1. Introduction
Triple-negative breast cancer (TNBC) is an aggressive subtype that accounts for ca. 15% of all breast tumors and is associated with poor prognosis and a high rate of recurrence. TNBC is characterized by the absence of estrogen receptors, progesterone receptors, and HER2 receptors, which limits the efficacy of targeted therapeutic agents [
1]. To address these challenges, novel strategies are being explored, such as the development of rapid breast cancer screening using Raman-based analysis [
2,
3]. Additionally, the development of innovative drugs targeting the epigenome of TNBC cells is also under investigation. Among these, compounds targeting the bromo and extra terminal domain proteins (BET), such as JQ1, have emerged as promising candidates and are currently under clinical development [
4,
5]. However, its widespread use is restricted by various factors. These include the development of acquired resistance due to compensatory pathways, limited long-term tolerability, and off-target effects on non-tumor tissues expressing the target protein. Furthermore, the highly lipophilia related to its chemical structure causes poor solubility which hinders cell permeability and therefore could require the use of higher doses for the treatment of patients. Such events might result in significant non-tumor toxicity and adverse effects on patients [
6]. In this context, nanomedicine offers a promising approach, potentially overcoming several of such existing drawbacks. By encapsulating JQ1 within nanoparticles (NPs), its delivery and pharmacokinetic profile can be enhanced, allowing for targeted delivery to tumor sites. This approach has the potential to overcome chemotherapy resistance, minimize systemic toxicity, and improve therapeutic outcomes. However, the development of drug delivery systems (DDS) remains in its early stages, requiring further validation and optimization. Key challenges include increased complexity, high costs associated with carrier materials and pharmaceutical excipients, and the need for rigorous assessment of safety and efficacy.
Designing innovative nanomedicines for rapid clinical translation remains a significant challenge. To date, only a limited number of nanomedicines have received clinical approval for cancer treatment. Among the various nanosystems, polymeric NPs have emerged as a leading drug delivery platform due to their exceptional biocompatibility, high bioavailability, and ability to carry multiple therapeutic payloads [
7]. One of the most promising materials for developing polymeric-based NPs is the Food and Drug Administration (FDA)-approved poly(lactic acid) (PLA) [
8]. Its advantageous physicochemical properties along with its low price made it an environmentally-friendly alternative to traditional petroleum-derived polyolefins. Furthermore, PLA is highly biocompatible, as its hydrolysis in physiological conditions generates lactic acid, an endogenous molecule that is naturally metabolized into water and carbon dioxide. These features position PLA as an ideal candidate for the development of advanced nanocarrier systems for cancer therapeutics [
9].
The physicochemical properties of PLA depend on different parameters such as stereoregularity architecture or molecular weight, which can be tuned by controlling the polymerization process, offering the possibility to tailor the properties of the final material for specific applications [
10]. In particular, the FDA approved the utilization of PLA for biomedical purposes back in 1960, reaching the clinical level for several applications such as sutures, dermal fillers, tissue engineering, or DDSs [
11,
12,
13]. The design of DDSs based on PLA derivatives allows tuning the encapsulation and loading efficiency, the release rate of the drug, etc. by modifying the synthesis and post-synthesis processing of PLA [
14]. Nowadays, more than 15 PLA-based microparticles have already reached the market to control drug delivery [
11] and in particular, PLA microparticles have been applied for the treatment of several pathologies such as cancer [
15,
16] or insulin-dependent diabetes [
17]. However, the absence of a clinical application for PLA nanoparticles despite the research abundance on the drug load/release might be due to the lack of know-how transfer between the fundamental research on the DDS design and clinical trials, which might be due to a lack of understanding over the formulation process. The lack of robustness and reproducibility during the nanoformulation of PLA at laboratory scale hampers the successful transfer from research to clinical application [
18,
19].
Numerous factors contribute to forming the properties of micro- and nanoparticles [
20] and several studies showed contradictory results for the same parameter-property relationship [
21,
22]. Particularly, the nanoparticle size was found to decrease [
23] or increase [
24] with increasing the PLA molecular weight. Therefore, novel formulation approaches focussing on a deeper understanding of the nanoparticle formation mechanism as well as its final thermodynamical state depending on the processing conditions are necessary to control the drug delivery process such as the initial burst release [
19,
20].
The conversion of JQ1 into nanomedicines has been scarcely explored. Previous studies have demonstrated that encapsulating JQ1 in polymeric NPs can reduce the expression of c-Myc mRNA in tumor tissues more effectively than free JQ1 [
25]. Moreover, liposomal NPs [
26] were also formulated to encapsulate JQ1, as well as poly(ethylene glycol)-poly(aspartic acid)-NPs [
27] and Zein NPs [
28]. Despite these advancements, the impact of formulation parameters on the properties of NPs is often overlooked. Inadequate control of these parameters can lead to significant variability in the properties of the final formulation [
20], potentially compromising its efficacy and reproducibility. Therefore, a deeper understanding of how formulation and process parameters influence NPs properties is essential for optimizing JQ1-based nanomedicines.
In this study, a series of PLA derivatives with varying molecular weights were synthesized via ring-opening polymerization (ROP) using an organometallic zinc-based initiator, allowing for tuning the properties of the polymer. The physicochemical characteristics of the PLA derivatives were analyzed using thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and infrared spectroscopy (FTIR). Subsequently, PLA-based NPs were formulated, and the influence of three key parameters—PLLA molecular weight, organic phase concentration, and excipient concentration—on the final NPs properties was systematically investigated. While the complexity of process development requires consideration of numerous factors, this study focused on these parameters and their effects on particle size, polydispersity index (PDI), and thermodynamic state. Additionally, the encapsulation efficiency, drug loading, and release profile were evaluated as critical variables.
This targeted approach provides insights into optimizing formulation parameters for the development of advanced polymeric nanocarrier systems.
4. Discussion
BET proteins were discovered in the early 1990s [
3,
43] and in the last decade BET inhibitors have emerged as a novel strategy to treat TNBC [
25]. Since then, several compounds have been developed and are currently under clinical development [
44]. Among BET inhibitors, JQ1 has demonstrated strong anti-cancer activity in preclinical models in hematological and solid tumors [
44,
45]. However, JQ1 presents a relatively short half-life in vivo (approximately 1 h in CD1 mice) due to a rapid clearance by cytochrome P450 enzymes that limits its effectiveness in long-term treatments requiring frequent dosing to maintain therapeutic levels. Moreover, JQ1 present a high LogP (6.41) value indicating a significant lipophilicity which might limit its solubility and distribution. In addition, JQ1 inhibits the BET family proteins in a non-selective manner which can result in off-target effects and resistance development [
45,
46]. Addressing these disadvantages requires a multi-disciplinary approach, including pharmacology, molecular biology, and material science. Different strategies have been proposed to improve the efficacy of JQ1 such as combination therapies [
27,
47], identifying biomarkers to predict selectivity to JQ1 [
4] and optimising pharmacokinetics [
26]. Optimizing the pharmacokinetics using polymeric NPs is a promising strategy that enables drug-controlled release of drugs and targeted delivery, improving efficacy while reducing side effects [
7]. Particularly, the use of FDA-approved polymers such as PLGA or PLA, is a well-established strategy to optimize DDS since, besides their biocompatibility and biodegradability properties, they assure prompt translation to the clinics [
8]. PLA is a more hydrophobic polymer than PLGA due to the lack of glycolic acid units and, therefore, is suitable for encapsulating hydrophobic drugs such as JQ1. Moreover, the hydrophobic character of PLA extends its degradation under physiological conditions and is preferable when a more sustained release is required. These features position PLA as an ideal candidate for the development of advanced nanocarrier systems for the encapsulation of JQ1 [
9]. However, contradictory results are often found in literature due to a lack of understanding of the formulation parameters effect on the final release profile. Moreover, the synthesis of PLA at the industrial level is usually accomplished using Sn-derived catalysts, which is potentially harmful due to its accumulation in brain and lung tissue, limiting its application in the biomedical field. Therefore, the synthesis of well-controlled PLA macrostructures using biocompatible catalytic systems, which enables good control over the polymerization process is required.
The main objective of this study was to optimize formulations for converting JQ1 into nanomedicines. This was achieved by evaluating the impact of various parameters on the RH, PdI, and Z-potential of the formulations. Additionally, the physicochemical state of the NPs was assessed, as it plays a critical role in determining the release profile of JQ1.
In the first part of the article PLA derivatives presenting different molecular weights were synthesized through a Zn-based initiator presenting high monomer conversion and good control of the molecular weight (
Figure 1 and
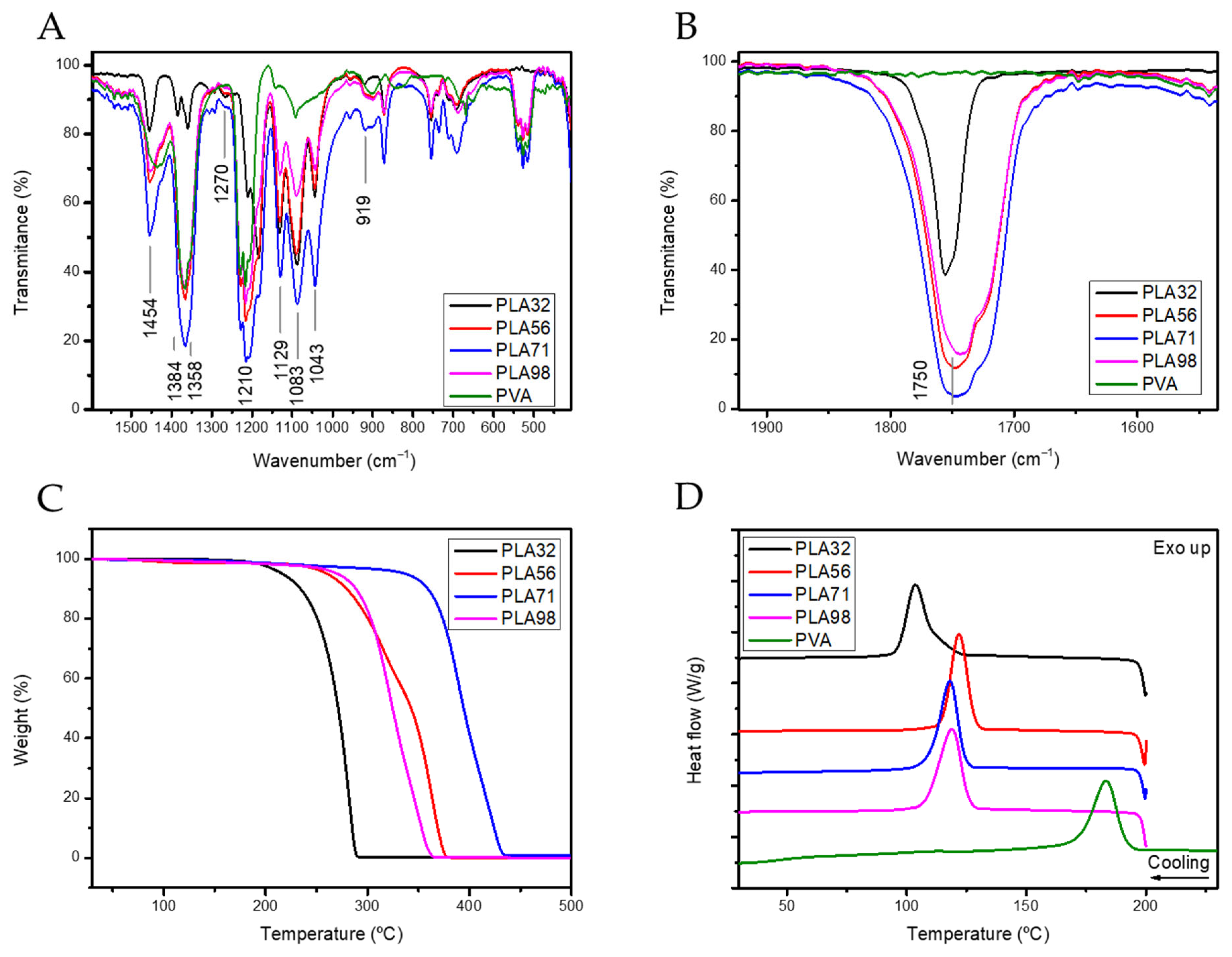
Table 1). The organometallic compound containing Zn as metal was selected attending the biocompatibility characteristics of Zn, since traces of the catalyst might be trapped within the polymer structure. Then, the influence of the molecular weight on the physicochemical properties of the polymers was evaluated by TGA and DSC (
Figure 2). The molecular weight significantly affected the degradation temperature, likely due to the entrapment of the catalyst and unreacted monomers within the polymer matrix. In addition, the T
g temperature remained mainly constant with the increase of the molecular weight. However, the increase of the PLA molecular weight affected to physicochemical state of the polymer as evidenced by the double melting peak observed by DSC, suggesting that the crystallization of PLA is hampered by the increase of the molecular weight. The double melting peak observed could be ascribed to the partial crystallization of the polymer in a defective crystal phase, which may affect the release rate (
Table 2).
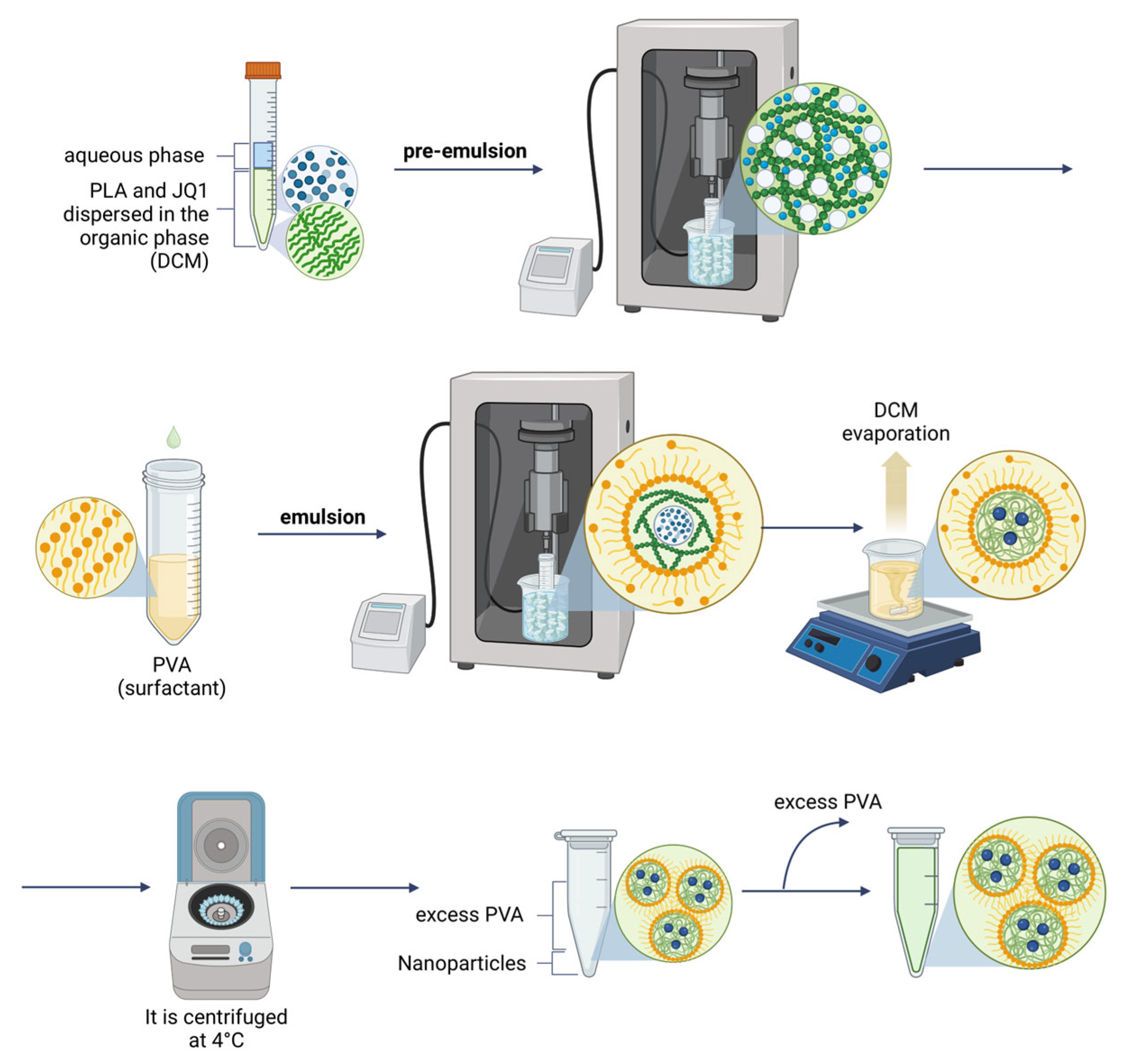
Subsequently, polymeric NPs were formulated by a double emulsion method (
Figure 3), obtaining an R
H of ca. 150 nm and monodispersed distributions (
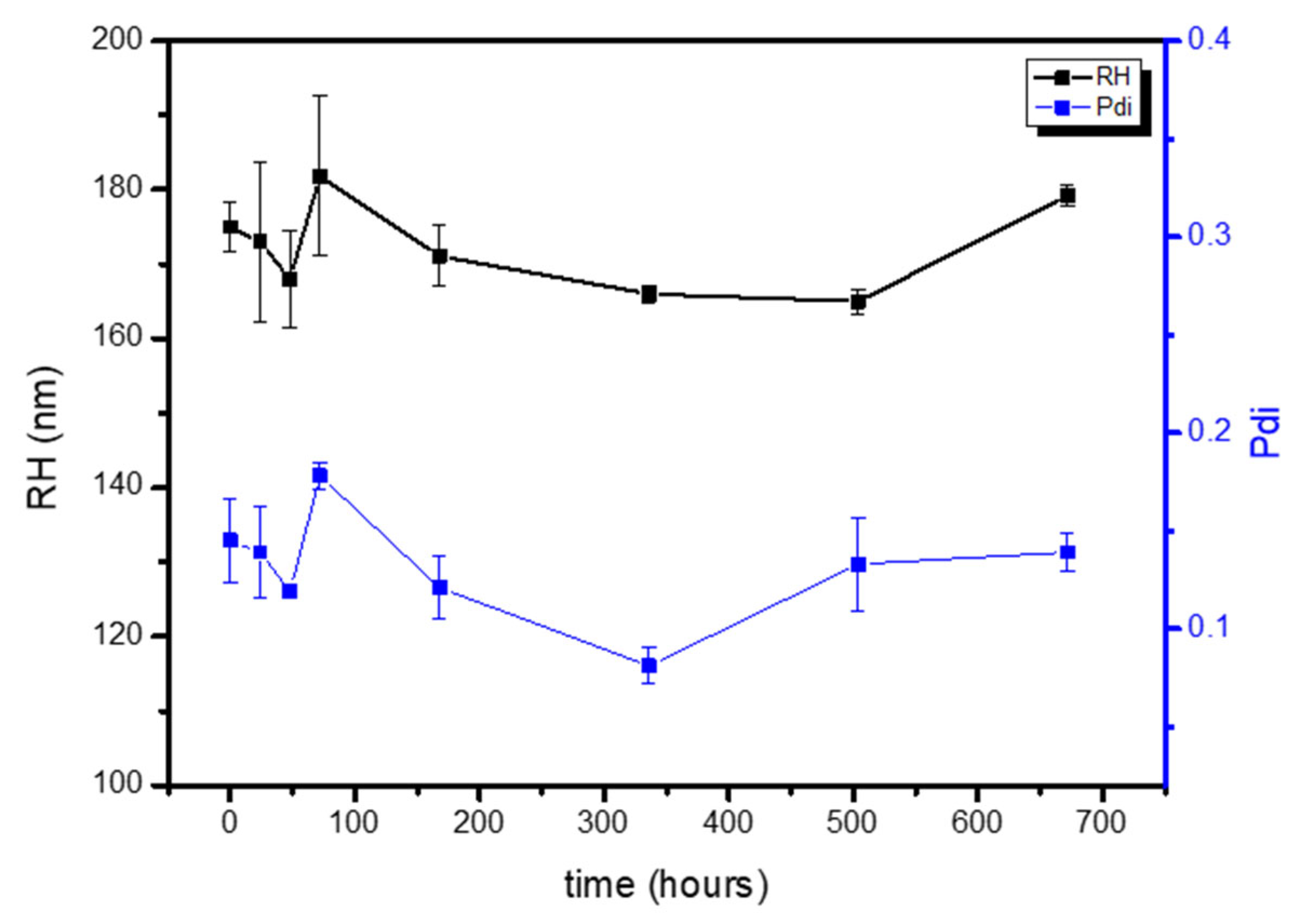
Table 3), which remained constant for 4 weeks stored at 4 °C (
Figure 4). Three parameters of the formulation process were analyzed to understand their effect on the final physicochemical state: the PLA molecular weight (PLA-Mw), PLA concentration in the organic phase, and finally the PVA concentration in the aqueous phase.
The variation in PLA-Mw had a significant impact on the size of the NPs, with lower PLA-Mw proving to be the most suitable raw material for formulation. This is because it resulted in smaller and more homogeneously dispersed nanodevices (
Table 3). DSC analysis of the different polymeric NPs (
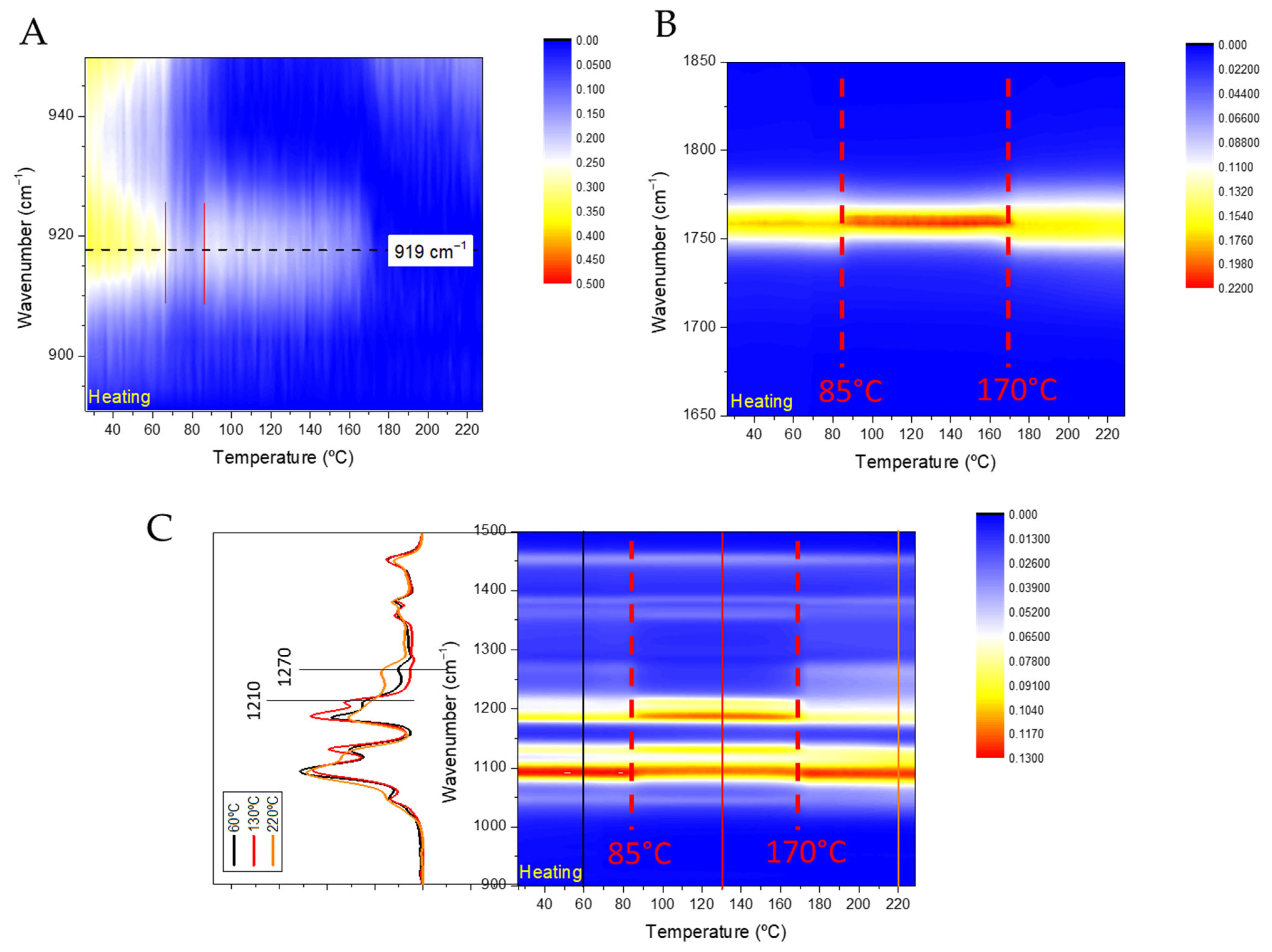
Figure 5A) showed negligible differences compared to the raw PLA (
Table 4), regardless of molecular weight, which follows previous reports [
48]. The effect of PLA concentration on the formulation process was less pronounced. However, the lower PLA concentration (2.5 g/L) proved optimal for fine-tuning the polymeric formulations (
Table 5). The thermodynamic state of the NPs after formulation was found to be independent of the polymer concentration used, as evidenced by DSC analysis (
Table 6 and
Figure 5B). On the other hand, the PVA concentration had a definitive impact on the formulation process. While PDI and Z-potential values remained unaffected by variations in PVA concentration, the size of the NPs decreased with increasing PVA concentration (
Table 7), which is attributed to the high surface/volume ratio obtained as the PVA concentration increases. The effect of PVA concentration was further analyzed through the thermodynamic state of the NPs (
Table 8 and
Figure 5C), which suggested that higher PVA concentrations hamper the crystallization of PLA, forcing the formation of the defective α’-phase (
Figure 5C).
Once the formulation process was optimized, we proceeded with the encapsulation of the BETi inhibitor JQ1. The main challenge was to tailor the release of the drug and loading by fine-tuning three key variables identified as critical for encapsulation: PLA-Mw, PLA concentration, and PVA concentration. Based on our results, we determined that a lower PLA Mw, combined with a polymer concentration of 2.5 g/L, was optimal for further encapsulation processes. However, the PVA concentration appeared to be a decisive factor for drug loading efficiency and release. Therefore, various PVA concentrations were systematically studied, 1–3% PVA. Due to the high hydrophobic character of JQ1, a PVA concentration of 1% resulted in higher EE% and LE%. The results suggest that higher PVA concentrations disrupt interactions between the PLA core and JQ1, potentially reducing both EE% and LE% (
Table 9).
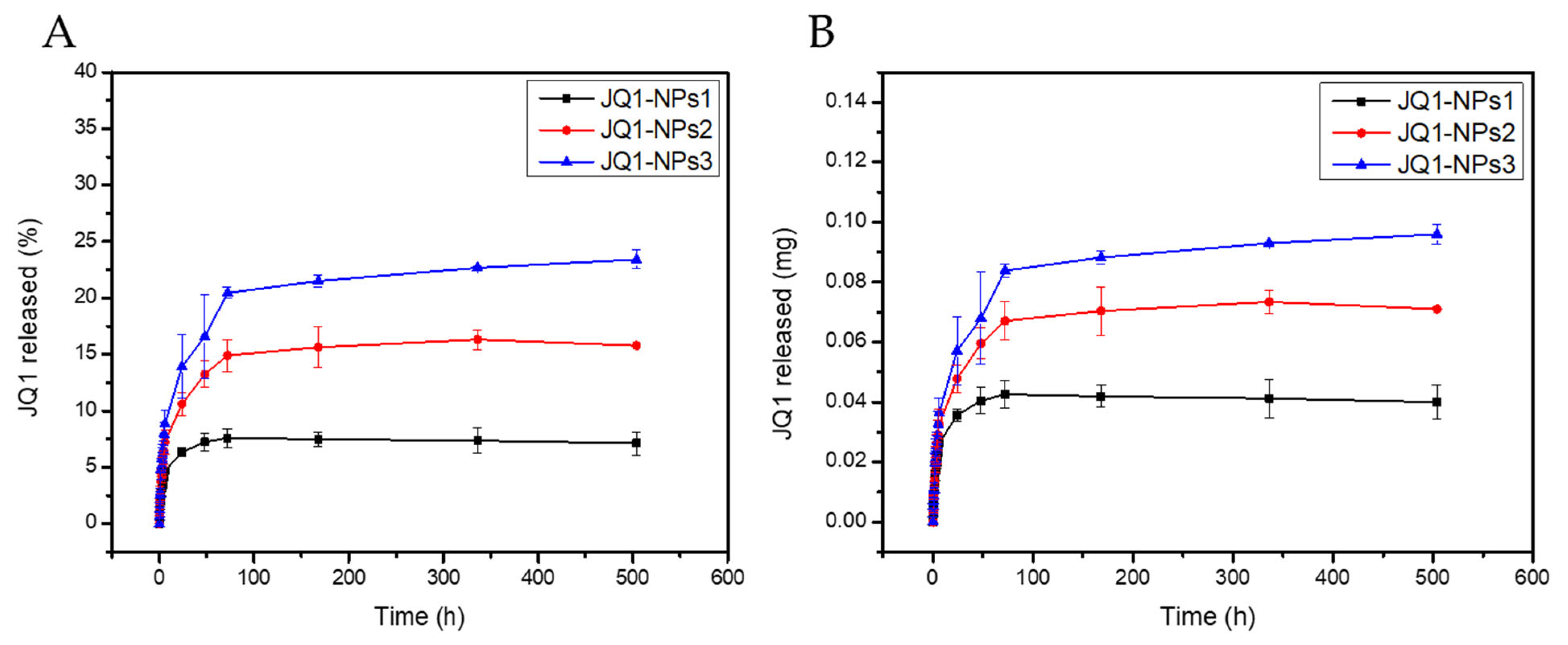
Finally, a triphasic release profile of JQ1 was observed for the three PVA-dependent formulations, indicating successful optimization (
Figure 7). The negligible burst release further supports the stability of the formulations. Additionally, the results demonstrated that the release rate of the drug could be modulated by varying the PVA concentration. Higher PVA concentrations led to faster drug release, likely due to the hydrophilic nature of PVA, which enhances water diffusion into the nanoparticle core.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}