of the Melt Mass-Flow Rate (MFR) and Melt Volume-Flow Rate (MVR) of Thermoplastics Part 1: Standard Method. ISO: Geneva, Switzerland, 2012.
- Gilkey, R.; Caldwell, J.R. Polyesters of Hydroxybenzoic Acids. J. Appl. Polym. Sci. 1959, 2, 198–202. [Google Scholar] [CrossRef]
- Economy, J. Liquid-Crystalline Aromatic Polyesters. J. Macromol. Sci.-Chem. 1984, 21, 1705–1724. [Google Scholar] [CrossRef]
- Calundann, G.W. 6-Hydroxy-2-Naphthoic Acid, Para-Hydroxy Benzoic Acid, Aromatic Diol, and Aromatic Diacid Capable of Readily Undergoing Melt Processing. U.S. Patent 4,219,461, 26 August 1980. [Google Scholar]
- Kricheldorf, H.R.; Schwarz, G. Phase Transitions and Melting of Poly(4-Hydroxybenzoate). Polymer 1990, 31, 481–485. [Google Scholar] [CrossRef]
- Schwarz, G.; Kricheldorf, H.R. New Polymer Syntheses, 23. Poly(Oxy-4,4′-Biphenylylenecarbonyl). Die Makromol. Chem. Rapid Commun. 1988, 9, 717–720. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Ruhser, F.; Schwarz, G. New Polymer Syntheses. 48. Thermotropic Copolyesters of 4′-Hydroxybiphenyl-4-Carboxylic Acid and 4-Hydroxybenzoic Acid or 3-Chloro-4-Hydroxybenzoic Acid. Macromolecules 1991, 24, 4990–4996. [Google Scholar] [CrossRef]
- Mikhaylov, P.A.; Zuev, K.V.; Filatova, M.P.; Strelets, B.K.h.; Kulichikhin, V.G. Synthesis and Properties of Thermotropic Copolyesters Based on Poly(Ethylene Terephthalate) and 4′-Acetoxy-4-Biphenyl-Carboxylic Acid. Polymers 2021, 13, 1720. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SSP Temperature, °C | SSP duration, h | IV in PFP |
|---|---|---|---|
| SI | - | - | 0.5 |
| ST | - | - | 3.0 |
| ST-250-6 | 250 | 6 | 4.9 |
| ST-250-12 | 250 | 12 | 6.3 |
| ST-250-18 | 250 | 18 | Insoluble |
| ST-270-6 | 270 | 6 | 4.5 |
| ST-270-12 | 270 | 12 | Insoluble |
| ST-270-18 | 270 | 18 | Insoluble |
| Sample | T, °C | Load, kg | MFI, g/10 min |
|---|---|---|---|
| ST-270-18 | 320 | 2.16 | 0.4 |
| ST-270-18 | 320 | 21.6 | 12.4 |
| ST-250-18 | 320 | 2.16 | 5.7 |
| ST-250-18 | 320 | 21.6 | 93 |
| ST | 320 | 2.16 | 102 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhaylov, P.A.; Mityukov, A.V.; Dudka, D.V.; Golubev, Y.V.; Kulichikhin, V.G.; Malkin, A.Y. Processing of Thermotropic Fully Aromatic Polyesters by Powder Molding Accompanied by Solid-State Post-Polymerization. Polymers 2025, 17, 1358. https://doi.org/10.3390/polym17101358
Mikhaylov PA, Mityukov AV, Dudka DV, Golubev YV, Kulichikhin VG, Malkin AY. Processing of Thermotropic Fully Aromatic Polyesters by Powder Molding Accompanied by Solid-State Post-Polymerization. Polymers. 2025; 17(10):1358. https://doi.org/10.3390/polym17101358
Chicago/Turabian StyleMikhaylov, Pavel A., Anton V. Mityukov, Dmitry V. Dudka, Yaroslav V. Golubev, Valery G. Kulichikhin, and Alexander Ya. Malkin. 2025. "Processing of Thermotropic Fully Aromatic Polyesters by Powder Molding Accompanied by Solid-State Post-Polymerization" Polymers 17, no. 10: 1358. https://doi.org/10.3390/polym17101358
APA StyleMikhaylov, P. A., Mityukov, A. V., Dudka, D. V., Golubev, Y. V., Kulichikhin, V. G., & Malkin, A. Y. (2025). Processing of Thermotropic Fully Aromatic Polyesters by Powder Molding Accompanied by Solid-State Post-Polymerization. Polymers, 17(10), 1358. https://doi.org/10.3390/polym17101358