
Cellulose Acetate–Ionic Liquid Blends as Potential Polymers for Efficient CO2 Separation Membranes




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Instruments
2.2. Synthesis of
2.3. CA-IL Film Preparation
2.4. CO2 and N2 Sorption Measurements and Estimation of Diffusion Coefficients and Permeability
3. Results and Discussion
3.1. CA-IL Membrane Characterization
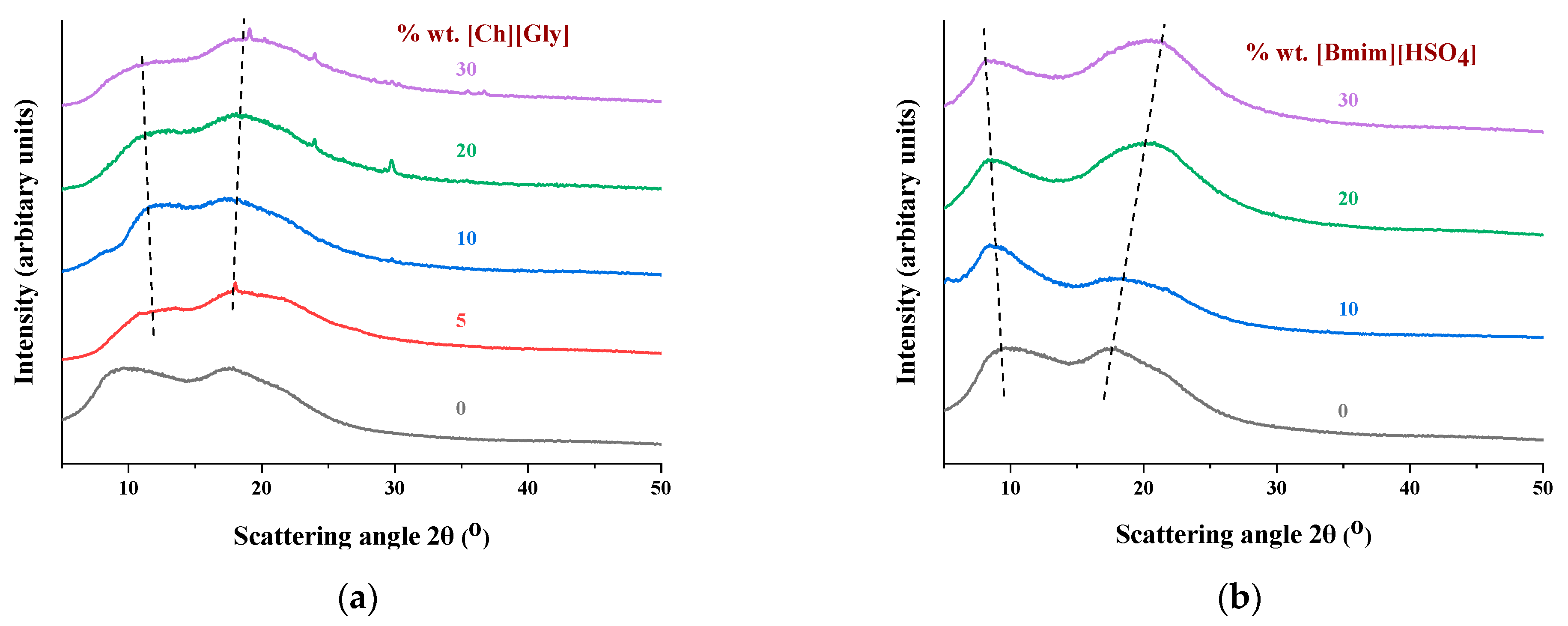
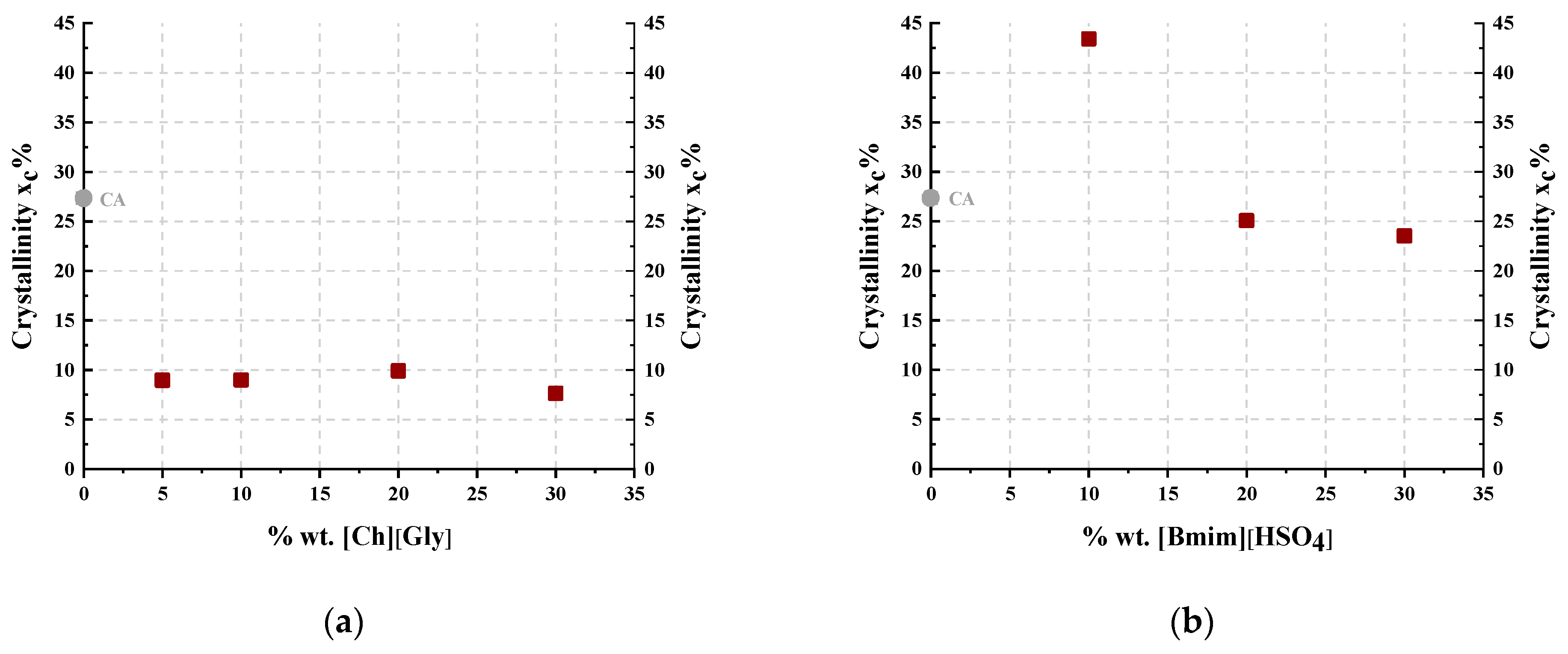
3.1.1. Membrane Structural Properties by X-ray Diffraction Studies
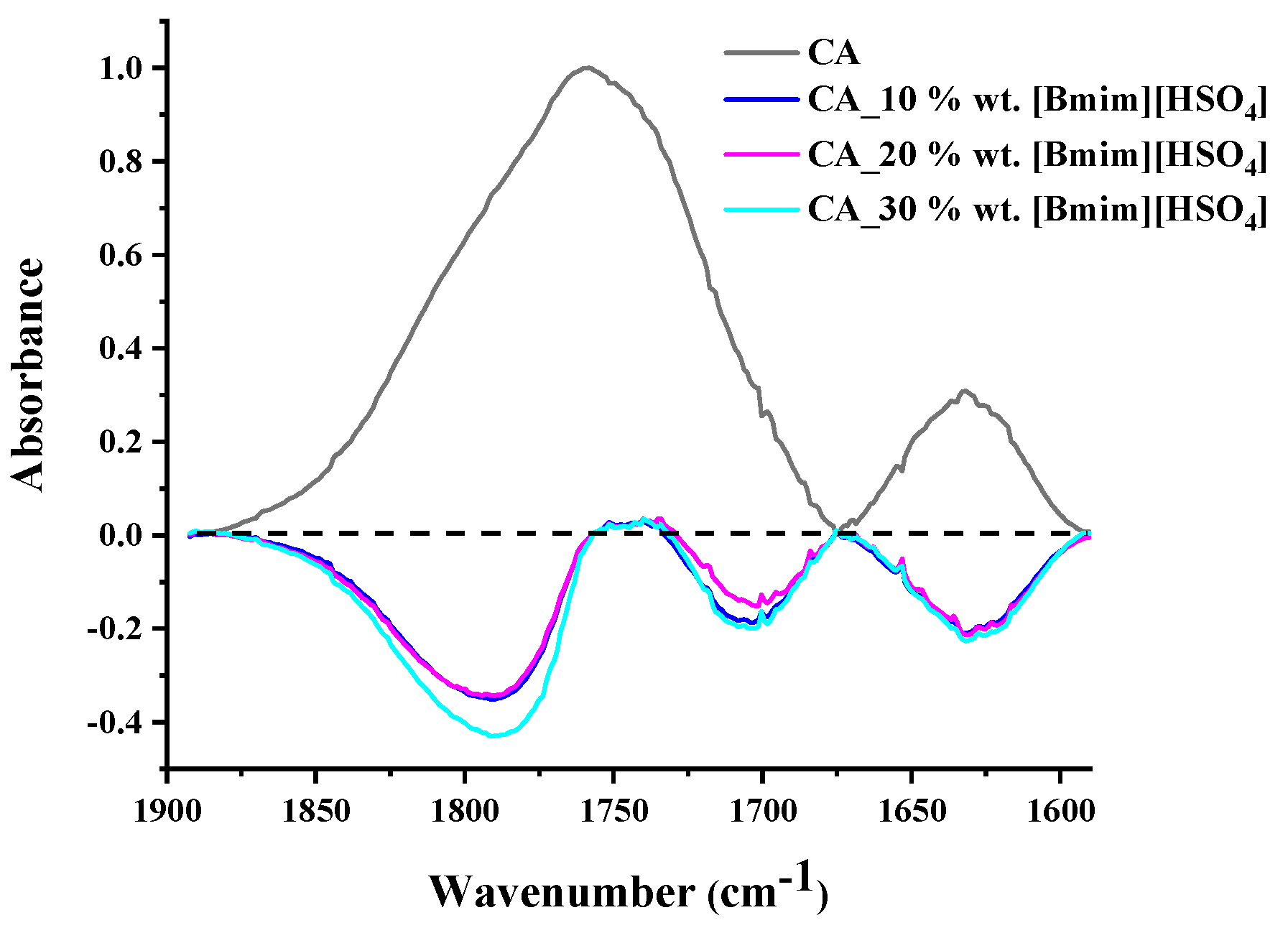
3.1.2. FTIR Analysis
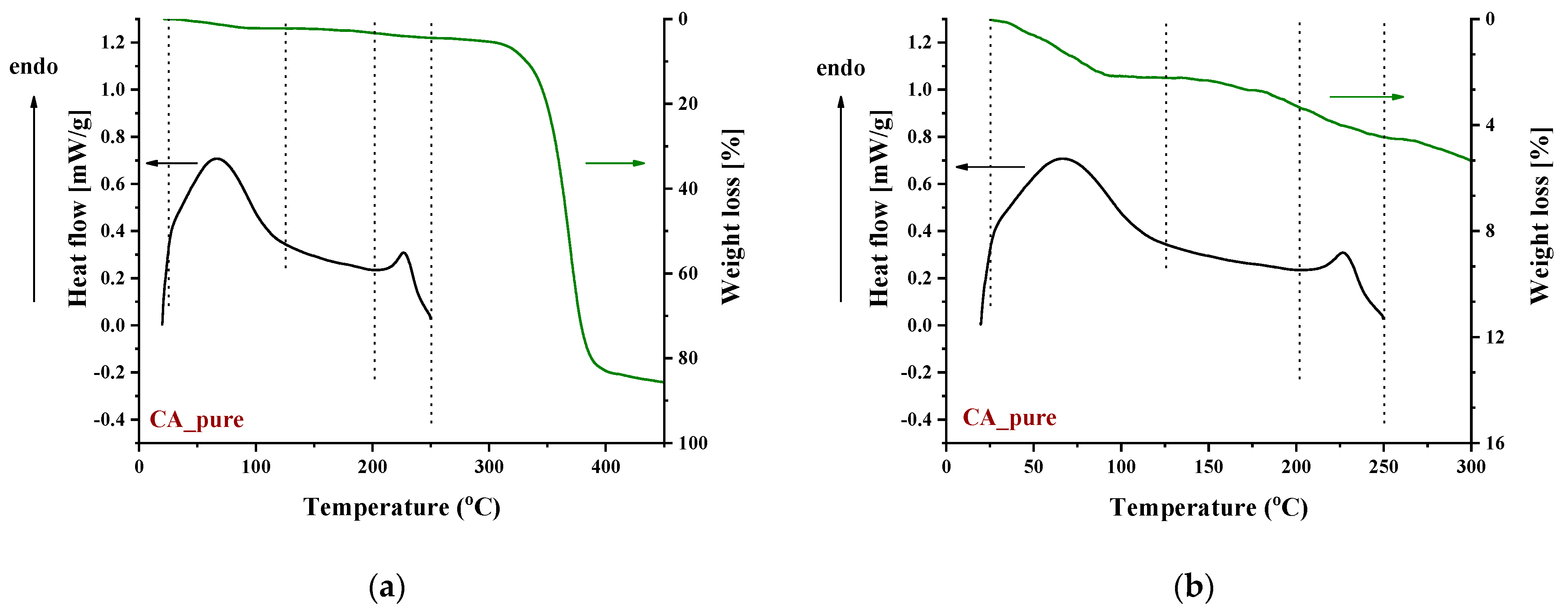
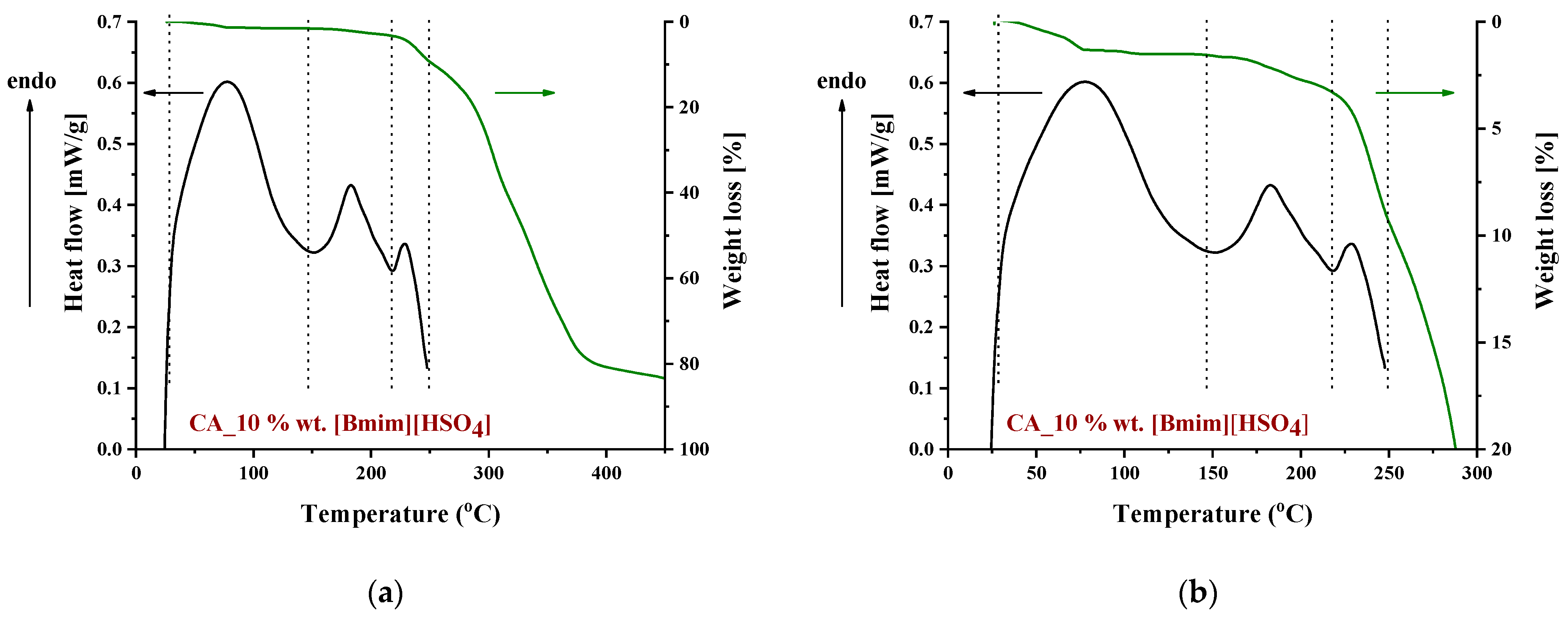
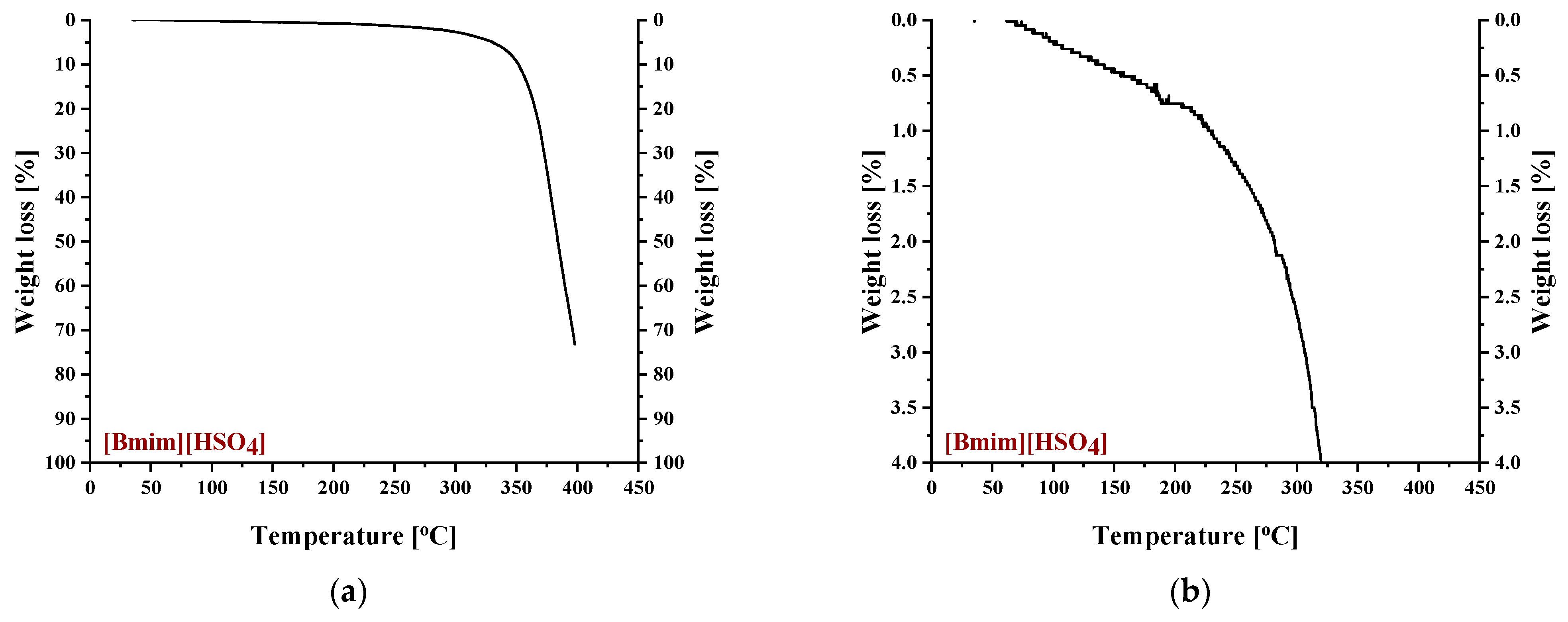
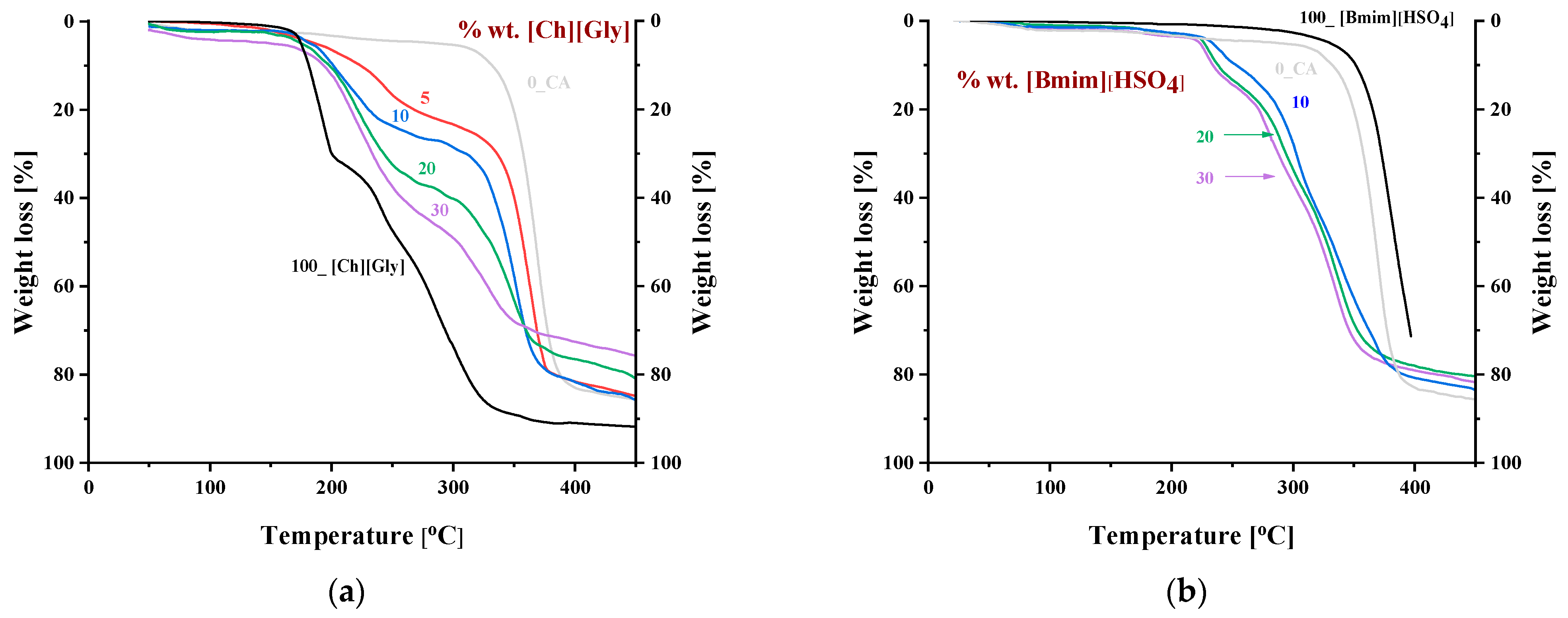
3.1.3. Thermal Behavior
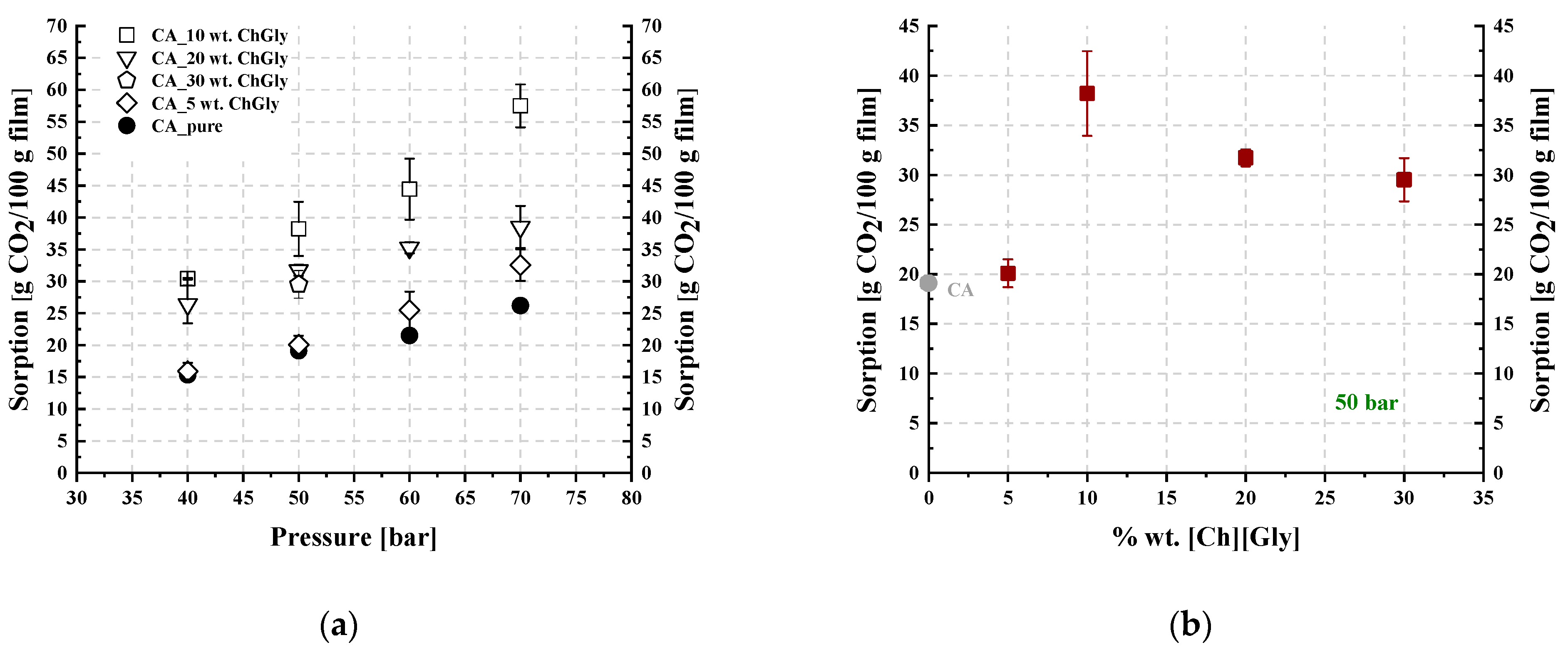
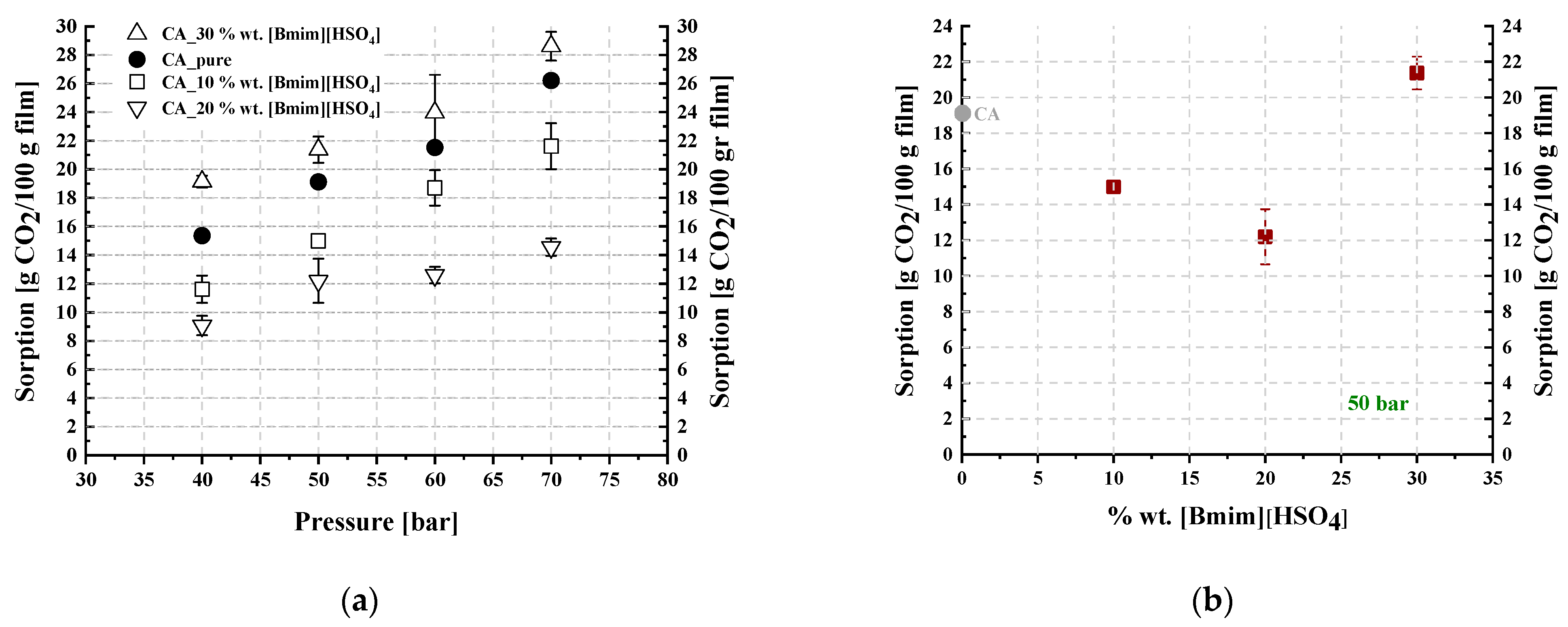
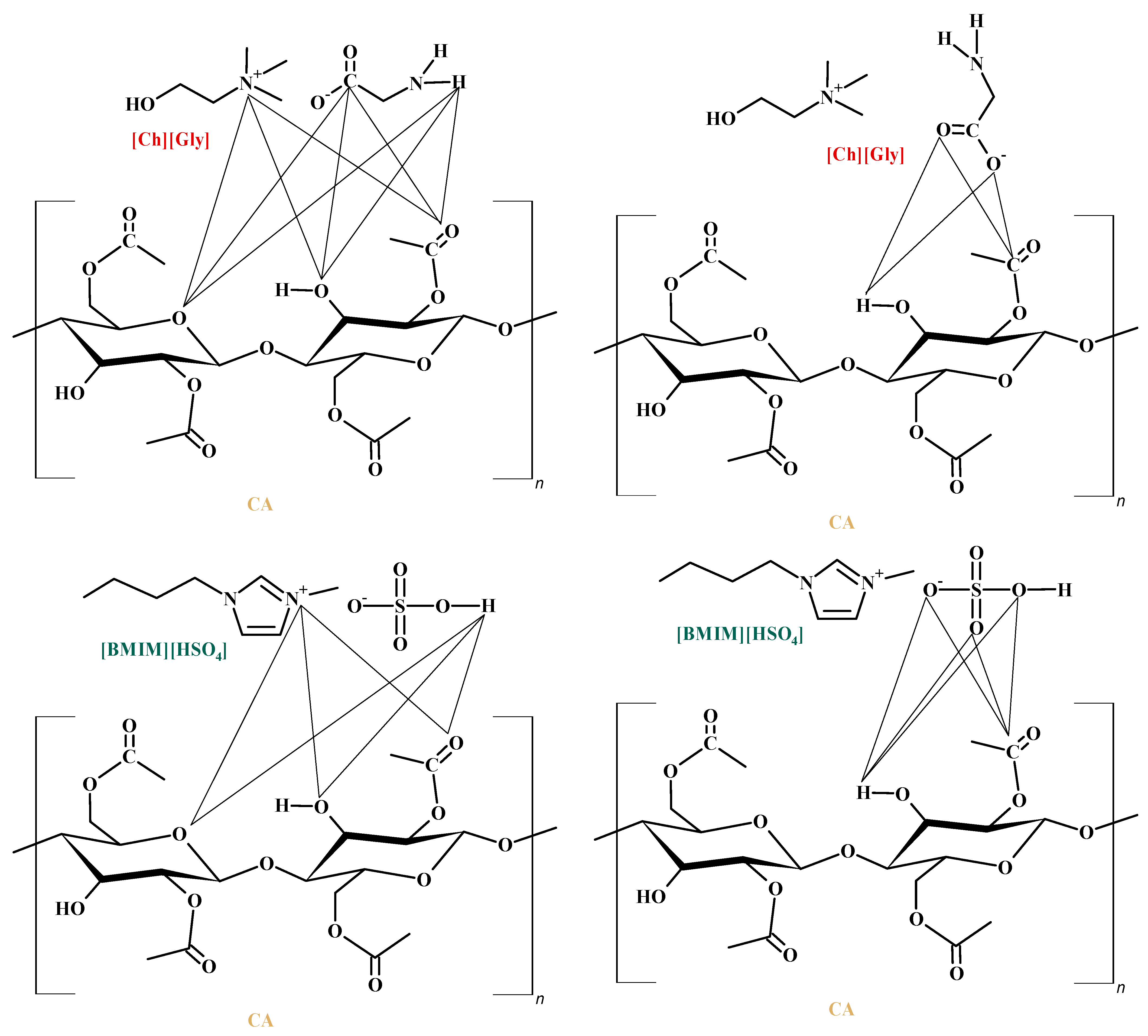
3.2. CO2 Sorption Measurements in CA-IL Membranes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohl, A.L.; Nielsen, R. Gas Purification; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Bernardo, P.; Drioli, E.; Golemme, G. Membrane gas separation: A review/state of the art. Ind. Eng. Chem. Res. 2009, 48, 4638–4663. [Google Scholar] [CrossRef]
- Tomé, L.C.; Marrucho, I.M. Ionic liquid-based materials: A platform to design engineered CO2 separation membranes. Chem. Soc. Rev. 2016, 45, 2785–2824. [Google Scholar] [CrossRef]
- IEA. World Energy Outlook 2022; IEA: Paris, France, 2022. [Google Scholar]
- Turner, J.A. Sustainable hydrogen production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef]
- Yang, S.-I.; Choi, D.-Y.; Jang, S.-C.; Kim, S.-H.; Choi, D.-K. Hydrogen separation by multi-bed pressure swing adsorption of synthesis gas. Adsorption 2008, 14, 583–590. [Google Scholar] [CrossRef]
- Davidson, R.M. Post-Combustion Carbon Capture from Coal Fired Plants-Solvent Scrubbing; IEA Clean Coal Centre: London, UK, 2007. [Google Scholar]
- Haszeldine, R.S. Carbon capture and storage: How green can black be? Science 2009, 325, 1647–1652. [Google Scholar] [CrossRef]
- Perry, J.D.; Nagai, K.; Koros, W.J. Polymer membranes for hydrogen separations. MRS Bull. 2006, 31, 745–749. [Google Scholar] [CrossRef]
- Haggin, J. New generation of membranes developed for industrial separations. Chem. Eng. News 1988, 66, 7–16. [Google Scholar] [CrossRef]
- Sanders, D.F.; Smith, Z.P.; Guo, R.; Robeson, L.M.; McGrath, J.E.; Paul, D.R.; Freeman, B.D. Energy-efficient polymeric gas separation membranes for a sustainable future: A review. Polymer 2013, 54, 4729–4761. [Google Scholar] [CrossRef]
- Scholes, C.A.; Stevens, G.W.; Kentish, S.E. Membrane gas separation applications in natural gas processing. Fuel 2012, 96, 15–28. [Google Scholar] [CrossRef]
- Stern, S.A. Polymers for gas separations: The next decade. J. Membr. Sci. 1994, 94, 1–65. [Google Scholar] [CrossRef]
- Lin, H.; Freeman, B.D. Gas solubility, diffusivity and permeability in poly (ethylene oxide). J. Membr. Sci. 2004, 239, 105–117. [Google Scholar] [CrossRef]
- Puleo, A.; Paul, D.R.; Kelley, S. The effect of degree of acetylation on gas sorption and transport behavior in cellulose acetate. J. Membr. Sci. 1989, 47, 301–332. [Google Scholar] [CrossRef]
- Bos, A.; Pünt, I.; Wessling, M.; Strathmann, H. CO2-induced plasticization phenomena in glassy polymers. J. Membr. Sci. 1999, 155, 67–78. [Google Scholar] [CrossRef]
- White, L.S.; Blinka, T.A.; Kloczewski, H.A.; Wang, I.-F. Properties of a polyimide gas separation membrane in natural gas streams. J. Membr. Sci. 1995, 103, 73–82. [Google Scholar] [CrossRef]
- Buchtová, N.; Guyomard-Lack, A.; Le Bideau, J. Biopolymer based nanocomposite ionogels: High performance, sustainable and solid electrolytes. Green Chem. 2014, 16, 1149–1152. [Google Scholar] [CrossRef]
- Guyomard-Lack, A.; Buchtová, N.; Humbert, B.; Le Bideau, J. Ion segregation in an ionic liquid confined within chitosan based chemical ionogels. Phys. Chem. Chem. Phys. 2015, 17, 23947–23951. [Google Scholar] [CrossRef]
- Tomé, L.C.; Isik, M.; Freire, C.S.; Mecerreyes, D.; Marrucho, I.M. Novel pyrrolidinium-based polymeric ionic liquids with cyano counter-anions: High performance membrane materials for post-combustion CO2 separation. J. Membr. Sci. 2015, 483, 155–165. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Nagai, K.; Nakagawa, T.; Mau, A.W. Effect of polyethyleneglycol (PEG) on gas permeabilities and permselectivities in its cellulose acetate (CA) blend membranes. J. Membr. Sci. 1998, 138, 143–152. [Google Scholar] [CrossRef]
- Yates, S.; Zaki, R.; Arzadon, A.; Liu, C.; Chiou, J. Thin Film Gas Separation Membranes. U.S. Patent US8016124B2, 13 September 2011. [Google Scholar]
- Chakma, A. Separation of CO2 and SO2 from flue gas streams by liquid membranes. Energy Convers. Manag. 1995, 36, 405–410. [Google Scholar] [CrossRef]
- Kemperman, A.J.; Bargeman, D.; Van Den Boomgaard, T.; Strathmann, H. Stability of supported liquid membranes: State of the art. Sep. Sci. Technol. 1996, 31, 2733–2762. [Google Scholar] [CrossRef]
- Teramoto, M.; Sakaida, Y.; Fu, S.S.; Ohnishi, N.; Matsuyama, H.; Maki, T.; Fukui, T.; Arai, K. An attempt for the stabilization of supported liquid membrane. Sep. Purif. Technol. 2000, 21, 137–144. [Google Scholar] [CrossRef]
- Kocherginsky, N.; Yang, Q.; Seelam, L. Recent advances in supported liquid membrane technology. Sep. Purif. Technol. 2007, 53, 171–177. [Google Scholar] [CrossRef]
- San Román, M.; Bringas, E.; Ibañez, R.; Ortiz, I. Liquid membrane technology: Fundamentals and review of its applications. J. Chem. Technol. Biotechnol. 2010, 85, 2–10. [Google Scholar] [CrossRef]
- Brennecke, J.F.; Maginn, E.J. Ionic liquids: Innovative fluids for chemical processing. Am. Inst. Chem. Eng. AIChE J. 2001, 47, 2384. [Google Scholar] [CrossRef]
- Maiyoh, G.K.; Njoroge, R.W.; Tuei, V.C. Effects and mechanisms of kerosene use-related toxicity. Environ. Toxicol. Pharmacol. 2015, 40, 57–70. [Google Scholar] [CrossRef]
- Takeuchi, H.; Takahashi, K.; Goto, W. Some observations on the stability of supported liquid membranes. J. Membr. Sci. 1987, 34, 19–31. [Google Scholar] [CrossRef]
- Tormoehlen, L.; Tekulve, K.; Nañagas, K. Hydrocarbon toxicity: A review. Clin. Toxicol. 2014, 52, 479–489. [Google Scholar] [CrossRef]
- Endres, F.; Abbott, A.P.; MacFarlane, D.R. Electrodeposition from Ionic Liquids; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Wasserscheid, P.; Welton, T. Ionic Liquids in Synthesis; Wiley Online Library: Hoboken, NJ, USA, 2008; Volume 1. [Google Scholar]
- Papaiconomou, N.; Estager, J.; Traore, Y.; Bauduin, P.; Bas, C.; Legeai, S.; Viboud, S.; Draye, M. Synthesis, physicochemical properties, and toxicity data of new hydrophobic ionic liquids containing dimethylpyridinium and trimethylpyridinium cations. J. Chem. Eng. Data 2010, 55, 1971–1979. [Google Scholar] [CrossRef]
- Luis, P.; Van Gerven, T.; Van der Bruggen, B. Recent developments in membrane-based technologies for CO2 capture. Prog. Energy Combust. Sci. 2012, 38, 419–448. [Google Scholar] [CrossRef]
- Wang, C.; Mahurin, S.M.; Luo, H.; Baker, G.A.; Li, H.; Dai, S. Reversible and robust CO2 capture by equimolar task-specific ionic liquid–superbase mixtures. Green Chem. 2010, 12, 870–874. [Google Scholar] [CrossRef]
- Akhmetshina, A.I.; Gumerova, O.R.; Atlaskin, A.A.; Petukhov, A.N.; Sazanova, T.S.; Yanbikov, N.R.; Nyuchev, A.V.; Razov, E.N.; Vorotyntsev, I.V. Permeability and selectivity of acid gases in supported conventional and novel imidazolium-based ionic liquid membranes. Sep. Purif. Technol. 2017, 176, 92–106. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, X.-M.; Li, Y.-X.; Wu, Y.-T.; Hu, X.-B. Facilitated separation of CO2 and SO2 through supported liquid membranes using carboxylate-based ionic liquids. J. Membr. Sci. 2014, 471, 227–236. [Google Scholar] [CrossRef]
- Teramoto, M.; Takeuchi, N.; Maki, T.; Matsuyama, H. Gas separation by liquid membrane accompanied by permeation of membrane liquid through membrane physical transport. Sep. Purif. Technol. 2001, 24, 101–112. [Google Scholar] [CrossRef]
- Cascon, H.R.; Choudhari, S.K. 1-Butanol pervaporation performance and intrinsic stability of phosphonium and ammonium ionic liquid-based supported liquid membranes. J. Membr. Sci. 2013, 429, 214–224. [Google Scholar] [CrossRef]
- Rynkowska, E.; Fatyeyeva, K.; Kujawski, W. Application of polymer-based membranes containing ionic liquids in membrane separation processes: A critical review. Rev. Chem. Eng. 2018, 34, 341–363. [Google Scholar] [CrossRef]
- Kanehashi, S.; Kishida, M.; Kidesaki, T.; Shindo, R.; Sato, S.; Miyakoshi, T.; Nagai, K. CO2 separation properties of a glassy aromatic polyimide composite membranes containing high-content 1-butyl-3-methylimidazolium bis (trifluoromethylsulfonyl) imide ionic liquid. J. Membr. Sci. 2013, 430, 211–222. [Google Scholar] [CrossRef]
- Chen, H.Z.; Li, P.; Chung, T.-S. PVDF/ionic liquid polymer blends with superior separation performance for removing CO2 from hydrogen and flue gas. Int. J. Hydrogen Energy 2012, 37, 11796–11804. [Google Scholar] [CrossRef]
- Lam, B.; Wei, M.; Zhu, L.; Luo, S.; Guo, R.; Morisato, A.; Alexandridis, P.; Lin, H. Cellulose triacetate doped with ionic liquids for membrane gas separation. Polymer 2016, 89, 1–11. [Google Scholar] [CrossRef]
- Lin, H.; White, L.S.; Lokhandwala, K.; Baker, R.W. Natural gas purification. Encycl. Membr. Sci. Technol. 2013, 1–25. [Google Scholar] [CrossRef]
- Swatloski, R.P.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Dissolution of cellose with ionic liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. [Google Scholar] [CrossRef]
- Higdon. Choline. Available online: https://lpi.oregonstate.edu/mic/other-nutrients/choline (accessed on 3 September 2023).
- Blusztajn, J.K. Choline, a vital amine. Science 1998, 281, 794–795. [Google Scholar] [CrossRef]
- Bernot, R.J.; Brueseke, M.A.; Evans-White, M.A.; Lamberti, G.A. Acute and chronic toxicity of imidazolium-based ionic liquids on Daphnia magna. Environ. Toxicol. Chem. Int. J. 2005, 24, 87–92. [Google Scholar] [CrossRef]
- Li, X.; Hou, M.; Zhang, Z.; Han, B.; Yang, G.; Wang, X.; Zou, L. Absorption of CO 2 by ionic liquid/polyethylene glycol mixture and the thermodynamic parameters. Green Chem. 2008, 10, 879–884. [Google Scholar] [CrossRef]
- Noorani, N.; Mehrdad, A. CO2 solubility in some amino acid-based ionic liquids: Measurement, correlation and DFT studies. Fluid Phase Equilibria 2020, 517, 112591. [Google Scholar] [CrossRef]
- Saravanamurugan, S.; Kunov-Kruse, A.J.; Fehrmann, R.; Riisager, A. Amine-functionalized amino acid-based ionic liquids as efficient and high-capacity absorbents for CO2. ChemSusChem 2014, 7, 897–902. [Google Scholar] [CrossRef]
- Van Valkenburg, M.E.; Vaughn, R.L.; Williams, M.; Wilkes, J.S. Thermochemistry of ionic liquid heat-transfer fluids. Thermochim. Acta 2005, 425, 181–188. [Google Scholar] [CrossRef]
- Yuan, S.; Yang, Z.; Ji, X.; Chen, Y.; Sun, Y.; Lu, X. CO2 absorption in mixed aqueous solution of MDEA and cholinium glycinate. Energy Fuels 2017, 31, 7325–7333. [Google Scholar] [CrossRef]
- Anderson, J.L.; Dixon, J.K.; Brennecke, J.F. Solubility of CO2, CH4, C2H6, C2H4, O2, and N2 in 1-Hexyl-3-methylpyridinium Bis (trifluoromethylsulfonyl) imide: Comparison to Other Ionic Liquids. Acc. Chem. Res. 2007, 40, 1208–1216. [Google Scholar] [CrossRef]
- Raveendran, P.; Wallen, S.L. Cooperative C−H···O Hydrogen Bonding in CO2−Lewis Base Complexes: Implications for Solvation in Supercritical CO2. J. Am. Chem. Soc. 2002, 124, 12590–12599. [Google Scholar] [CrossRef]
- Mejía, I.; Stanley, K.; Canales, R.; Brennecke, J.F. On the high-pressure solubilities of carbon dioxide in several ionic liquids. J. Chem. Eng. Data 2013, 58, 2642–2653. [Google Scholar] [CrossRef]
- Liu, Q.-P.; Hou, X.-D.; Li, N.; Zong, M.-H. Ionic liquids from renewable biomaterials: Synthesis, characterization and application in the pretreatment of biomass. Green Chem. 2012, 14, 304–307. [Google Scholar] [CrossRef]
- Moriel, P.; García-Suárez, E.J.; Martínez, M.; García, A.B.; Montes-Morán, M.A.; Calvino-Casilda, V.; Bañares, M.A. Synthesis, characterization, and catalytic activity of ionic liquids based on biosources. Tetrahedron Lett. 2010, 51, 4877–4881. [Google Scholar] [CrossRef]
- Kiran, E.; Sarver, J.A.; Hassler, J.C. Solubility and diffusivity of CO2 and N2 in polymers and polymer swelling, glass transition, melting, and crystallization at high pressure: A critical review and perspectives on experimental methods, data, and modeling. J. Supercrit. Fluids 2022, 185, 105378. [Google Scholar] [CrossRef]
- Felder, R.; Huvard, G. 17. Permeation, diffusion, and sorption of gases and vapors. In Methods in Experimental Physics; Elsevier: Amsterdam, The Netherlands, 1980; Volume 16, pp. 315–377. [Google Scholar]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- WebBook, N.C. Available online: https://webbook.nist.gov/chemistry/ (accessed on 10 February 2024).
- Yang, Y. Polymer Data Handbook; Mark, J.E., Ed.; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Reed, D.G.; Dowson, G.R.; Styring, P. Cellulose-supported ionic liquids for low-cost pressure swing CO2 capture. Front. Energy Res. 2017, 5, 13. [Google Scholar] [CrossRef]
- Barud, H.S.; de Araújo Júnior, A.M.; Santos, D.B.; de Assunção, R.M.; Meireles, C.S.; Cerqueira, D.A.; Rodrigues Filho, G.; Ribeiro, C.A.; Messaddeq, Y.; Ribeiro, S.J. Thermal behavior of cellulose acetate produced from homogeneous acetylation of bacterial cellulose. Thermochim. Acta 2008, 471, 61–69. [Google Scholar] [CrossRef]
- Kamide, K.; Saito, M. Thermal analysis of cellulose acetate solids with total degrees of substitution of 0.49, 1.75, 2.46, and 2.92. Polym. J. 1985, 17, 919–928. [Google Scholar] [CrossRef]
- Doyle, S.E.; Pethrick, R.A. Structure of fibrous cellulose acetate: X-ray diffraction, positron annihilation and electron microscopy investigations. J. Appl. Polym. Sci. 1987, 33, 95–106. [Google Scholar] [CrossRef]
- Rodrigues Filho, G.; da Cruz, S.F.; Pasquini, D.; Cerqueira, D.A.; de Souza Prado, V.; de Assunção, R.M.N. Water flux through cellulose triacetate films produced from heterogeneous acetylation of sugar cane bagasse. J. Membr. Sci. 2000, 177, 225–231. [Google Scholar] [CrossRef]
- Stuart, B.H. Infrared Spectroscopy: Fundamentals and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Ahmad, I.R.; Cane, D.; Townsend, J.H.; Triana, C.; Mazzei, L.; Curran, K. Are we overestimating the permanence of cellulose triacetate cinematographic films? A mathematical model for the vinegar syndrome. Polym. Degrad. Stab. 2020, 172, 109050. [Google Scholar] [CrossRef]
- Tsioptsias, C.; Foukas, G.-R.P.; Papaioannou, S.-M.; Tzimpilis, E.; Tsivintzelis, I. On the Thermochemical Transition Depression of Cellulose Acetate Composite Membranes. Polymers 2022, 14, 3434. [Google Scholar] [CrossRef]
- Tsioptsias, C.; Karabinaki, O.; Christofilos, D.; Tzimpilis, E.; Tsivintzelis, I.; Panayiotou, C. On polymer-polymer miscibility and cellulose ester blends: A case study. Thermochim. Acta 2022, 714, 179265. [Google Scholar] [CrossRef]
- Tsioptsias, C.; Nikolaidou, E.G.; Ntampou, X.; Tsivintzelis, I.; Panayiotou, C. Thermo-chemical transition in cellulose esters and other polymers. Thermochim. Acta 2022, 707, 179106. [Google Scholar] [CrossRef]
- Tsioptsias, C. Thermodynamic explanation and criterion for the exhibition of melting inability in molecular species. AIMS Mater. Sci. 2023, 10, 618–636. [Google Scholar] [CrossRef]
- Kazarian, S.G.; Vincent, M.F.; Bright, F.V.; Liotta, C.L.; Eckert, C.A. Specific intermolecular interaction of carbon dioxide with polymers. J. Am. Chem. Soc. 1996, 118, 1729–1736. [Google Scholar] [CrossRef]
- Finotello, A.; Bara, J.E.; Narayan, S.; Camper, D.; Noble, R.D. Ideal gas solubilities and solubility selectivities in a binary mixture of room-temperature ionic liquids. J. Phys. Chem. B 2008, 112, 2335–2339. [Google Scholar] [CrossRef] [PubMed]
- Shannon, M.S.; Tedstone, J.M.; Danielsen, S.P.; Hindman, M.S.; Irvin, A.C.; Bara, J.E. Free volume as the basis of gas solubility and selectivity in imidazolium-based ionic liquids. Ind. Eng. Chem. Res. 2012, 51, 5565–5576. [Google Scholar] [CrossRef]
- Pantoula, M.; Panayiotou, C. Sorption and swelling in glassy polymer/carbon dioxide systems: Part I. Sorption. J. Supercrit. Fluids 2006, 37, 254–262. [Google Scholar] [CrossRef]
- Sanders, E.S.; Koros, W.J.; Hopfenberg, H.B.; Stannett, V.T. Mixed gas sorption in glassy polymers: Equipment design considerations and preliminary results. J. Membr. Sci. 1983, 13, 161–174. [Google Scholar] [CrossRef]
- Sato, Y.; Takikawa, T.; Yamane, M.; Takishima, S.; Masuoka, H. Solubility of carbon dioxide in PPO and PPO/PS blends. Fluid Phase Equilibria 2002, 194–197, 847–858. [Google Scholar] [CrossRef]
- Tsioptsias, C.; Panayiotou, C. Simultaneous determination of sorption, heat of sorption, diffusion coefficient and glass transition depression in polymer–CO2 systems. Thermochim. Acta 2011, 521, 98–106. [Google Scholar] [CrossRef]
- Hermans, P.; Weidinger, A. Estimation of crystallinity of some polymers from x-ray intensity measurements. J. Polym. Sci. 1949, 4, 709–723. [Google Scholar] [CrossRef]
- Krimm, S.; Tobolsky, A.V. Quantitative x-ray studies of order in amorphous and crystalline polymers. Quantitative x-ray determination of crystallinity in polyethylene. J. Polym. Sci. 1951, 7, 57–76. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymeric Base | |
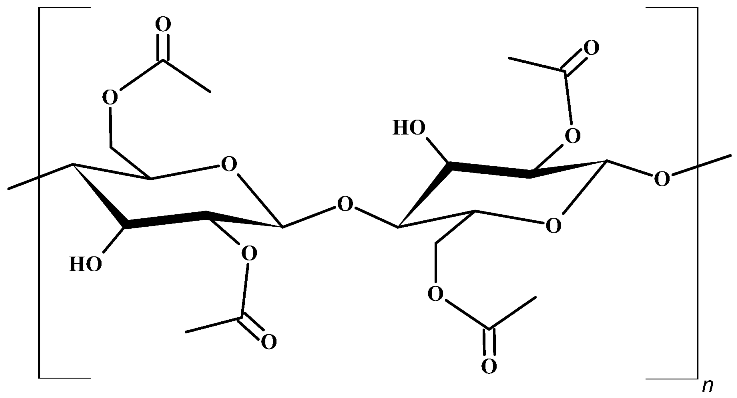 Cellulose acetate, CA | |
| Ionic Liquids | |
 Choline glycine, |  1-Butyl-3-methylimidazolium hydrogen sulfate, [] |
| IL %wt. | CA- | CA-] |
|---|---|---|
| 0 | 27 | 27 |
| 5 | 9 | -- |
| 10 | 9 | 43 |
| 20 | 10 | 25 |
| 30 | 8 | 24 |
| 100 | -- | -- |
| IL Content/ | Pressure/Bar | |||
|---|---|---|---|---|
| %wt. | 40 | 50 | 60 | 70 |
| 0 | 15.4 ± 0.4 | 19.1 ± 0.2 | 21.5 ± 0.5 | 26.2 ± 0.2 |
| 5 | 15.9 ± 1.3 | 20.1 ± 1.4 | 25.5 ± 2.9 | 32.6 ± 2.5 |
| 10 | 30.4 ± 0.1 | 38.2 ± 4.3 | 44.5 ± 4.8 | 57.5 ± 3.4 |
| 20 | 26.4 ± 3.0 | 31.7 ± 0.9 | 35.3 ± 0.9 | 38.5 ± 3.3 |
| 30 | -- | 29.5 ± 2.2 | -- | -- |
| IL Content/ | Pressure/Bar | |||
|---|---|---|---|---|
| %wt. | 40 | 50 | 60 | 70 |
| 0 | 15.4 ± 0.4 | 19.1 ± 0.2 | 21.5 ± 0.5 | 26.2 ± 0.2 |
| 10 | 11.6 ± 1.0 | 15.0 ± 0.2 | 18.7 ± 1.2 | 21.6 ± 1.6 |
| 20 | 9.1 ± 0.7 | 12.2 ± 1.5 | 12.6 ± 0.6 | 14.6 ± 0.6 |
| 30 | 19.1 ± 0.4 | 21.4 ± 0.9 | 24.0 ± 2.6 | 28.6 ± 1.0 |
| IL Content/ | D/10−7 cm2 s−1 | |||
|---|---|---|---|---|
| %wt. | 40 Bar | 50 Bar | 60 Bar | 70 Bar |
| 0 | 0.2 ± 0.1 | 0.4 ± 0.2 | 0.2 ± 0.1 | 0.3 ± 0.1 |
| 5 | 2.0 ± 0.4 | 1.9 ± 0.5 | 2.6 ± 0.4 | 2.6 ± 0.7 |
| 10 | 10.6 ± 0.2 | 11.1 ± 0.6 | 10.5 ± 0.6 | 9.0 ± 1.1 |
| 20 | 13.0 ± 1.0 | 14.2 ± 0.3 | 12.1 ± 1.0 | 11.1 ± 0.7 |
| 30 | -- | 30.2 ± 6.2 | -- | -- |
| IL Content/ | D/10−7 cm2 s−1 | |||
|---|---|---|---|---|
| %wt. | 40 Bar | 50 Bar | 60 Bar | 70 Bar |
| 0 | 0.2 ± 0.1 | 0.4 ± 0.2 | 0.2 ± 0.1 | 0.3 ± 0.1 |
| 10 | 0.1 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 |
| 20 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.1 |
| 30 | 0.5 ± 0.1 | 0.5 ± 0.1 | 0.6 ± 0.2 | 0.3 ± 0.1 |
| IL Content/ | ) | |||
|---|---|---|---|---|
| %wt. | 40 Bar | 50 Bar | 60 Bar | 70 Bar |
| 0 | 6.5 | 13.2 | 7.4 | 8.5 |
| 5 | 68.6 | 68.3 | 95.5 | 104.9 |
| 10 | 706.5 | 741.9 | 681.9 | 634.9 |
| 20 | 752.3 | 790 | 622.5 | 536.4 |
| 30 | -- | 1564.2 | -- | -- |
| IL Content/ | ) | |||
|---|---|---|---|---|
| %wt. | 40 Bar | 50 Bar | 60 Bar | 70 Bar |
| 0 | 6.5 | 13.2 | 7.4 | 8.5 |
| 10 | 2.4 | 9.8 | 12.2 | 13.0 |
| 20 | 3.8 | 4.2 | 2.9 | 3.4 |
| 30 | 21.2 | 20.6 | 22.3 | 9.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kontos, G.; Tsioptsias, C.; Tsivintzelis, I. Cellulose Acetate–Ionic Liquid Blends as Potential Polymers for Efficient CO2 Separation Membranes. Polymers 2024, 16, 554. https://doi.org/10.3390/polym16040554
Kontos G, Tsioptsias C, Tsivintzelis I. Cellulose Acetate–Ionic Liquid Blends as Potential Polymers for Efficient CO2 Separation Membranes. Polymers. 2024; 16(4):554. https://doi.org/10.3390/polym16040554
Chicago/Turabian StyleKontos, Giannis, Costas Tsioptsias, and Ioannis Tsivintzelis. 2024. "Cellulose Acetate–Ionic Liquid Blends as Potential Polymers for Efficient CO2 Separation Membranes" Polymers 16, no. 4: 554. https://doi.org/10.3390/polym16040554
APA StyleKontos, G., Tsioptsias, C., & Tsivintzelis, I. (2024). Cellulose Acetate–Ionic Liquid Blends as Potential Polymers for Efficient CO2 Separation Membranes. Polymers, 16(4), 554. https://doi.org/10.3390/polym16040554