
Kraft (Nano)Lignin as Reactive Additive in Epoxy Polymer Bio-Composites
 ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Treatment of Lignin
2.2.1. Ball Milling of Initial Kraft Lignin (BMKL)
2.2.2. Ultrasonication of Kraft Lignin towards Nano-Lignin (NLH)
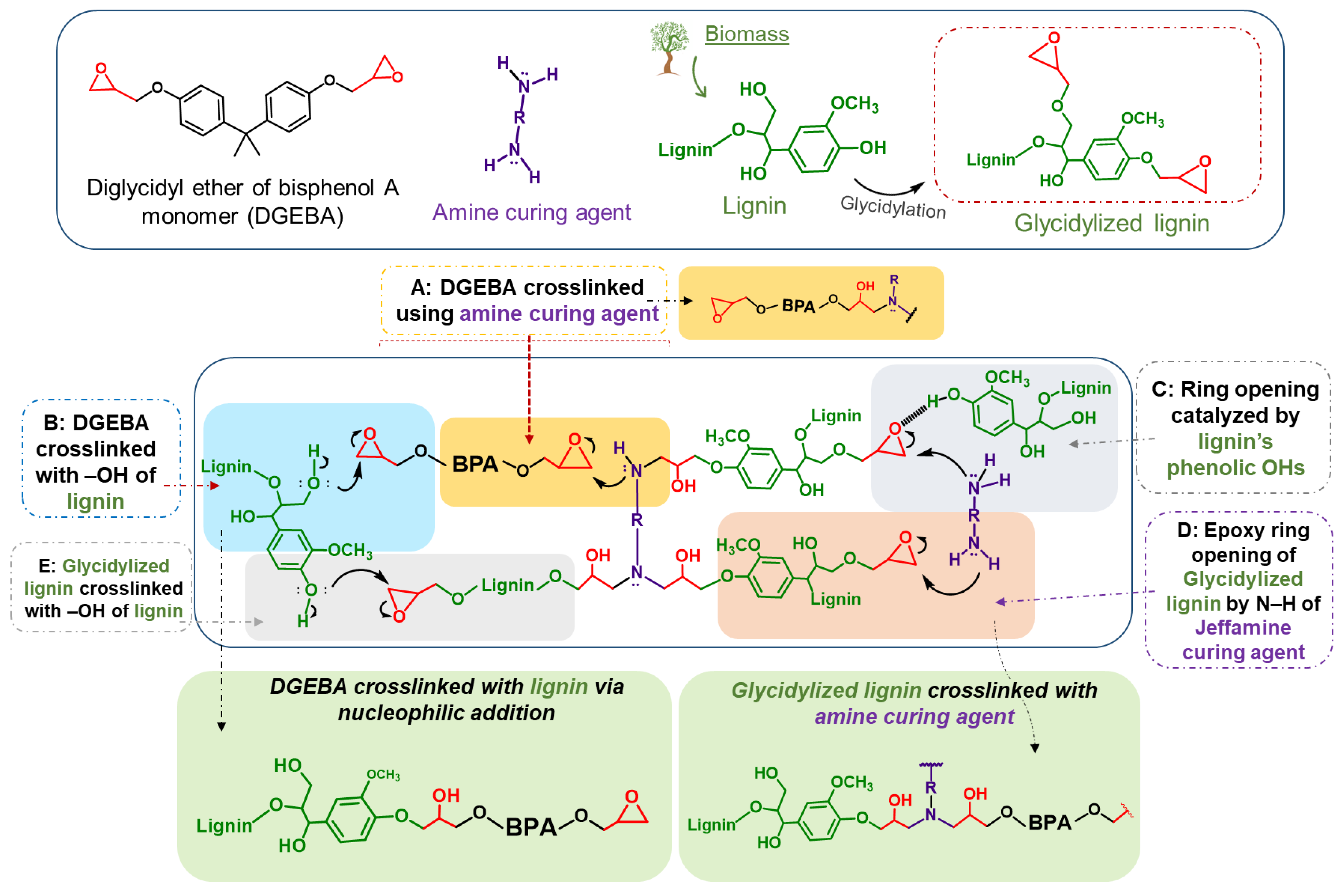
2.2.3. Synthesis of Glycidylized Lignin (GKL)
2.3. Preparation of Lignin–Epoxy Composites
2.4. Characterization of Lignins and Lignin–Epoxy Composites
3. Results and Discussion
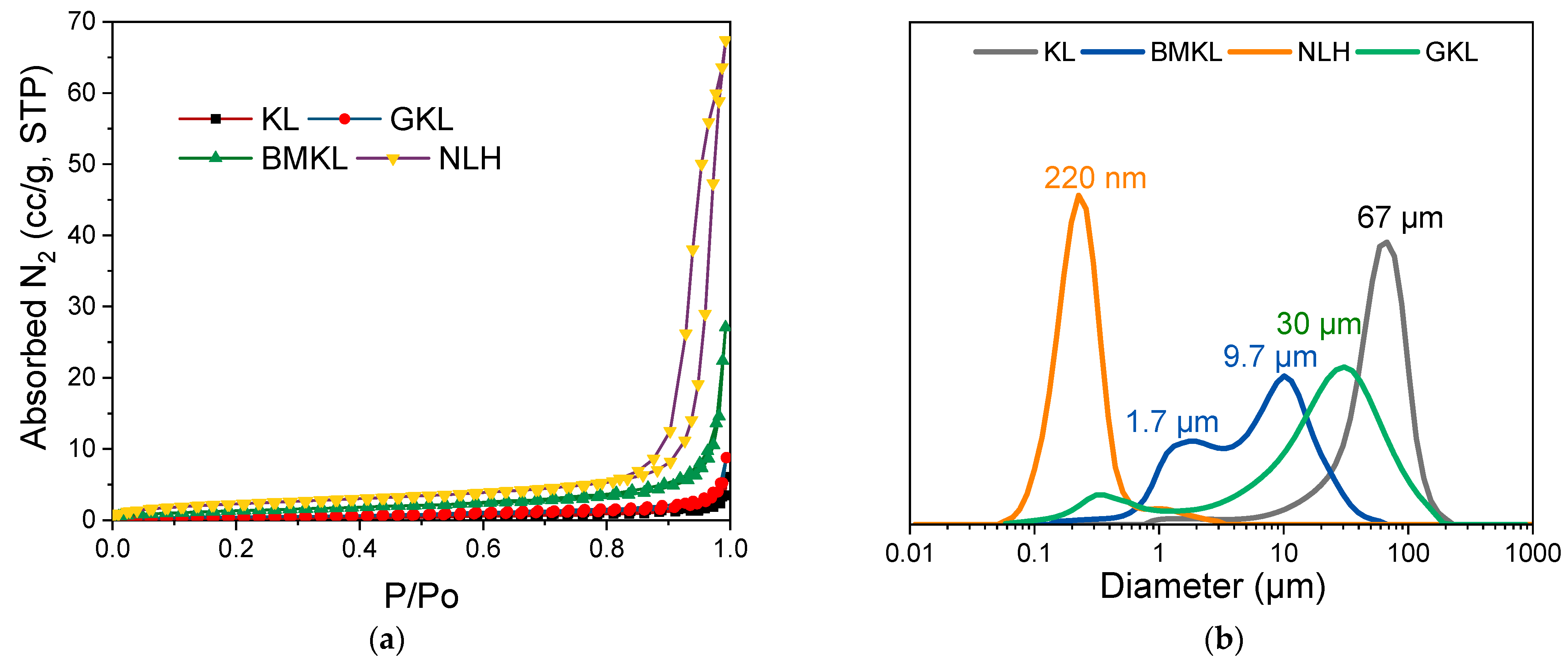
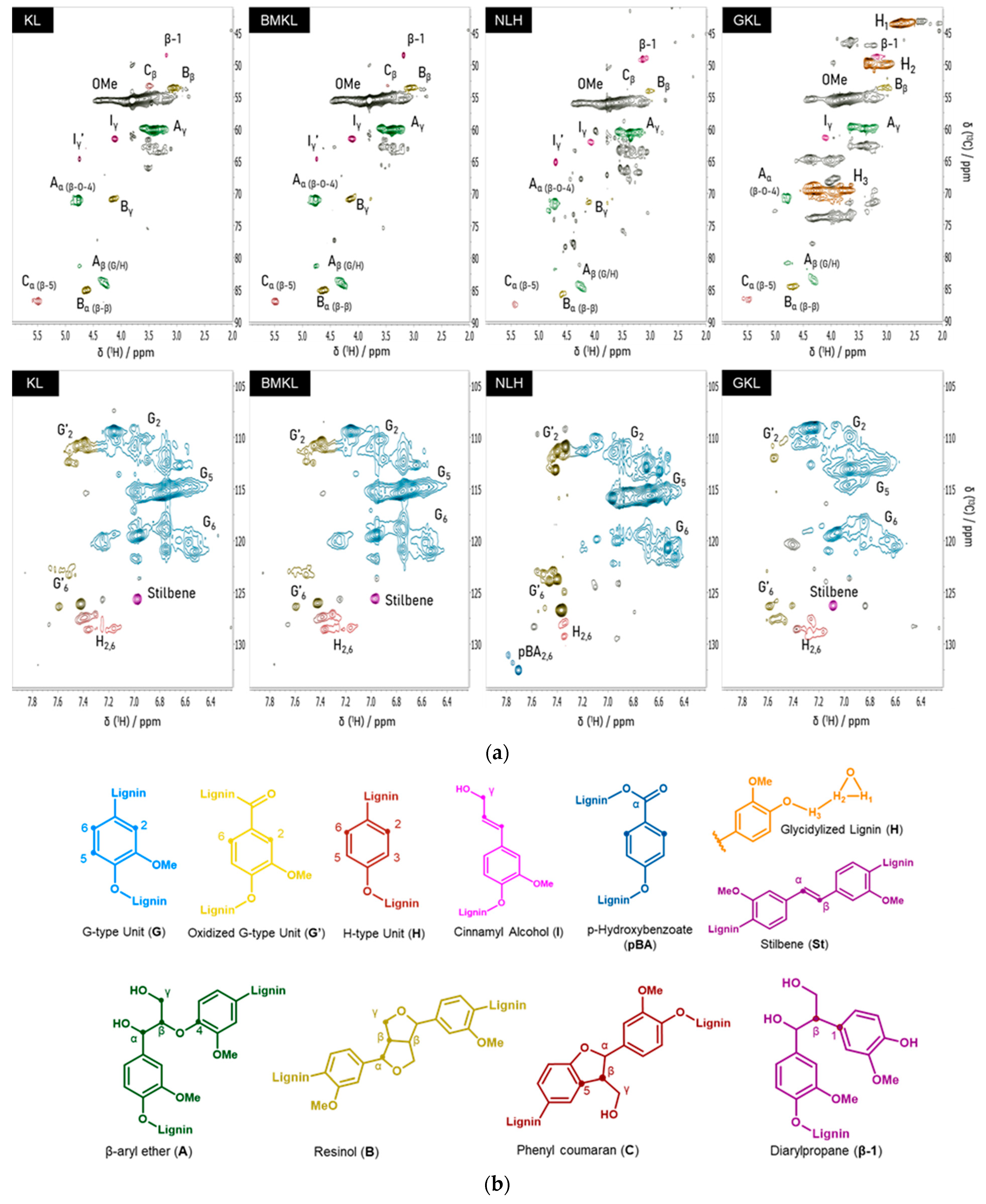
3.1. Physicochemical Characterization of Initial and Modified Lignins
3.2. Lignin–Epoxy Composites
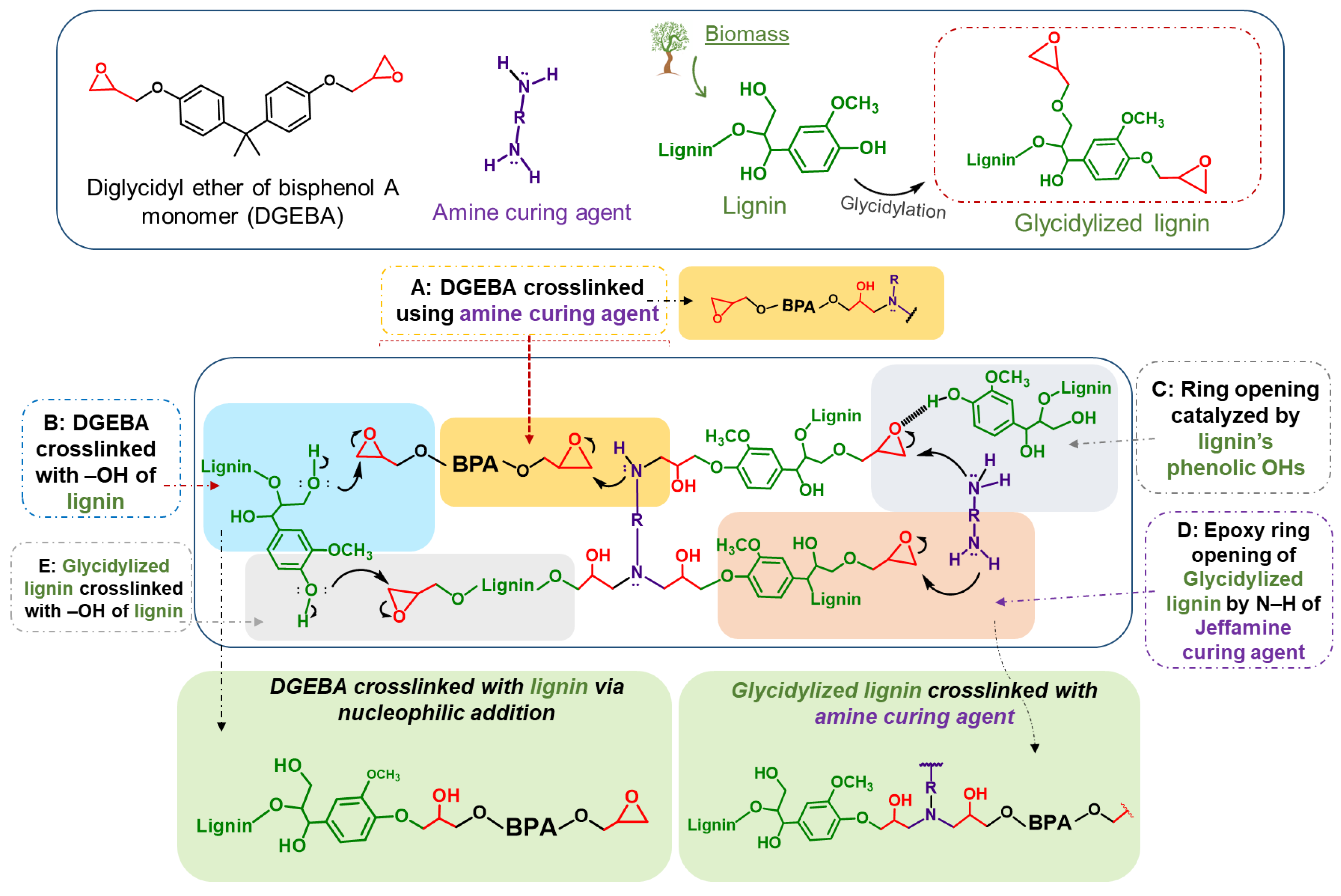
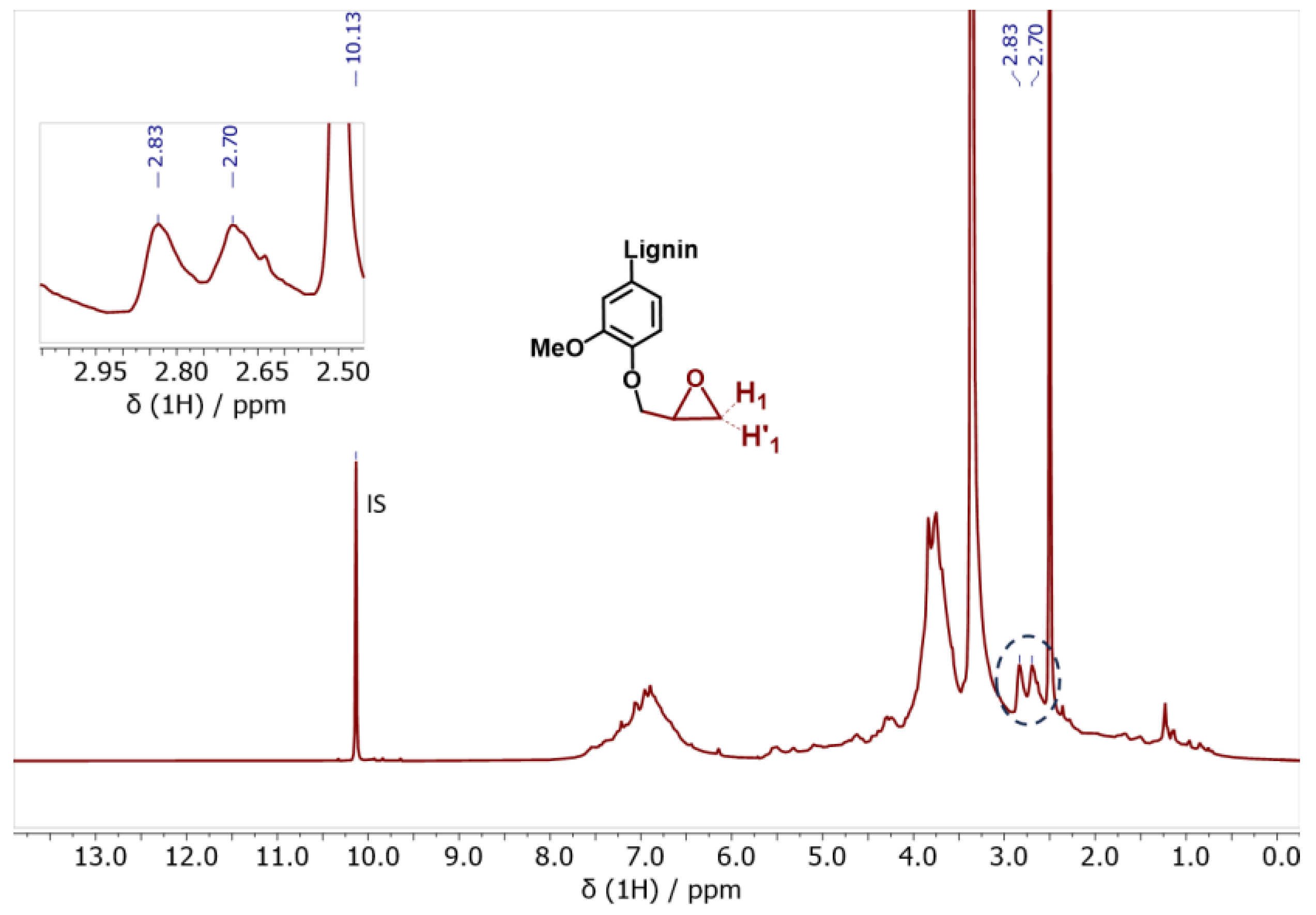
3.2.1. Reactivity of Kraft Lignin Ohs towards Epoxy Ring Opening
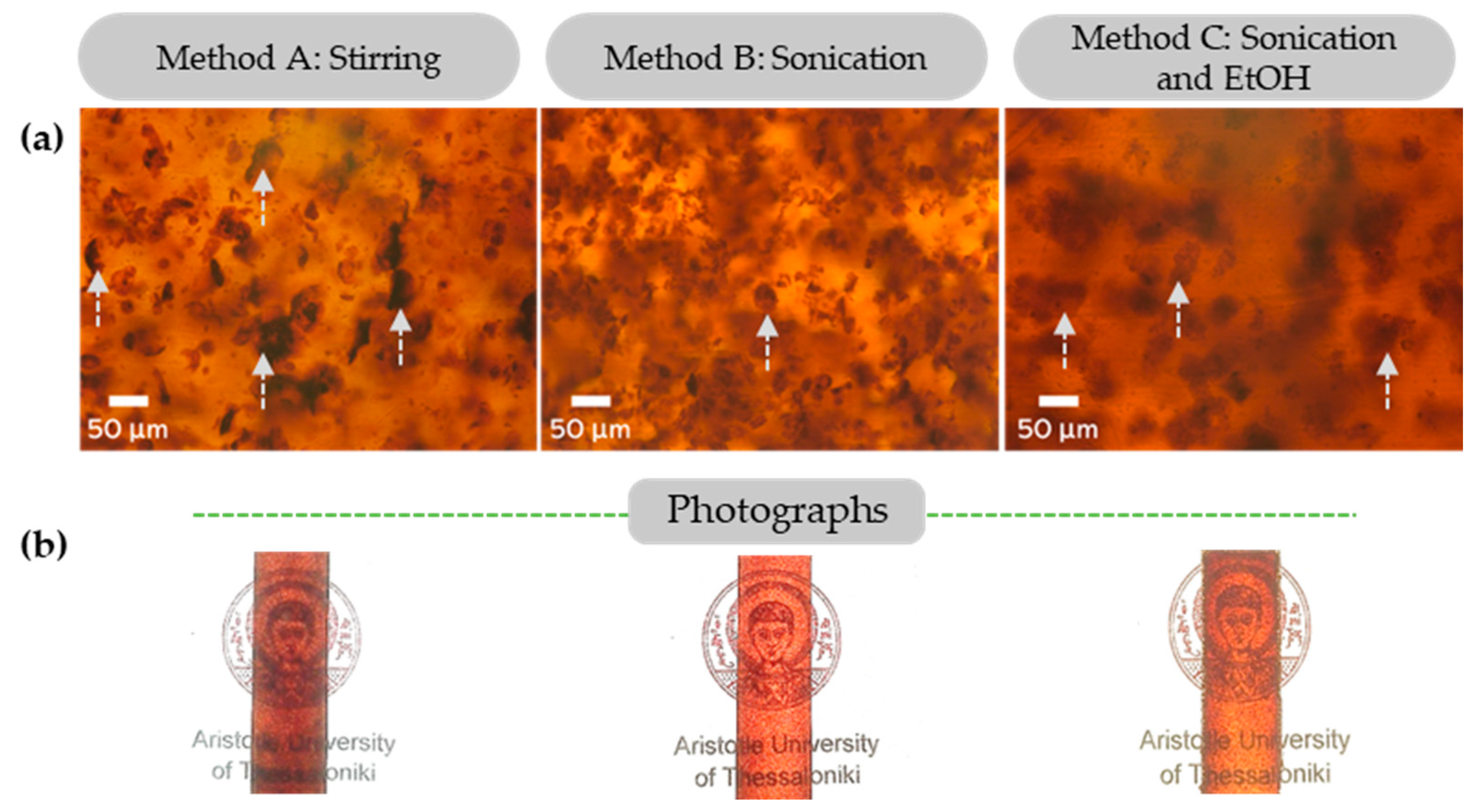
3.2.2. Dispersion of Kraft Lignin and Modified Lignins in Epoxy Composites
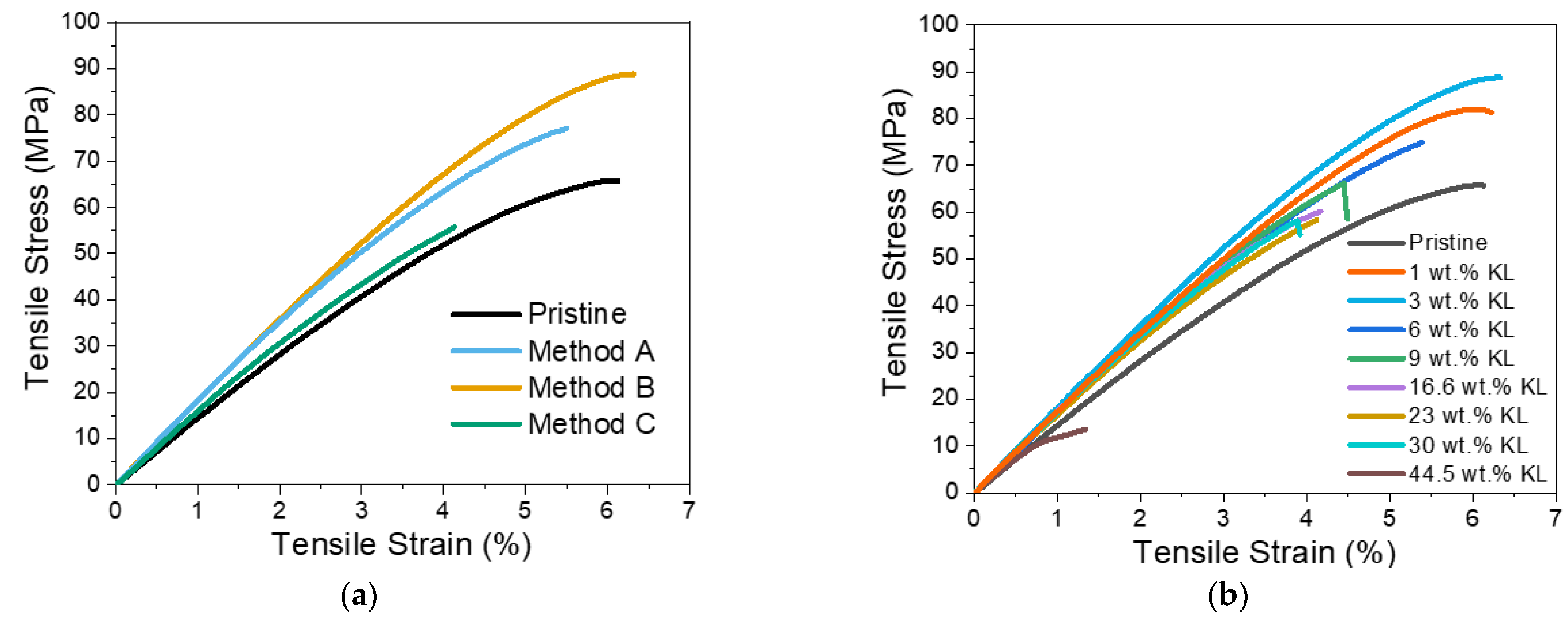
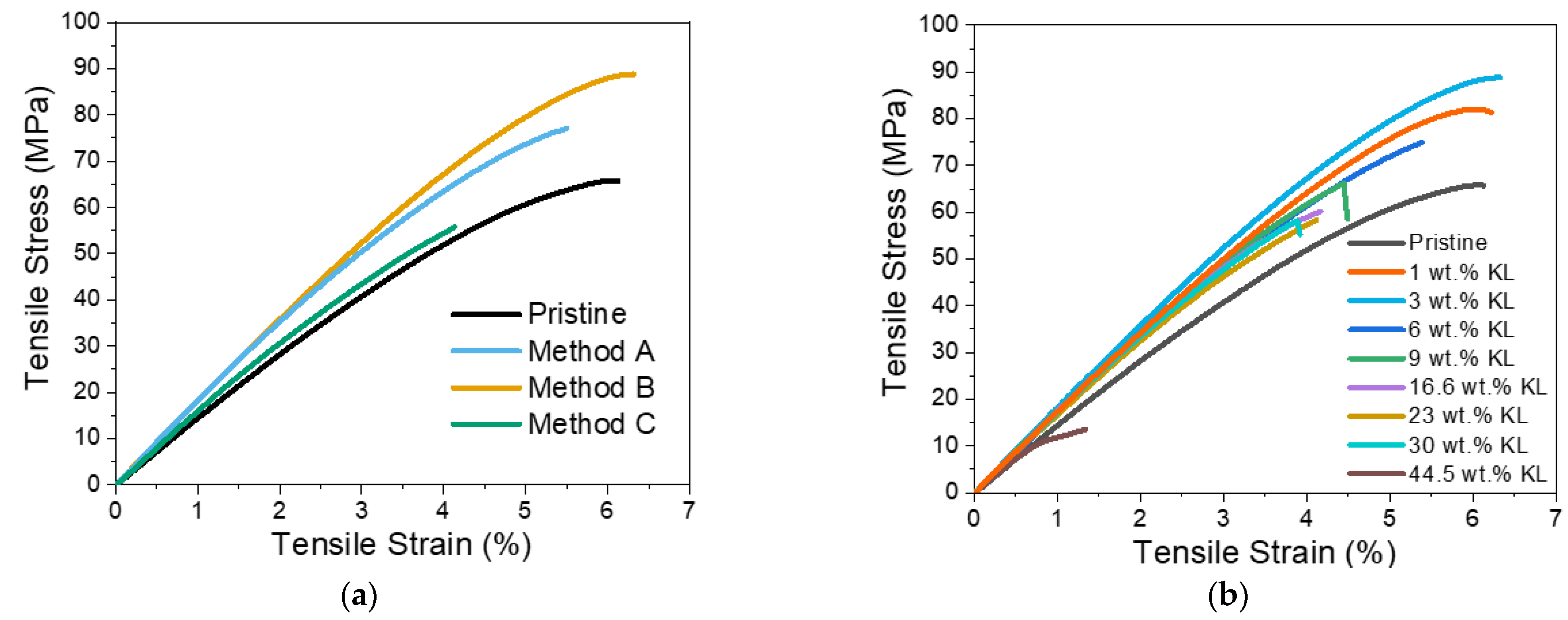
3.2.3. Kraft Lignin as a Curing Agent—Thermo-Mechanical Properties
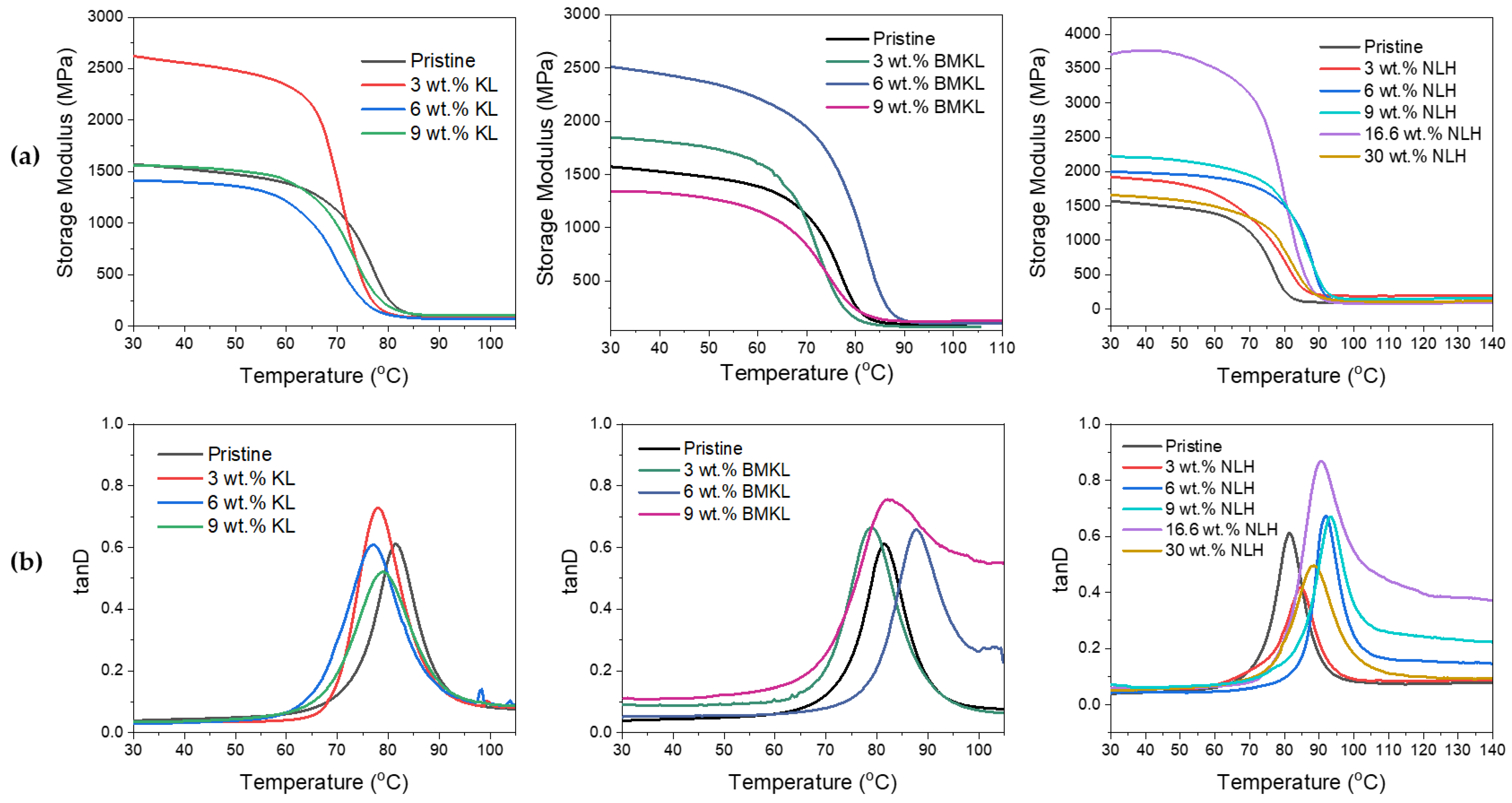
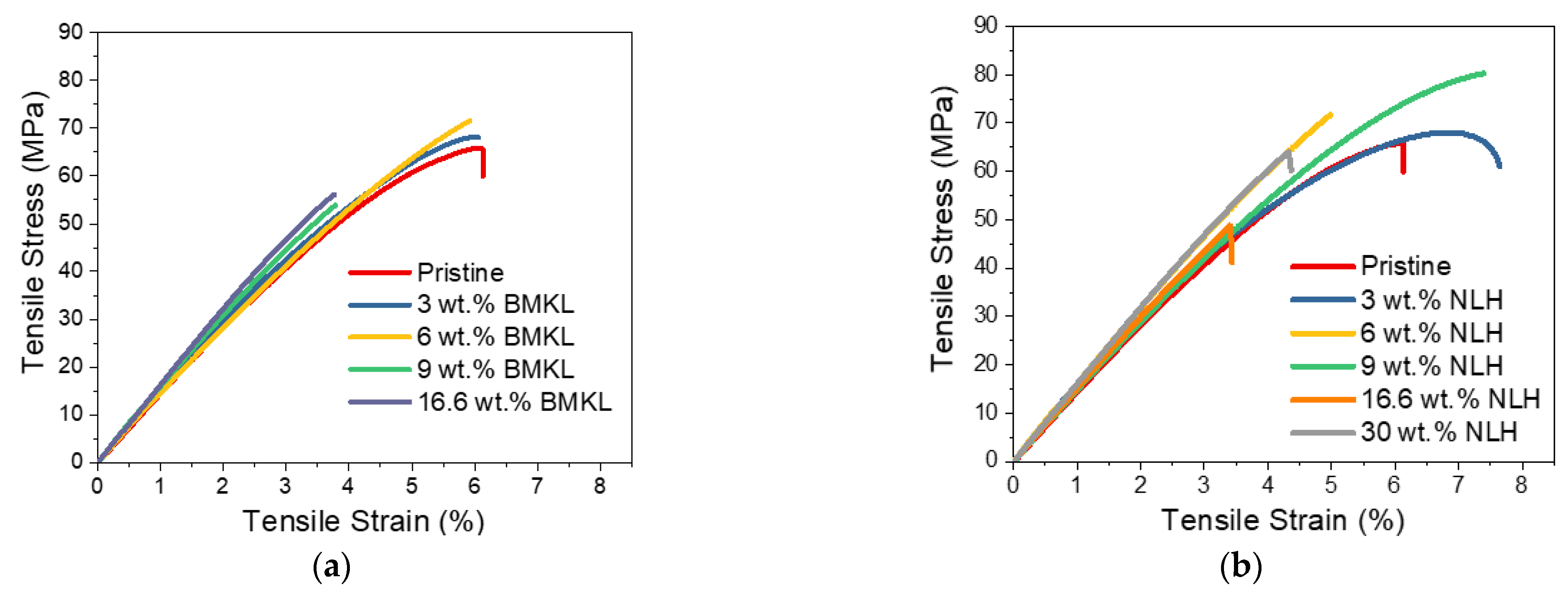
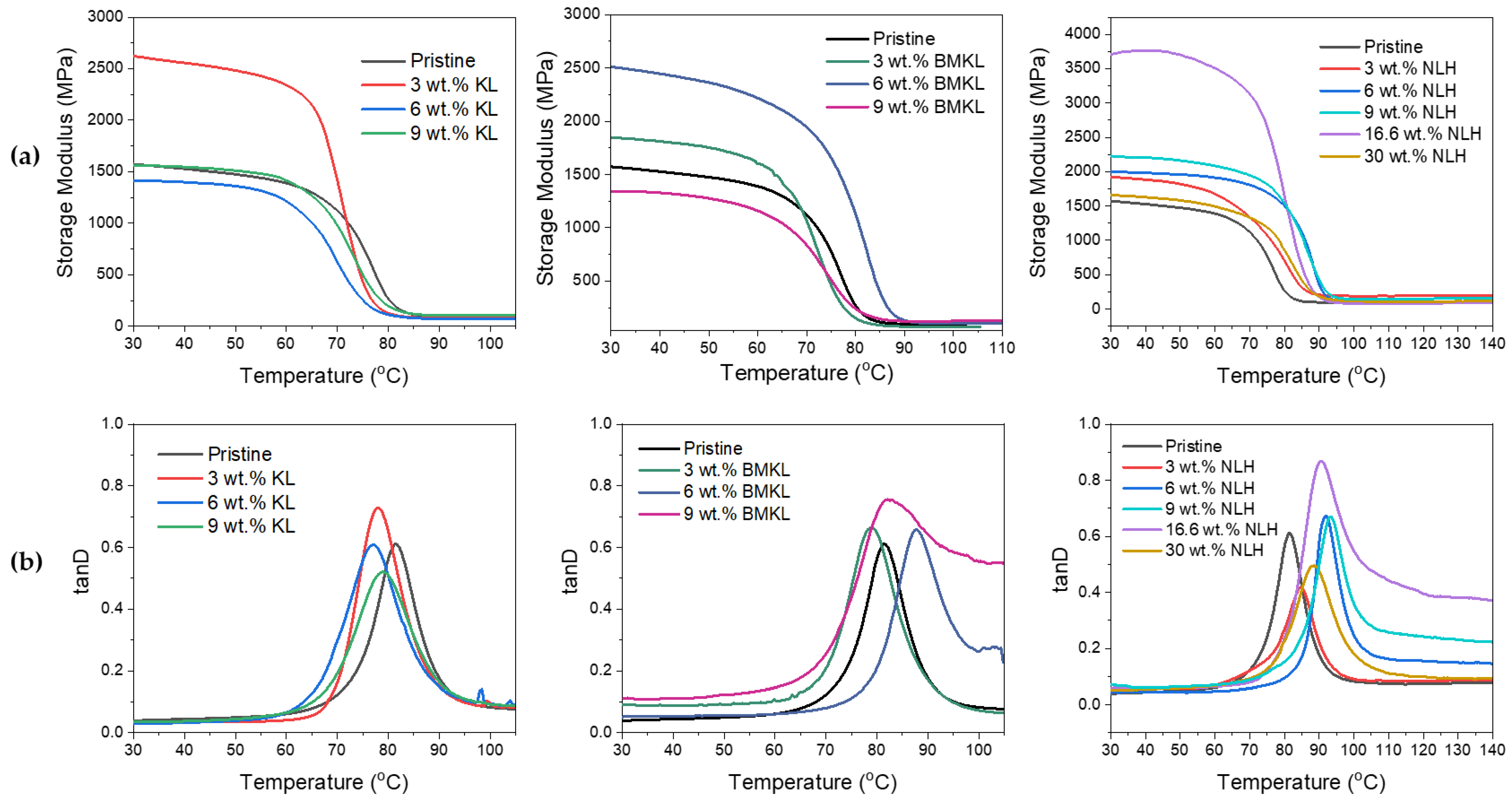
3.2.4. Lignins as Additives in Lignin–Epoxy Composites—Thermo-Mechanical Properties
- Kraft Lignin as an Additive
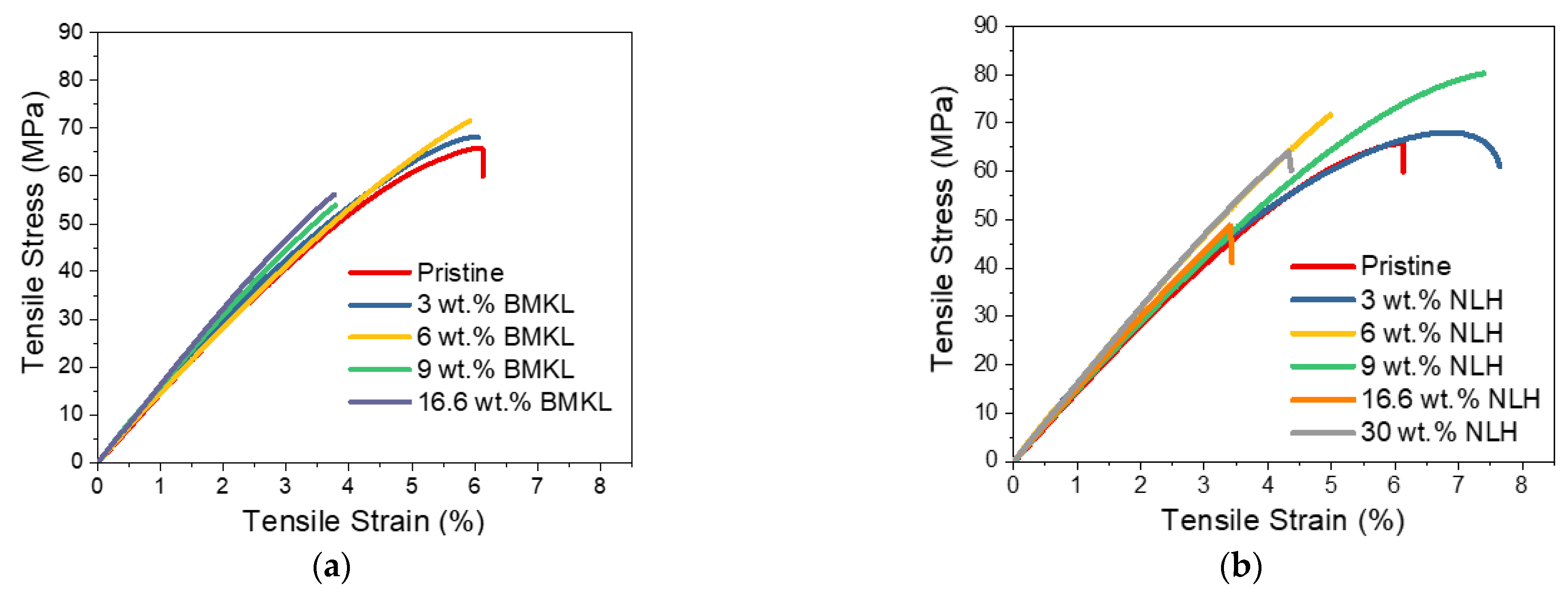
- Modified Lignins as Additives
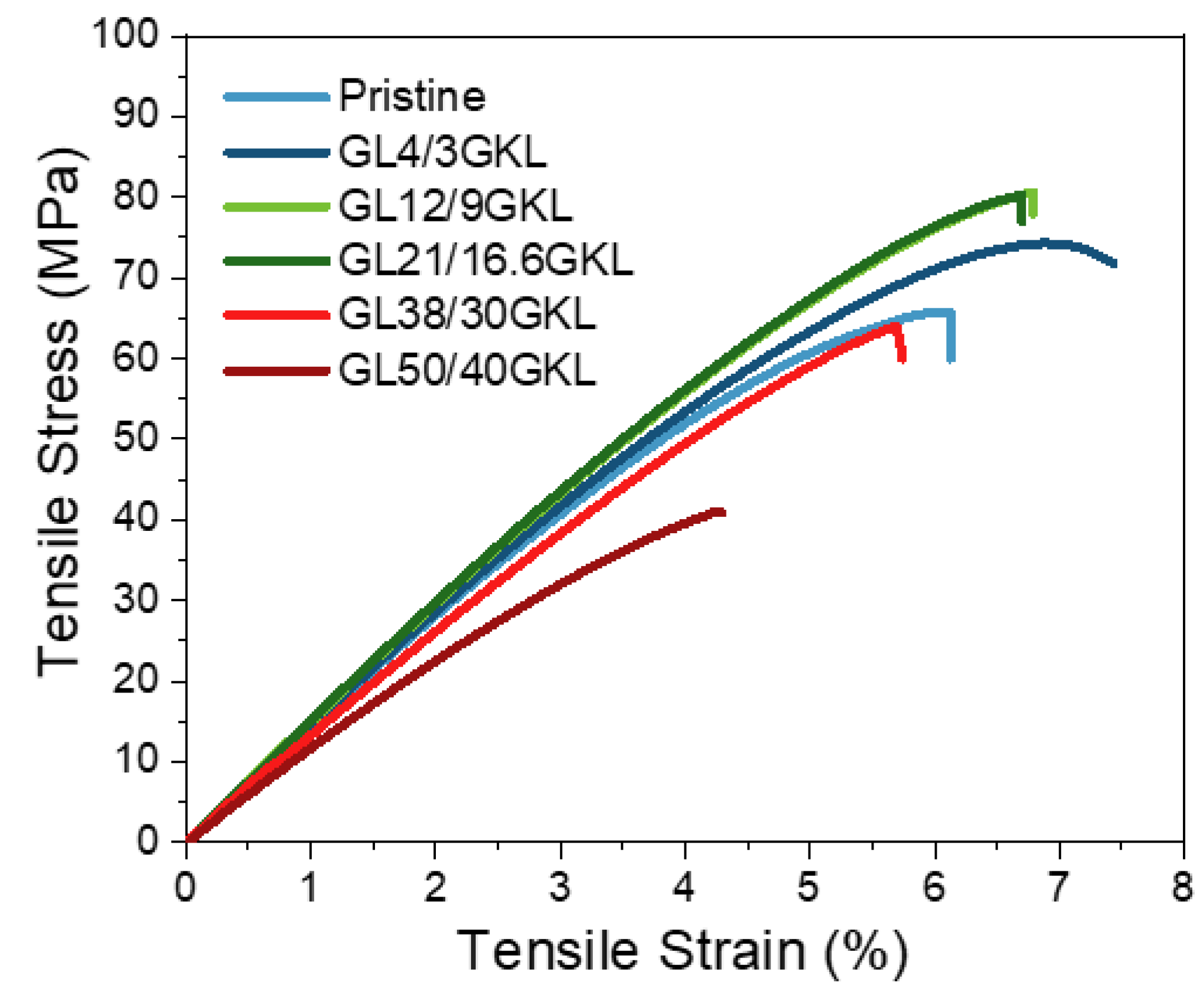
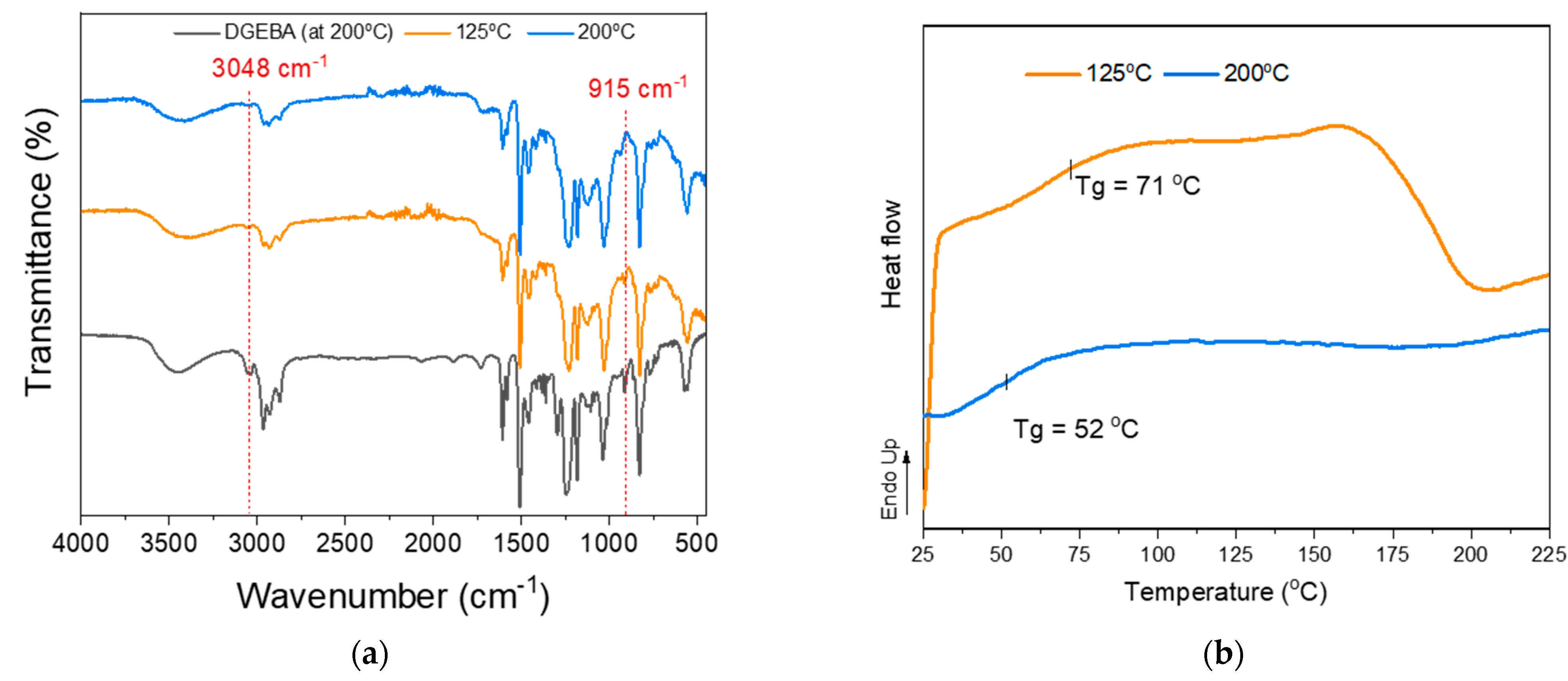
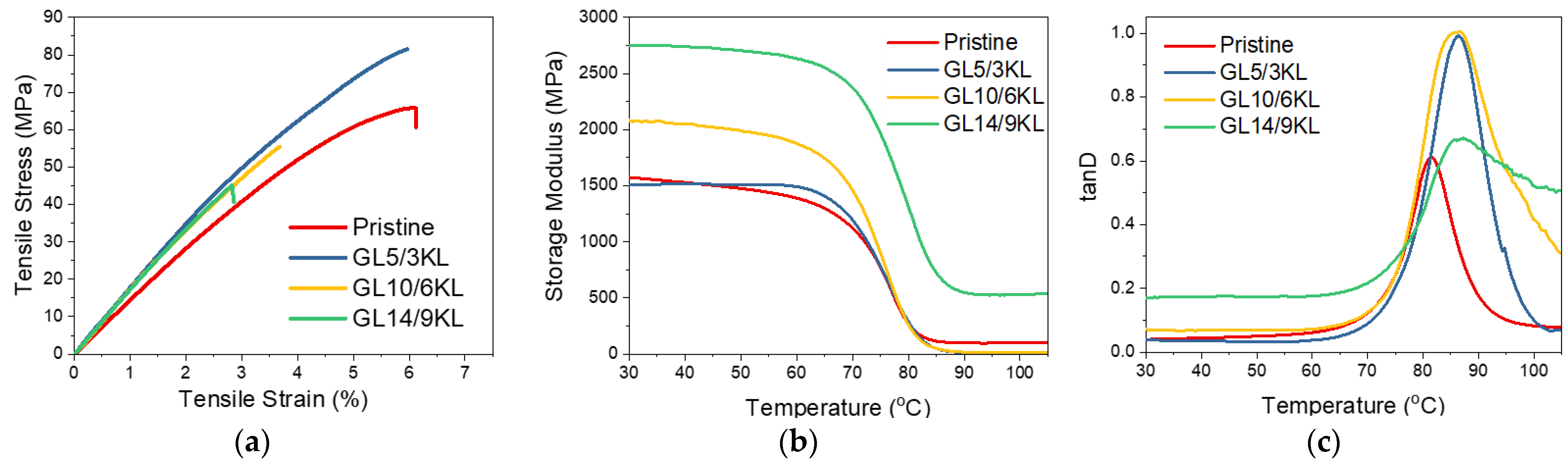
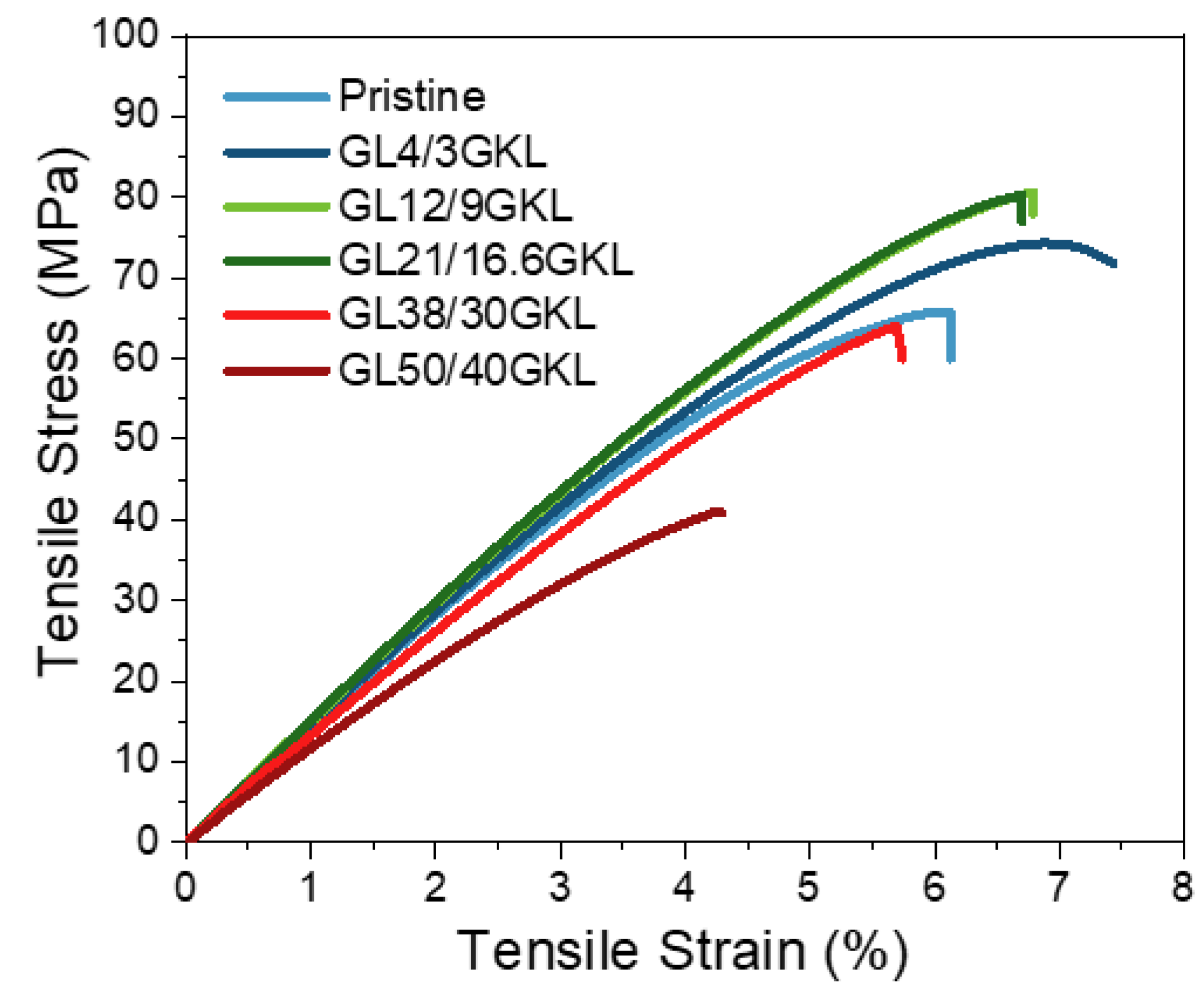
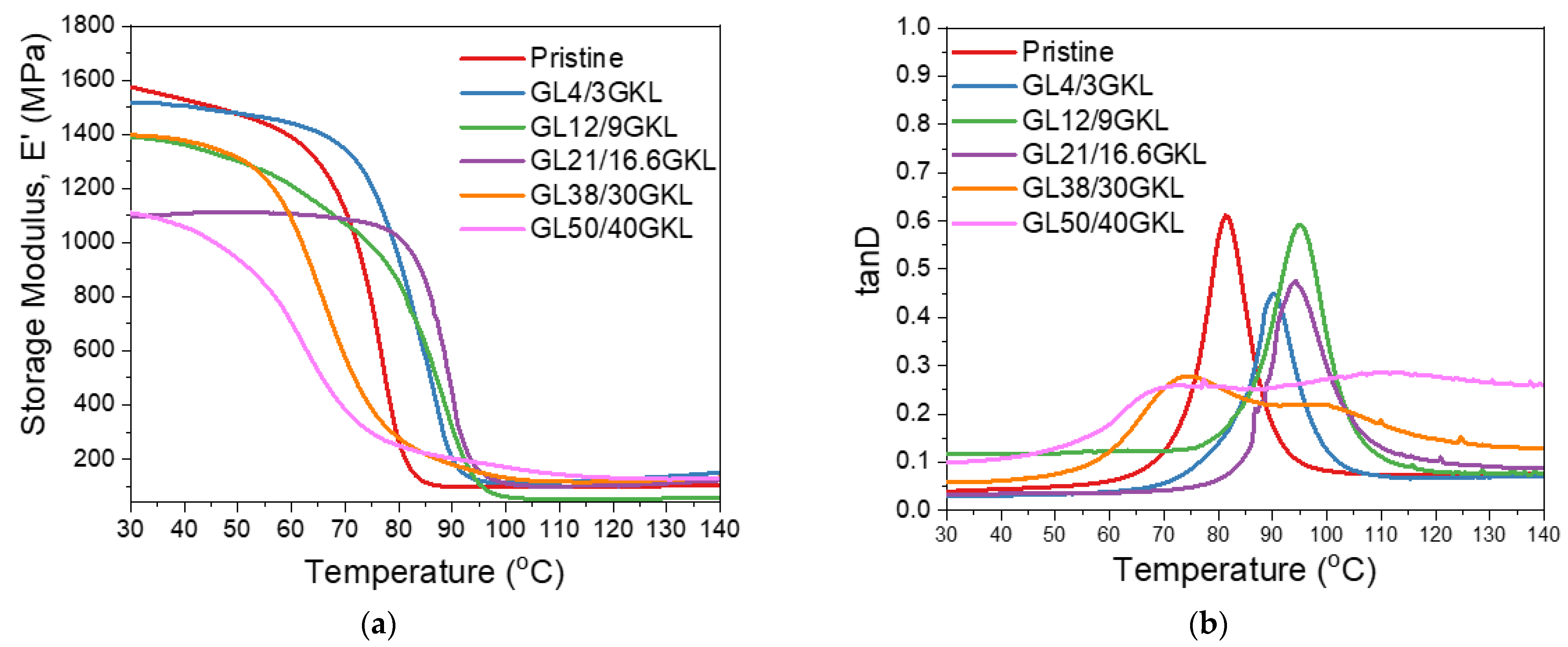
3.2.5. Glycidylized Lignin as a DGEBA Replacement—Thermo-Mechanical Properties
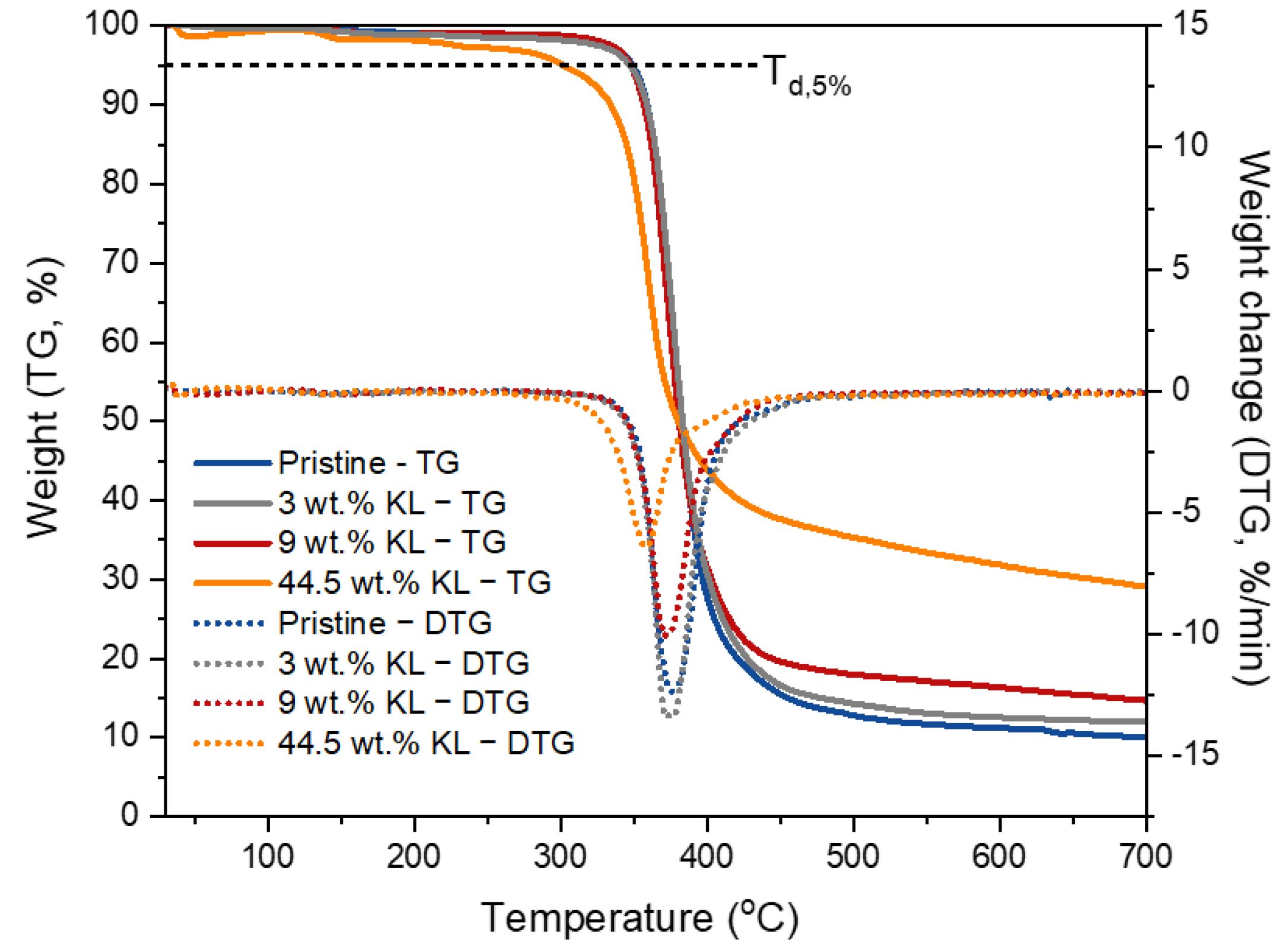
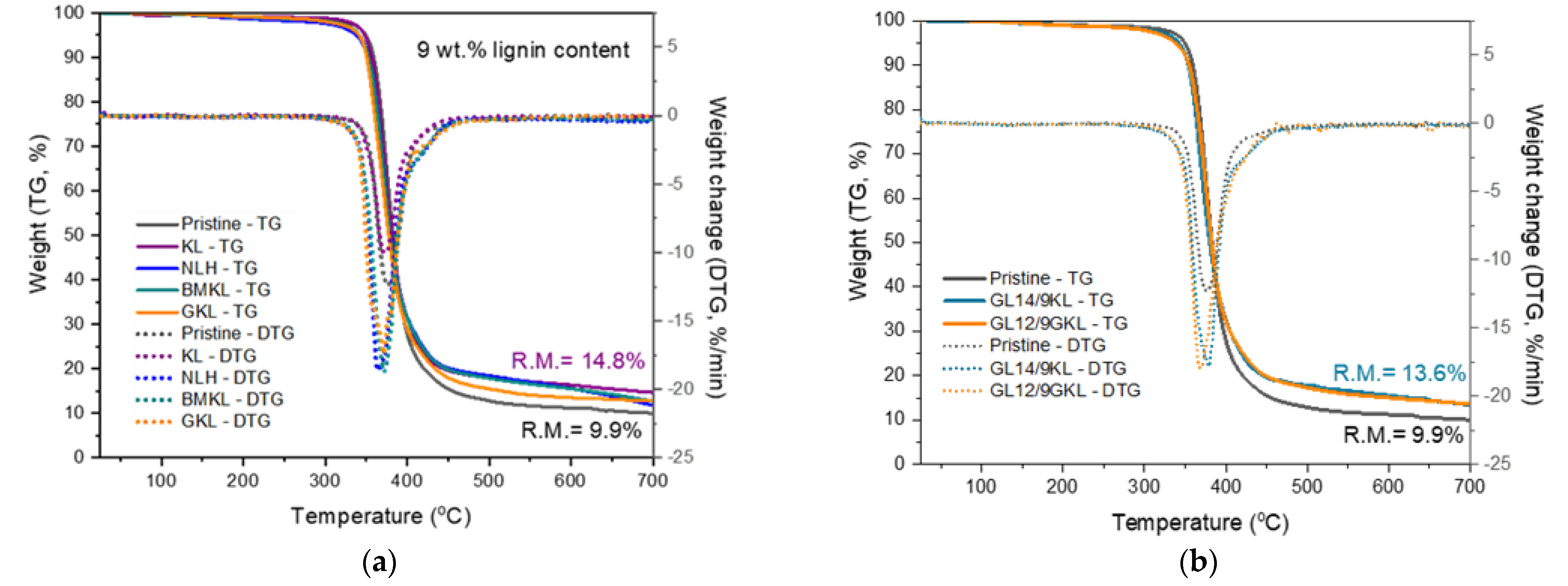
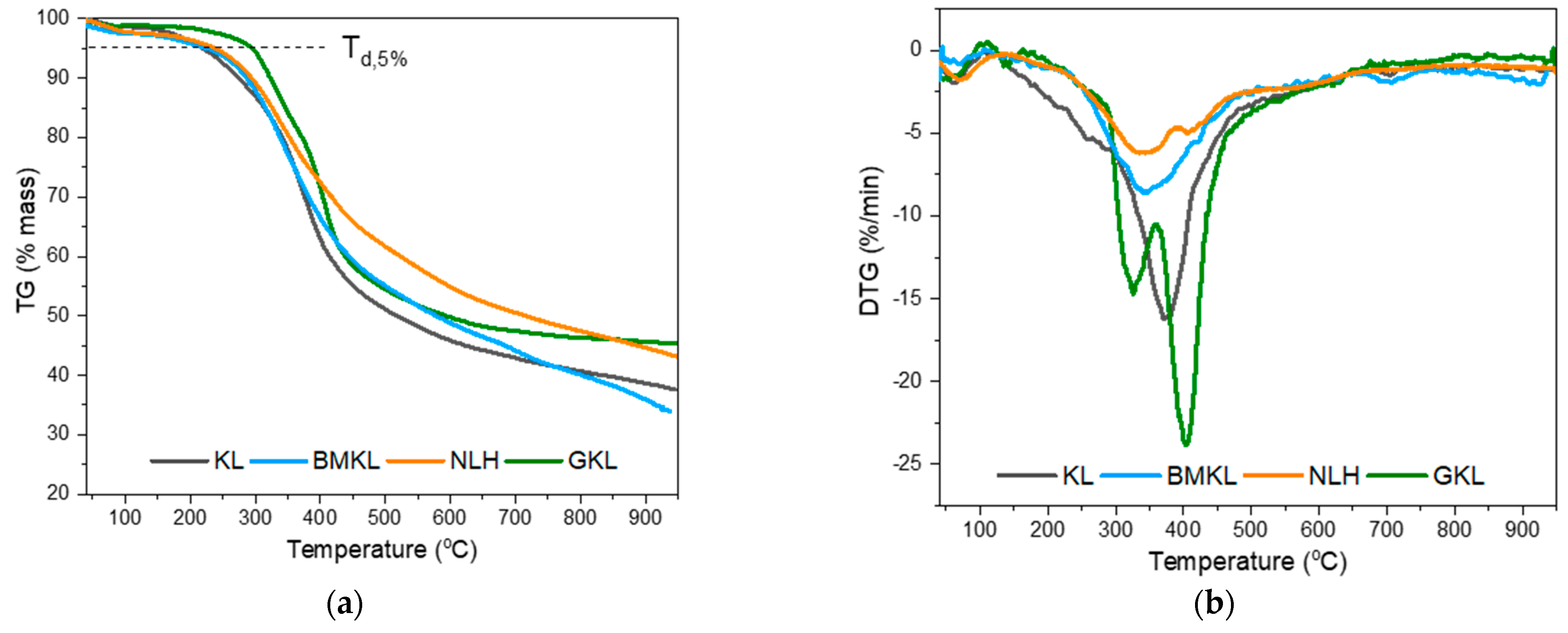
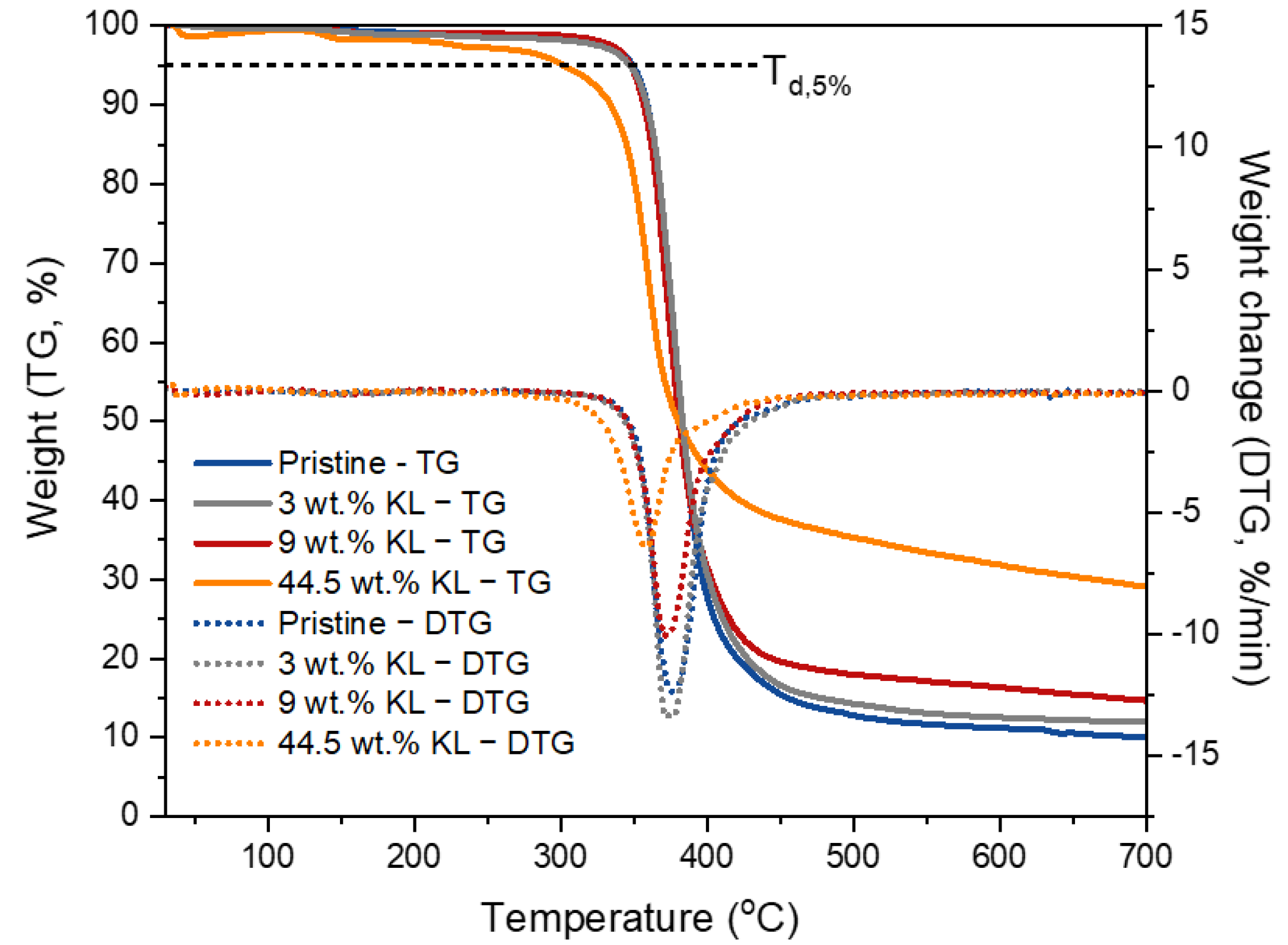
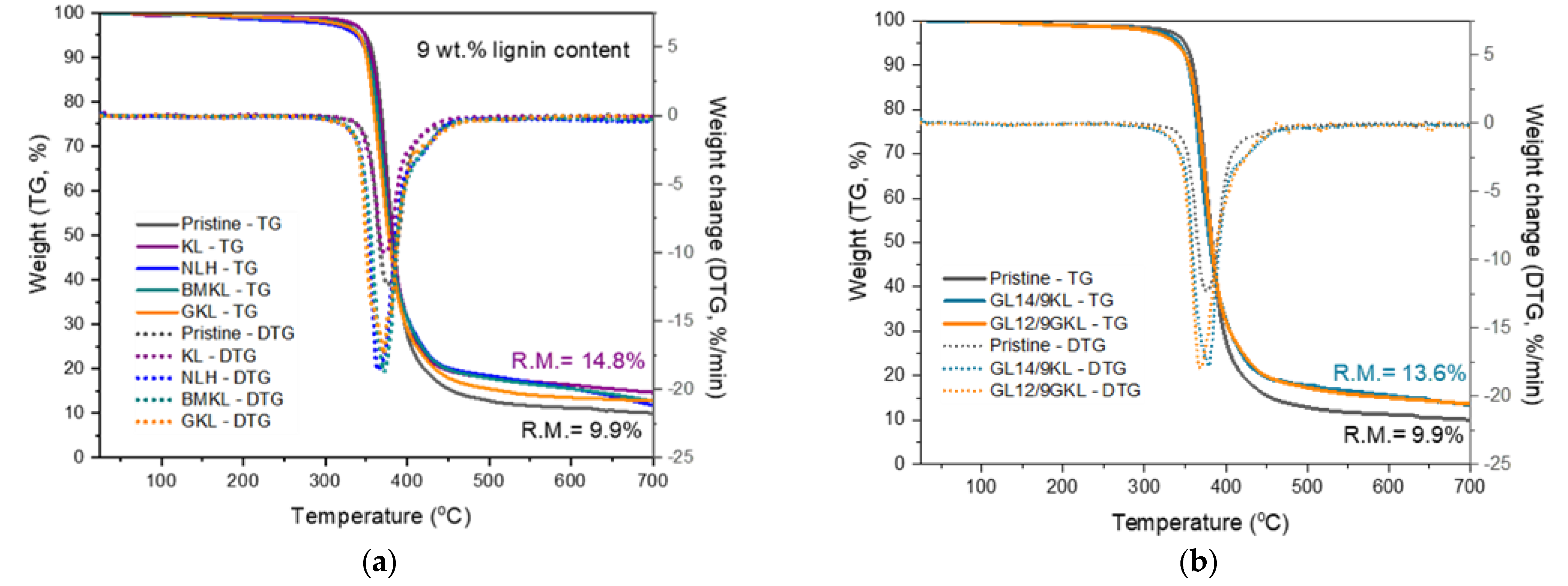
3.2.6. Thermal Properties of Lignin–Epoxy Composites
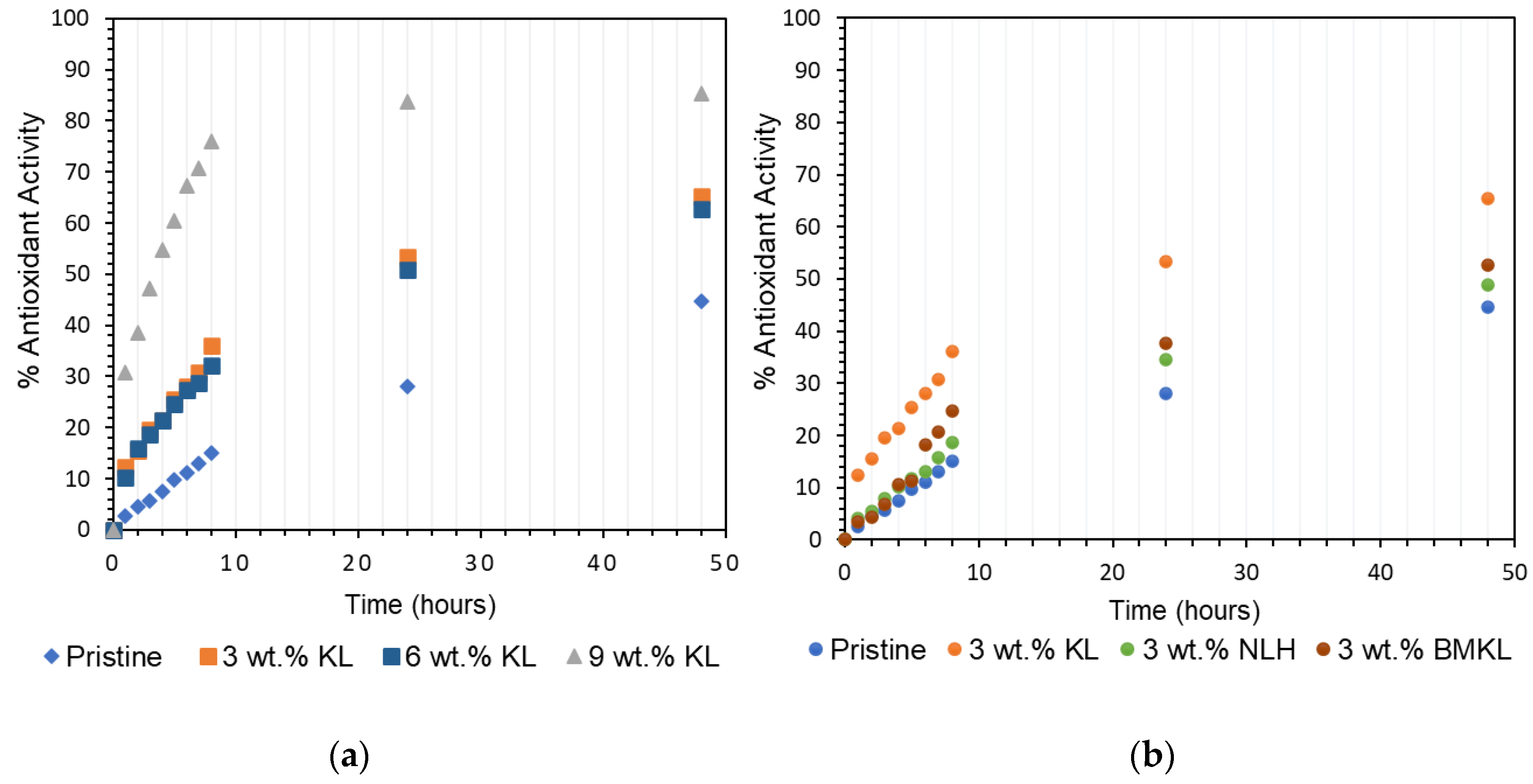
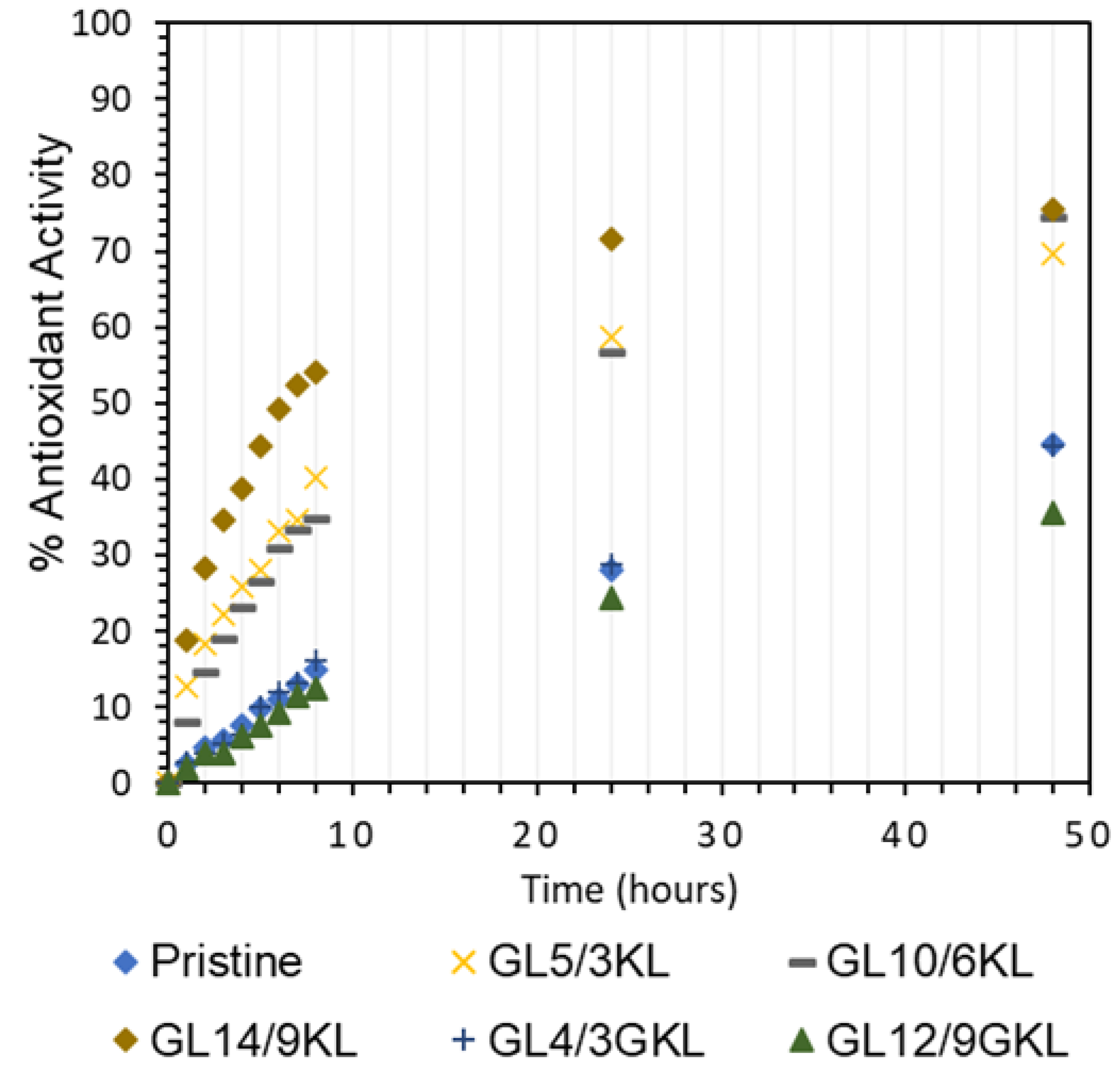
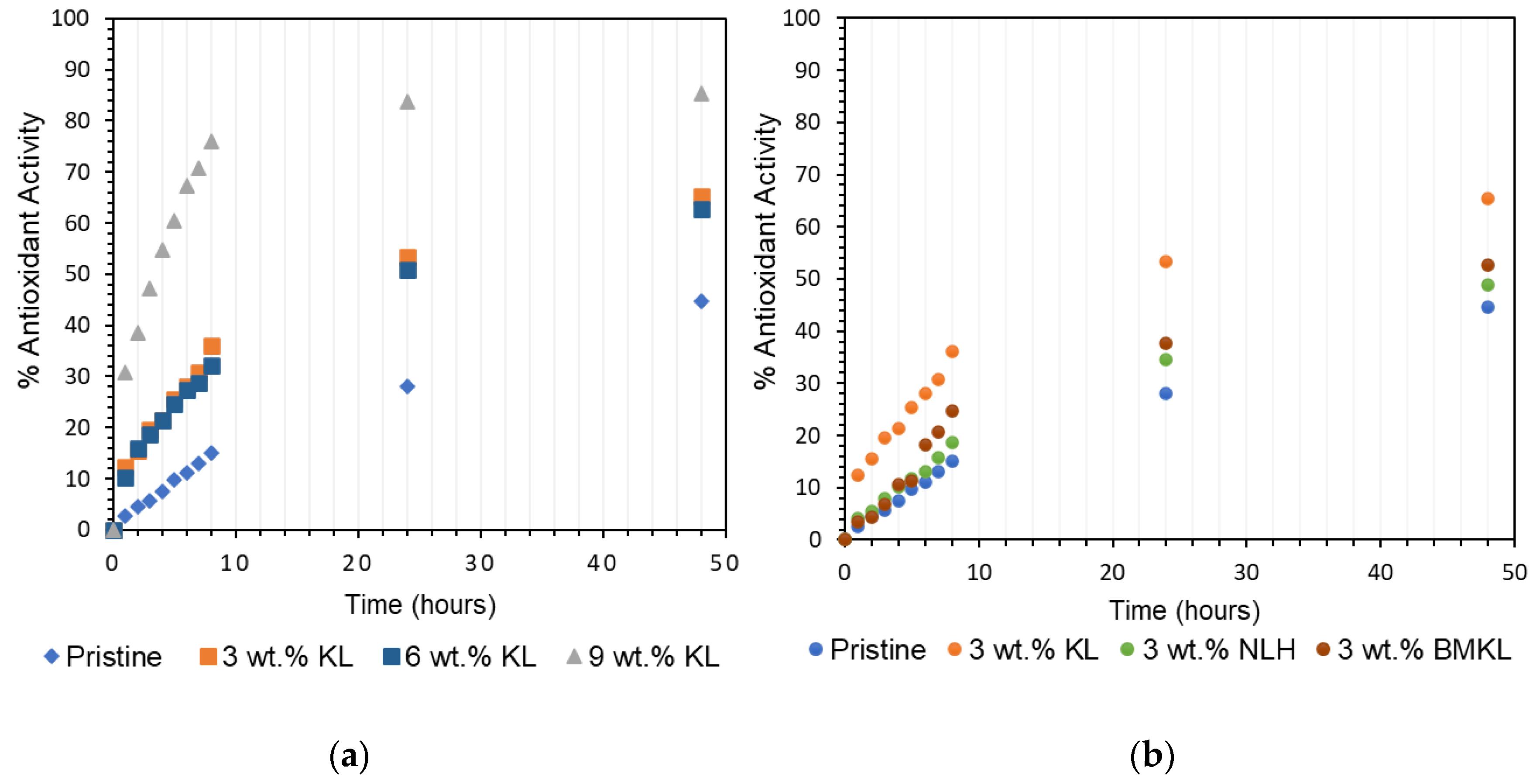
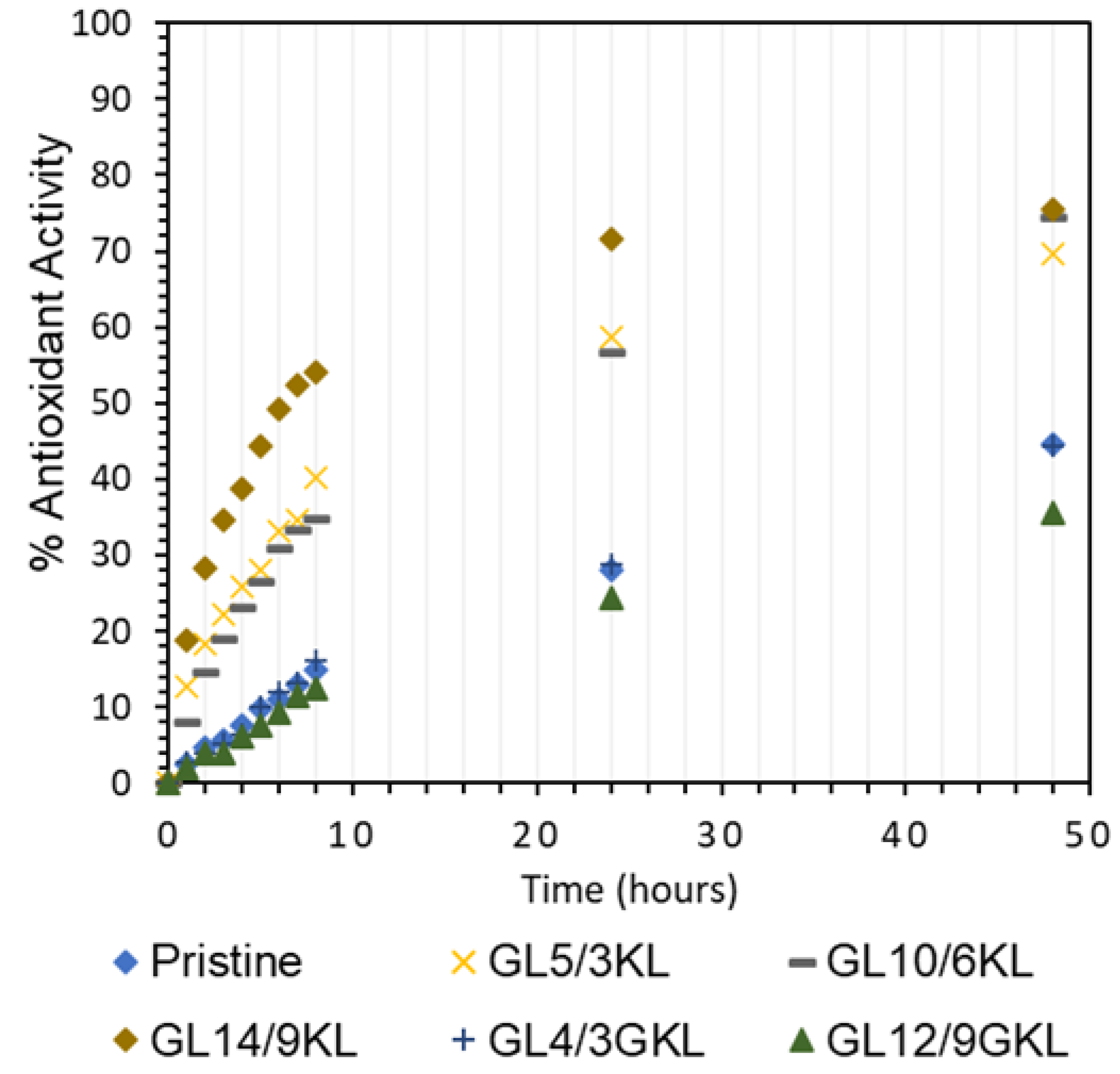
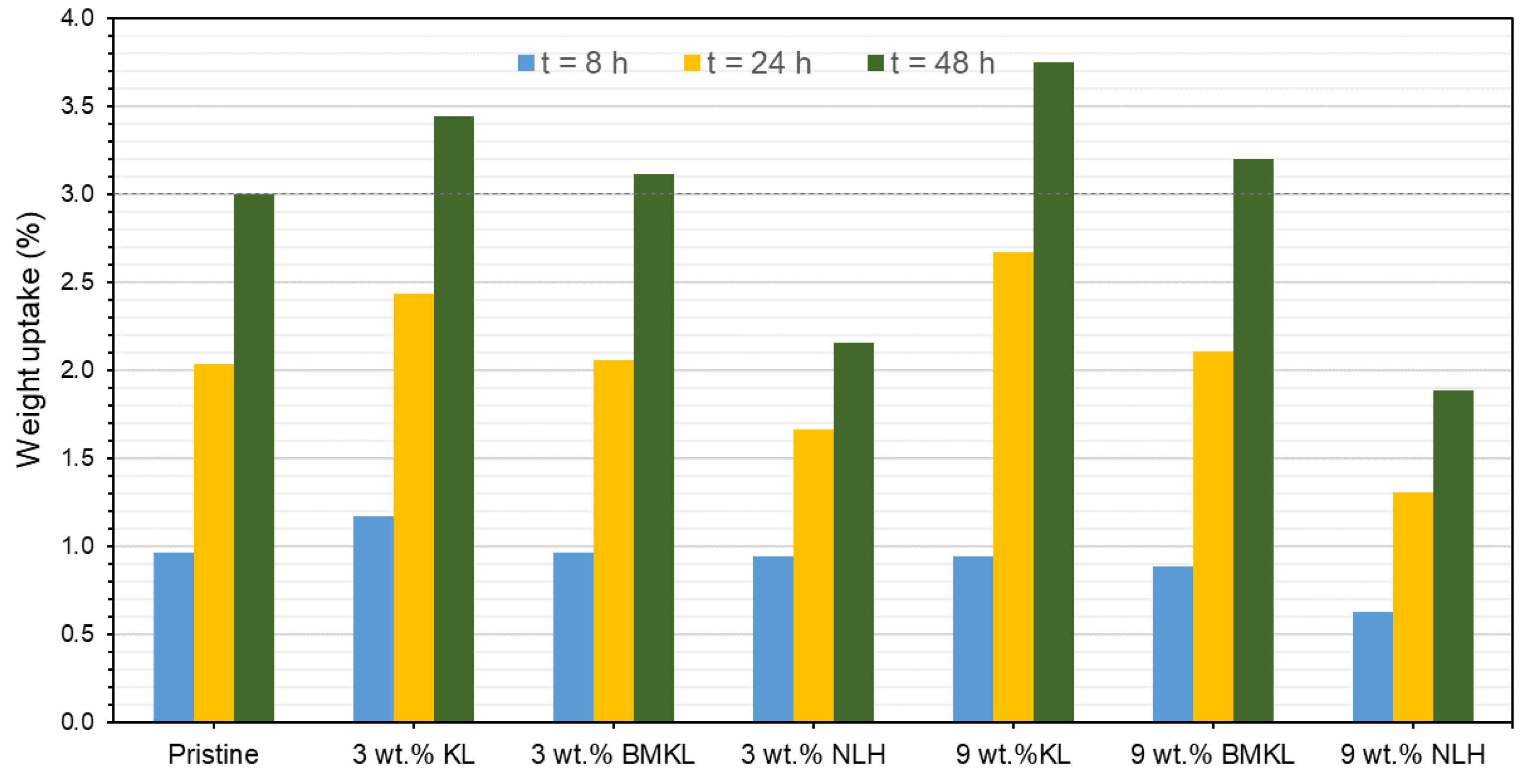
3.2.7. Antioxidant Properties and Solvent Resistance of Lignin–Epoxy Composites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lange, J.P. Towards circular carbo-chemicals—The metamorphosis of petrochemicals. Energy Environ. Sci. 2021, 14, 4358–4376. [Google Scholar] [CrossRef]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Shabanov, N.S.; Rabadanov, K.S.; Gafurov, M.M.; Isaev, A.B.; Sobola, D.S.; Suleimanov, S.I.; Amirov, A.M.; Asvarov, A.S. Lignin-Based Gel Polymer Electrolyte for Cationic Conductivity. Polymers 2021, 13, 2306. [Google Scholar] [CrossRef] [PubMed]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Getzler, Y.D.Y.L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 2020, 5, 501–516. [Google Scholar] [CrossRef]
- Soroudi, A.; Jakubowicz, I. Recycling of bioplastics, their blends and biocomposites: A review. Eur. Polym. J. 2013, 49, 2839–2858. [Google Scholar] [CrossRef]
- Yan, T.; Balzer, A.H.; Herbert, K.M.; Epps, T.H.; Korley, L.T.J. Circularity in polymers: Addressing performance and sustainability challenges using dynamic covalent chemistries. Chem. Sci. 2023, 14, 5243–5265. [Google Scholar] [CrossRef]
- Luo, Z.; Qian, Q.; Sun, H.; Wei, Q.; Zhou, J.; Wang, K. Lignin-First Biorefinery for Converting Lignocellulosic Biomass into Fuels and Chemicals. Energies 2023, 16, 125. [Google Scholar] [CrossRef]
- Mahajan, J.S.; O’Dea, R.M.; Norris, J.B.; Korley, L.T.J.; Epps, T.H., III. Aromatics from Lignocellulosic Biomass: A Platform for High-Performance Thermosets. ACS Sustain. Chem. Eng. 2020, 8, 15072–15096. [Google Scholar] [CrossRef]
- Chundawat, S.P.S.; Beckham, G.T.; Himmel, M.E.; Dale, B.E. Deconstruction of Lignocellulosic Biomass to Fuels and Chemicals. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 121–145. [Google Scholar] [CrossRef]
- Losito, O.; Casiello, M.; Fusco, C.; Mateos Cuadrado, H.; Monopoli, A.; Nacci, A.; D’Accolti, L. Eco-Friendly Catalytic Synthesis of Top Value Chemicals from Valorization of Cellulose Waste. Polymers 2023, 15, 1501. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Feng, Y.; Fu, J.; Guo, H.; Guo, Y.; Han, B.; Jiang, Z.; Kong, L.; Li, C.; Liu, H.; et al. Catalytic conversion of lignocellulosic biomass into chemicals and fuels. Green Energy Environ. 2023, 8, 10–114. [Google Scholar] [CrossRef]
- Patel, R.; Dhar, P.; Babaei-Ghazvini, A.; Nikkhah Dafchahi, M.; Acharya, B. Transforming lignin into renewable fuels, chemicals, and materials: A review. Bioresour. Technol. Rep. 2023, 22, 101463. [Google Scholar] [CrossRef]
- Bruijnincx, P.C.A.; Rinaldi, R.; Weckhuysen, B.M. Unlocking the potential of a sleeping giant: Lignins as sustainable raw materials for renewable fuels, chemicals and materials. Green Chem. 2015, 17, 4860–4861. [Google Scholar] [CrossRef]
- Sternberg, J.; Sequerth, O.; Pilla, S. Green chemistry design in polymers derived from lignin: Review and perspective. Prog. Polym. Sci. 2021, 113, 101344. [Google Scholar] [CrossRef]
- Argyropoulos, D.D.S.; Crestini, C.; Dahlstrand, C.; Furusjö, E.; Gioia, C.; Jedvert, K.; Henriksson, G.; Hulteberg, C.; Lawoko, M.; Pierrou, C.; et al. Kraft Lignin: A Valuable, Sustainable Resource, Opportunities and Challenges. ChemSusChem 2023, 16, e202300492. [Google Scholar] [CrossRef]
- Bajwa, D.S.; Pourhashem, G.; Ullah, A.H.; Bajwa, S.G. A concise review of current lignin production, applications, products and their environmental impact. Ind. Crops Prod. 2019, 139, 111526. [Google Scholar] [CrossRef]
- Dessbesell, L.; Paleologou, M.; Leitch, M.; Pulkki, R.; Xu, C. Global lignin supply overview and kraft lignin potential as an alternative for petroleum-based polymers. Renew. Sustain. Energy Rev. 2020, 123, 109768. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. Int. Ed. 2016, 55, 8164–8215. [Google Scholar] [CrossRef] [PubMed]
- Constant, S.; Wienk, H.L.J.; Frissen, A.E.; Peinder, P.D.; Boelens, R.; van Es, D.S.; Grisel, R.J.H.; Weckhuysen, B.M.; Huijgen, W.J.J.; Gosselink, R.J.A.; et al. New insights into the structure and composition of technical lignins: A comparative characterisation study. Green Chem. 2016, 18, 2651–2665. [Google Scholar] [CrossRef]
- Sen, S.; Patil, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Yang, J.; Ching, Y.C.; Chuah, C.H. Applications of Lignocellulosic Fibers and Lignin in Bioplastics: A Review. Polymers 2019, 11, 751. [Google Scholar] [CrossRef]
- Maldhure, A.V.; Ekhe, J.D.; Deenadayalan, E. Mechanical properties of polypropylene blended with esterified and alkylated lignin. J. Appl. Polym. Sci. 2012, 125, 1701–1712. [Google Scholar] [CrossRef]
- Chen, F.; Dai, H.; Dong, X.; Yang, J.; Zhong, M. Physical properties of lignin-based polypropylene blends. Polym. Compos. 2011, 32, 1019–1025. [Google Scholar] [CrossRef]
- Lu, S.; Li, S.; Yu, J.; Guo, D.; Ling, R.; Huang, B. The effect of hyperbranched polymer lubricant as a compatibilizer on the structure and properties of lignin/polypropylene composites. Wood Mater. Sci. Eng. 2013, 8, 159–165. [Google Scholar] [CrossRef]
- Auvergne, R.; Caillol, S.; David, G.; Boutevin, B.; Pascault, J.-P. Biobased Thermosetting Epoxy: Present and Future. Chem. Rev. 2014, 114, 1082–1115. [Google Scholar] [CrossRef] [PubMed]
- Sadeghifar, H.; Argyropoulos, D.S. Macroscopic Behavior of Kraft Lignin Fractions: Melt Stability Considerations for Lignin–Polyethylene Blends. ACS Sustain. Chem. Eng. 2016, 4, 5160–5166. [Google Scholar] [CrossRef]
- Toriz, G.; Denes, F.; Young, R.A. Lignin-polypropylene composites. Part 1: Composites from unmodified lignin and polypropylene. Polym. Compos. 2002, 23, 806–813. [Google Scholar] [CrossRef]
- Fiorani, G.; Crestini, C.; Selva, M.; Perosa, A. Advancements and Complexities in the Conversion of Lignocellulose Into Chemicals and Materials. Front. Chem. 2020, 8, 797. [Google Scholar] [CrossRef]
- Argyropoulos, D.S.; Crestini, C. A Perspective on Lignin Refining, Functionalization, and Utilization. ACS Sustain. Chem. Eng. 2016, 4, 5089. [Google Scholar] [CrossRef]
- Pappa, C.; Feghali, E.; Vanbroekhoven, K.; Triantafyllidis, K.S. Recent advances in epoxy resins and composites derived from lignin and related bio-oils. Curr. Opin. Green Sustain. Chem. 2022, 38, 100687. [Google Scholar] [CrossRef]
- Gioia, C.; Lo Re, G.; Lawoko, M.; Berglund, L. Tunable Thermosetting Epoxies Based on Fractionated and Well-Characterized Lignins. J. Am. Chem. Soc. 2018, 140, 4054–4061. [Google Scholar] [CrossRef] [PubMed]
- Nikafshar, S.; Wang, J.; Dunne, K.; Sangthonganotai, P.; Nejad, M. Choosing the Right Lignin to Fully Replace Bisphenol A in Epoxy Resin Formulation. ChemSusChem 2021, 14, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Over, L.C.; Grau, E.; Grelier, S.; Meier, M.A.R.; Cramail, H. Synthesis and Characterization of Epoxy Thermosetting Polymers from Glycidylated Organosolv Lignin and Bisphenol A. Macromol. Chem. Phys. 2017, 218, 1600411. [Google Scholar] [CrossRef]
- Pappa, C.P.; Torofias, S.; Triantafyllidis, K.S. Sub-Micro Organosolv Lignin as Bio-Based Epoxy Polymer Component: A Sustainable Curing Agent and Additive. ChemSusChem 2023, 16, e202300076. [Google Scholar] [CrossRef]
- Giannì, P.; Lange, H.; Crestini, C. Functionalized Organosolv Lignins Suitable for Modifications of Hard Surfaces. ACS Sustain. Chem. Eng. 2020, 8, 7628–7638. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.-L.; Li, X.; Park, S.-J. Synthesis and application of epoxy resins: A review. J. Ind. Eng. Chem. 2015, 29, 1–11. [Google Scholar] [CrossRef]
- Kumar, S.; Krishnan, S.; Samal, S.K.; Mohanty, S.; Nayak, S.K. Toughening of Petroleum Based (DGEBA) Epoxy Resins with Various Renewable Resources Based Flexible Chains for High Performance Applications: A Review. Ind. Eng. Chem. Res. 2018, 57, 2711–2726. [Google Scholar] [CrossRef]
- Hirose, S.; Hatakeyama, T.; Hatakeyama, H. Novel Epoxy Resins Derived from Biomass Components. Procedia Chem. 2012, 4, 26–33. [Google Scholar] [CrossRef]
- Nikafshar, S.; Zabihi, O.; Hamidi, S.; Moradi, Y.; Barzegar, S.; Ahmadi, M.; Naebe, M. A renewable bio-based epoxy resin with improved mechanical performance that can compete with DGEBA. RSC Adv. 2017, 7, 8694–8701. [Google Scholar] [CrossRef]
- Tsai, H.-Y.; Fujita, T.; Wang, S.; Naito, M. Environmentally friendly recycling system for epoxy resin with dynamic covalent bonding. Sci. Technol. Adv. Mater. 2021, 22, 532–542. [Google Scholar] [CrossRef]
- Khan, N.G.; Correia, J.; Adiga, D.; Rai, P.S.; Dsouza, H.S.; Chakrabarty, S.; Kabekkodu, S.P. A comprehensive review on the carcinogenic potential of bisphenol A: Clues and evidence. Environ. Sci. Pollut. Res. Int. 2021, 28, 19643–19663. [Google Scholar] [CrossRef]
- Yamini, G.; Shakeri, A.; Zohuriaan-Mehr, M.J.; Kabiri, K. Cyclocarbonated lignosulfonate as a bio-resourced reactive reinforcing agent for epoxy biocomposite: From natural waste to value-added bio-additive. J. CO2 Util. 2018, 24, 50–58. [Google Scholar] [CrossRef]
- Ortiz, P.; Vendamme, R.; Eevers, W. Fully Biobased Epoxy Resins from Fatty Acids and Lignin. Molecules 2020, 25, 1158. [Google Scholar] [CrossRef]
- Kong, X.; Xu, Z.; Guan, L.; Di, M. Study on polyblending epoxy resin adhesive with lignin I-curing temperature. Int. J. Adhes. Adhes. 2014, 48, 75–79. [Google Scholar] [CrossRef]
- Jablonskis, A.; Arshanitsa, A.; Arnautov, A.; Telysheva, G.; Evtuguin, D. Evaluation of Ligno Boost™ softwood kraft lignin epoxidation as an approach for its application in cured epoxy resins. Ind. Crops Prod. 2018, 112, 225–235. [Google Scholar] [CrossRef]
- Delmas, G.-H.; Benjelloun-Mlayah, B.; Bigot, Y.L.; Delmas, M. Biolignin™ based epoxy resins. J. Appl. Polym. Sci. 2013, 127, 1863–1872. [Google Scholar] [CrossRef]
- Abdul Khalil, H.P.S.; Marliana, M.M.; Alshammari, T. Material properties of epoxy-reinforced biocomposites with lignin from empty fruit bunch as curing agent. BioResources 2011, 6, 5206–5223. [Google Scholar] [CrossRef]
- Cui, C.; Sun, R.; Argyropoulos, D.S. Fractional Precipitation of Softwood Kraft Lignin: Isolation of Narrow Fractions Common to a Variety of Lignins. ACS Sustain. Chem. Eng. 2014, 2, 959–968. [Google Scholar] [CrossRef]
- Jääskeläinen, A.-S.; Liitiä, T.; Mikkelson, A.; Tamminen, T. Aqueous organic solvent fractionation as means to improve lignin homogeneity and purity. Ind. Crops Prod. 2017, 103, 51–58. [Google Scholar] [CrossRef]
- Gilca, I.A.; Popa, V.I.; Crestini, C. Obtaining lignin nanoparticles by sonication. Ultrason. Sonochem. 2015, 23, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Iravani, S.; Varma, R.S. Greener synthesis of lignin nanoparticles and their applications. Green Chem. 2020, 22, 612–636. [Google Scholar] [CrossRef]
- Gigli, M.; Crestini, C. Fractionation of industrial lignins: Opportunities and challenges. Green Chem. 2020, 22, 4722–4746. [Google Scholar] [CrossRef]
- Ebrahimi Majdar, R.; Ghasemian, A.; Resalati, H.; Saraeian, A.; Crestini, C.; Lange, H. Case Study in Kraft Lignin Fractionation: “Structurally Purified” Lignin Fractions—The Role of Solvent H-Bonding Affinity. ACS Sustain. Chem. Eng. 2020, 8, 16803–16813. [Google Scholar] [CrossRef]
- Majdar, R.E.; Crestini, C.; Lange, H. Lignin Fractionation in Segmented Continuous Flow. ChemSusChem 2020, 13, 4735–4742. [Google Scholar] [CrossRef] [PubMed]
- Karnaouri, A.; Lange, H.; Crestini, C.; Rova, U.; Christakopoulos, P. Chemoenzymatic Fractionation and Characterization of Pretreated Birch Outer Bark. ACS Sustain. Chem. Eng. 2016, 4, 5289–5302. [Google Scholar] [CrossRef]
- Duval, A.; Vilaplana, F.; Crestini, C.; Lawoko, M. Solvent screening for the fractionation of industrial kraft lignin. Holzforschung 2016, 70, 11–20. [Google Scholar] [CrossRef]
- Lange, H.; Schiffels, P.; Sette, M.; Sevastyanova, O.; Crestini, C. Fractional Precipitation of Wheat Straw Organosolv Lignin: Macroscopic Properties and Structural Insights. ACS Sustain. Chem. Eng. 2016, 4, 5136–5151. [Google Scholar] [CrossRef]
- Yin, Q.F.; Di, M.W. Preparation and Mechanical Properties of Lignin/Epoxy Resin Composites. Adv. Mater. Res. 2012, 482-484, 1959–1962. [Google Scholar] [CrossRef]
- Sasaki, C.; Wanaka, M.; Takagi, H.; Tamura, S.; Asada, C.; Nakamura, Y. Evaluation of epoxy resins synthesized from steam-exploded bamboo lignin. Ind. Crops Prod. 2013, 43, 757–761. [Google Scholar] [CrossRef]
- Du, X.; Li, J.; Lindström, M.E. Modification of industrial softwood kraft lignin using Mannich reaction with and without phenolation pretreatment. Ind. Crops Prod. 2014, 52, 729–735. [Google Scholar] [CrossRef]
- Pan, H.; Sun, G.; Zhao, T. Synthesis and characterization of aminated lignin. Int. J. Biol. Macromol. 2013, 59, 221–226. [Google Scholar] [CrossRef]
- Mendis, G.P.; Hua, I.; Youngblood, J.P.; Howarter, J.A. Enhanced dispersion of lignin in epoxy composites through hydration and mannich functionalization. J. Appl. Polym. Sci. 2015, 132, 41263. [Google Scholar] [CrossRef]
- Pan, H.; Sun, G.; Zhao, T.; Wang, G. Thermal Properties of Epoxy Resins Crosslinked by an Aminated Lignin. Polym. Eng. Sci. 2014, 55, 924–932. [Google Scholar] [CrossRef]
- Asada, C.; Basnet, S.; Otsuka, M.; Sasaki, C.; Nakamura, Y. Epoxy resin synthesis using low molecular weight lignin separated from various lignocellulosic materials. Int. J. Biol. Macromol. 2015, 74, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Xiao, L.P.; Li, X.Y.; Xiao, W.Z.; Yang, Y.Q.; Sun, R.C. Renewable and flexible thermosetting epoxies based on functionalized biorefinery lignin fractions. Mater. Today Sustain. 2021, 15, 100083. [Google Scholar] [CrossRef]
- Gouveia, J.R.; Garcia, G.E.S.; Antonino, L.D.; Tavares, L.B.; Dos Santos, D.J. Epoxidation of Kraft Lignin as a Tool for Improving the Mechanical Properties of Epoxy Adhesive. Molecules 2020, 25, 2513. [Google Scholar] [CrossRef]
- Ferdosian, F.; Yuan, Z.; Anderson, M.; Xu, C. Synthesis of lignin-based epoxy resins: Optimization of reaction parameters using response surface methodology. RSC Adv. 2014, 4, 31745–31753. [Google Scholar] [CrossRef]
- Ferdosian, F.; Zhang, Y.; Yuan, Z.; Anderson, M.; Xu, C. Curing Kinetics and Mechanical Properties of Bio-based Epoxy Composites Comprising Lignin-Based Epoxy Resins. Eur. Polym. J. 2016, 82, 153–165. [Google Scholar] [CrossRef]
- Măluțan, T.; Nicu, R.; Popa, V. Lignin modification by epoxidation. BioResources 2008, 3, 1371–13767. [Google Scholar] [CrossRef]
- Mansouri, E.; Yuan, Q.; Huang, F. Synthesis and characterization of kraft lignin-based epoxy resins. BioResources 2011, 6, 2492–2503. [Google Scholar] [CrossRef]
- Sun, J.; Wang, C.; Yeo, J.C.C.; Yuan, D.; Li, H.; Stubbs, L.P.; He, C. Lignin Epoxy Composites: Preparation, Morphology, and Mechanical Properties. Macromol. Mater. Eng. 2016, 301, 328–336. [Google Scholar] [CrossRef]
- Sipponen, M.H.; Lange, H.; Ago, M.; Crestini, C. Understanding Lignin Aggregation Processes. A Case Study: Budesonide Entrapment and Stimuli Controlled Release from Lignin Nanoparticles. ACS Sustain. Chem. Eng. 2018, 6, 9342–9351. [Google Scholar] [CrossRef]
- Sipponen, M.H.; Lange, H.; Crestini, C.; Henn, A.; Österberg, M. Lignin for Nano- and Microscaled Carrier Systems: Applications, Trends, and Challenges. ChemSusChem 2019, 12, 2039–2054. [Google Scholar] [CrossRef]
- Cailotto, S.; Gigli, M.; Bonini, M.; Rigoni, F.; Crestini, C. Sustainable Strategies in the Synthesis of Lignin Nanoparticles for the Release of Active Compounds: A Comparison. ChemSusChem 2020, 13, 4759–4767. [Google Scholar] [CrossRef]
- Piombino, C.; Lange, H.; Sabuzi, F.; Galloni, P.; Conte, V.; Crestini, C. Lignosulfonate Microcapsules for Delivery and Controlled Release of Thymol and Derivatives. Molecules 2020, 25, 866. [Google Scholar] [CrossRef]
- Zongo, L.; Lange, H.; Crestini, C. A Study of the Effect of Kosmotropic and Chaotropic Ions on the Release Characteristics of Lignin Microcapsules under Stimuli-Responsive Conditions. ACS Omega 2019, 4, 6979–6993. [Google Scholar] [CrossRef] [PubMed]
- Garcia Gonzalez, M.N.; Levi, M.; Turri, S.; Griffini, G. Lignin nanoparticles by ultrasonication and their incorporation in waterborne polymer nanocomposites. J. Appl. Polym. Sci. 2017, 134, 45318. [Google Scholar] [CrossRef]
- Argyropoulos, D.S.; Pajer, N.; Crestini, C. Quantitative 31P NMR Analysis of Lignins and Tannins. J. Vis. Exp. JoVE 2021, 174, e62696. [Google Scholar] [CrossRef]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the structure of softwood kraft lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
- Margellou, A.; Triantafyllidis, K.S. Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts 2019, 9, 43. [Google Scholar] [CrossRef]
- Lazaridis, P.A.; Fotopoulos, A.P.; Karakoulia, S.A.; Triantafyllidis, K.S. Catalytic Fast Pyrolysis of Kraft Lignin With Conventional, Mesoporous and Nanosized ZSM-5 Zeolite for the Production of Alkyl-Phenols and Aromatics. Front. Chem. 2018, 6, 295. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; An, X.; Yang, S. The Effect of Ball Milling Time on the Isolation of Lignin in the Cell Wall of Different Biomass. Front. Bioeng. Biotechnol. 2021, 9, 807625. [Google Scholar] [CrossRef] [PubMed]
- Sapouna, I.; Lawoko, M. Deciphering lignin heterogeneity in ball milled softwood: Unravelling the synergy between the supramolecular cell wall structure and molecular events. Green Chem. 2021, 23, 3348–3364. [Google Scholar] [CrossRef]
- Lu, X.; Gu, X. A review on lignin-based epoxy resins: Lignin effects on their synthesis and properties. Int. J. Biol. Macromol. 2023, 229, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Jawerth, M.E.; Brett, C.J.; Terrier, C.; Larsson, P.T.; Lawoko, M.; Roth, S.V.; Lundmark, S.; Johansson, M. Mechanical and Morphological Properties of Lignin-Based Thermosets. ACS Appl. Polym. Mater. 2020, 2, 668–676. [Google Scholar] [CrossRef]
- Gioia, C.; Colonna, M.; Tagami, A.; Medina, L.; Sevastyanova, O.; Berglund, L.A.; Lawoko, M. Lignin-Based Epoxy Resins: Unravelling the Relationship between Structure and Material Properties. Biomacromolecules 2020, 21, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Feghali, E.; van de Pas, D.J.; Parrott, A.J.; Torr, K.M. Biobased Epoxy Thermoset Polymers from Depolymerized Native Hardwood Lignin. ACS Macro Lett. 2020, 9, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Ferdosian, F.; Yuan, Z.; Anderson, M.; Xu, C. Sustainable Lignin-based Epoxy Resins Cured with Aromatic and Aliphatic Amine Curing Agents: Curing Kinetics and Thermal Properties. Thermochim. Acta 2015, 618, 48–55. [Google Scholar] [CrossRef]
- Ding, J.; Gu, L. Epoxidation Modification of Renewable Lignin to Improve the Corrosion Performance of Epoxy Coating. Int. J. Electrochem. Sci. 2016, 11, 6256–6265. [Google Scholar] [CrossRef]
- Stark, N.M.; Yelle, D.J.; Agarwal, U.P. 4—Techniques for Characterizing Lignin. In Lignin in Polymer Composites; Faruk, O., Sain, M., Eds.; William Andrew Publishing: Norwich, NY, USA, 2016; pp. 49–66. [Google Scholar]
- Yin, Q.; Yang, W.; Sun, C.; Di, M. Preparation and properties of lignin-epoxy resin composite. Bioresources 2012, 7, 5737–5748. [Google Scholar] [CrossRef]
- Lauwaert, J.; Stals, I.; Lancefield, C.S.; Deschaumes, W.; Depuydt, D.; Vanlerberghe, B.; Devlamynck, T.; Bruijnincx, P.C.A.; Verberckmoes, A. Pilot scale recovery of lignin from black liquor and advanced characterization of the final product. Sep. Purif. Technol. 2019, 221, 226–235. [Google Scholar] [CrossRef]
- Haiping, Y.; Yan, R.; Chen, H.; Zheng, C.; Lee, D.; Liang, D. In-Depth Investigation of Biomass Pyrolysis Based on Three Major Components: Hemicellulose, Cellulose and Lignin. Energy Fuels 2005, 20, 388–393. [Google Scholar] [CrossRef]
- Fache, M.; Boutevin, B.; Caillol, S. Epoxy thermosets from model mixtures of the lignin-to-vanillin process. Green Chem. 2016, 18, 712–725. [Google Scholar] [CrossRef]
- Sbirrazzuoli, N.; Vincent, L.; Alzina, C. Isoconversional kinetic analysis of stoichiometric and off-stoichiometric epoxy-amine cures. Thermochim. Acta 2006, 447, 167–177. [Google Scholar] [CrossRef]
- Jiang, G.; Nowakowski, D.; Bridgwater, T. A systematic study of the kinetics of lignin pyrolysis. Thermochim. Acta 2010, 498, 61–66. [Google Scholar] [CrossRef]
- El Mansouri, N.E.; Yuan, Q.; Huang, F. Characterization of alkaline lignins for use in phenol-formaldehyde and epoxy resins. BioResources 2011, 6, 2647–2662. [Google Scholar] [CrossRef]
- Hatakeyama, H. Thermal Analysis; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar]
- Gordobil, O.; Moriana, R.; Zhang, L.; Labidi, J.; Sevastyanova, O. Assesment of technical lignins for uses in biofuels and biomaterials: Structure-related properties, proximate analysis and chemical modification. Ind. Crops Prod. 2016, 83, 155–165. [Google Scholar] [CrossRef]
- Zijlstra, D.S.; de Santi, A.; Oldenburger, B.; de Vries, J.; Barta, K.; Deuss, P.J. Extraction of Lignin with High β-O-4 Content by Mild Ethanol Extraction and Its Effect on the Depolymerization Yield. JoVE 2019, 143, e58575. [Google Scholar] [CrossRef]
- Gabov, K.; Gosselink, R.J.; Smeds, A.I.; Fardim, P. Characterization of lignin extracted from birch wood by a modified hydrotropic process. J. Agric. Food Chem. 2014, 62, 10759–10767. [Google Scholar] [CrossRef]
- Shundo, A.; Yamamoto, S.; Tanaka, K. Network Formation and Physical Properties of Epoxy Resins for Future Practical Applications. JACS Au 2022, 2, 1522–1542. [Google Scholar] [CrossRef]
- Mora, A.-S.; Tayouo, R.; Boutevin, B.; David, G.; Caillol, S. A perspective approach on the amine reactivity and the hydrogen bonds effect on epoxy-amine systems. Eur. Polym. J. 2020, 123, 109460. [Google Scholar] [CrossRef]
- Tcharkhtchi, A.; Nony, F.; Khelladi, S.; Fitoussi, J.; Farzaneh, S. 13—Epoxy/amine reactive systems for composites materials and their thermomechanical properties. In Advances in Composites Manufacturing and Process Design; Boisse, P., Ed.; Woodhead Publishing: Cambridge, UK, 2015; pp. 269–296. [Google Scholar]
- Nikolic, G.; Zlatkovic, S.; Cakic, M.; Cakic, S.; Lacnjevac, C.; Rajic, Z. Fast Fourier transform IR characterization of epoxy GY systems crosslinked with aliphatic and cycloaliphatic EH polyamine adducts. Sensors 2010, 10, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Fu, C.; Liang, C.; Ni, X.; Zhao, X.; Chen, M.; Ou, L. Crop Lodging and The Roles of Lignin, Cellulose, and Hemicellulose in Lodging Resistance. Agronomy 2022, 12, 1795. [Google Scholar] [CrossRef]
- Gibson, L.J. The hierarchical structure and mechanics of plant materials. J. R. Soc. Interface 2012, 9, 2749–2766. [Google Scholar] [CrossRef]
- Zhao, D.; Luan, Y.; Xia, X.; Shi, W.; Tang, Y.; Tao, J. Lignin provides mechanical support to herbaceous peony (Paeonia lactiflora Pall.) stems. Hortic. Res. 2020, 7, 213. [Google Scholar] [CrossRef] [PubMed]
- Ponyrko, S.; Donato, R.; Matějka, L. Tailored high performance shape memory epoxy–silica nanocomposites. Structure design. Polym. Chem. 2016, 7, 560–572. [Google Scholar] [CrossRef]
- Vásquez-Garay, F.; Carrillo-Varela, I.; Vidal, C.; Reyes-Contreras, P.; Faccini, M.; Teixeira Mendonça, R. A Review on the Lignin Biopolymer and Its Integration in the Elaboration of Sustainable Materials. Sustainability 2021, 13, 2697. [Google Scholar] [CrossRef]
- Putz, K.W.; Palmeri, M.J.; Cohn, R.B.; Andrews, R.; Brinson, L.C. Effect of Cross-Link Density on Interphase Creation in Polymer Nanocomposites. Macromolecules 2008, 41, 6752–6756. [Google Scholar] [CrossRef]
- Mat Yazik, M.H.; Sultan, M.T.H.; Jawaid, M.; Abu Talib, A.R.; Mazlan, N.; Md Shah, A.U.; Safri, S.N.A. Effect of Nanofiller Content on Dynamic Mechanical and Thermal Properties of Multi-Walled Carbon Nanotube and Montmorillonite Nanoclay Filler Hybrid Shape Memory Epoxy Composites. Polymers 2021, 13, 700. [Google Scholar] [CrossRef]
- Xidas, P.I.; Triantafyllidis, K.S. Effect of the type of alkylammonium ion clay modifier on the structure and thermal/mechanical properties of glassy and rubbery epoxy–clay nanocomposites. Eur. Polym. J. 2010, 46, 404–417. [Google Scholar] [CrossRef]
- Song, L.; Meng, Y.; Lv, P.; Liu, W.; Pang, H. Preparation of a Dmap-Catalysis Lignin Epoxide and the Study of Its High Mechanical-Strength Epoxy Resins with High-Biomass Content. Polymers 2021, 13, 750. [Google Scholar] [CrossRef]
- Qin, J.; Wolcott, M.; Zhang, J. Use of Polycarboxylic Acid Derived from Partially Depolymerized Lignin As a Curing Agent for Epoxy Application. ACS Sustain. Chem. Eng. 2013, 2, 188–193. [Google Scholar] [CrossRef]
- Ferdosian, F.; Yuan, Z.; Anderson, M.; Xu, C. Synthesis and characterization of hydrolysis lignin-based epoxy resins. Ind. Crops Prod. 2016, 91, 295–301. [Google Scholar] [CrossRef]
- Harvey, B.G.; Guenthner, A.J.; Lai, W.W.; Meylemans, H.A.; Davis, M.C.; Cambrea, L.R.; Reams, J.T.; Lamison, K.R. Effects of o-Methoxy Groups on the Properties and Thermal Stability of Renewable High-Temperature Cyanate Ester Resins. Macromolecules 2015, 48, 3173–3179. [Google Scholar] [CrossRef]
- Aniol, A.; Grosse, T.; Fischer, F.; Böhm, S. Evaluation of adhesion properties of lignin-epoxy adhesives in structural wood applications for automotive components. Proc. Inst. Mech. Eng. Part E J. Process Mech. Eng. 2020, 234, 511–519. [Google Scholar] [CrossRef]
- Li, K.; Zhong, W.; Li, P.; Ren, J.; Jiang, K.; Wu, W. Recent advances in lignin antioxidant: Antioxidant mechanism, evaluation methods, influence factors and various applications. Int. J. Biol. Macromol. 2023, 251, 125992. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Liu, W.; Huang, J.; Lou, H.; Qiu, X. Study on the Antioxidant Activity of Lignin and Its Application Performance in SBS Elastomer. Ind. Eng. Chem. Res. 2021, 60, 790–797. [Google Scholar] [CrossRef]
- Domenek, S.; Louaifi, A.; Guinault, A.; Baumberger, S. Potential of Lignins as Antioxidant Additive in Active Biodegradable Packaging Materials. J. Polym. Environ. 2013, 21, 692–701. [Google Scholar] [CrossRef]
- Domínguez-Robles, J.; Martin, N.K.; Fong, M.L.; Stewart, S.A.; Irwin, N.J.; Rial-Hermida, M.I.; Donnelly, R.F.; Larrañeta, E. Antioxidant PLA Composites Containing Lignin for 3D Printing Applications: A Potential Material for Healthcare Applications. Pharmaceutics 2019, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Boarino, A.; Schreier, A.; Leterrier, Y.; Klok, H.-A. Uniformly Dispersed Poly(lactic acid)-Grafted Lignin Nanoparticles Enhance Antioxidant Activity and UV-Barrier Properties of Poly(lactic acid) Packaging Films. ACS Appl. Polym. Mater. 2022, 4, 4808–4817. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
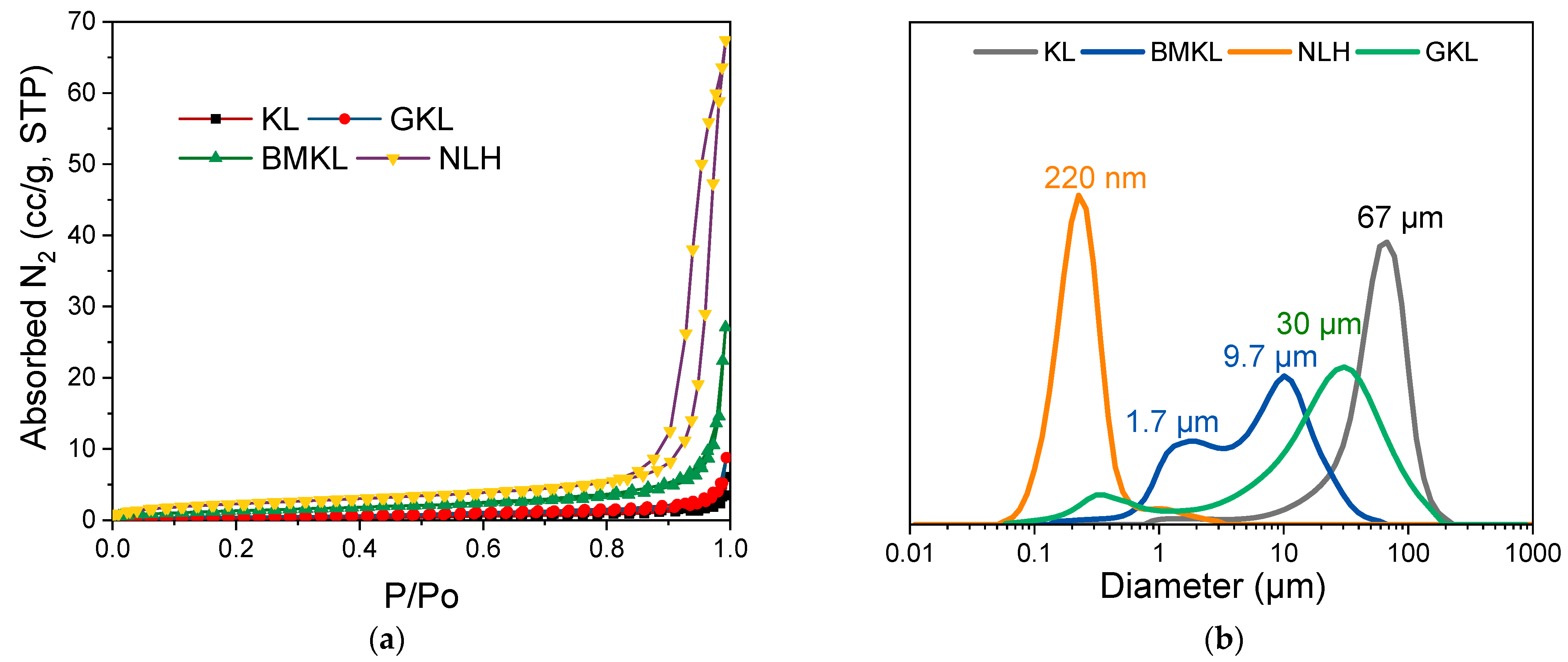
| GPC | N2 Porosimetry | Elemental Analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lignin | Mw (g/mol) | Mn (g/mol) | PDI | Total Pore Volume (cc/g) | Surface Area (m2/g) | N (wt.%) | C (wt.%) | H (wt.%) | S (wt.%) | O (wt.%) * |
| Kraft | 6500 | 1400 | 4.7 | 0.009 | 0.8 | 1.44 | 61.13 | 5.73 | 1.49 | 30.21 |
| BMKL | 3400 | 1000 | 3.4 | 0.042 | 5.1 | 2.97 | 62.67 | 5.29 | 1.27 | 27.80 |
| NLH | 3200 | 650 | 4.9 | 0.104 | 8.6 | 3.34 | 51.64 | 4.75 | 2.92 | 37.35 |
| GKL | 5400 | 1500 | 3.6 | 0.013 | 1.7 | 2.82 | 62.63 | 5.72 | 1.11 | 27.72 |
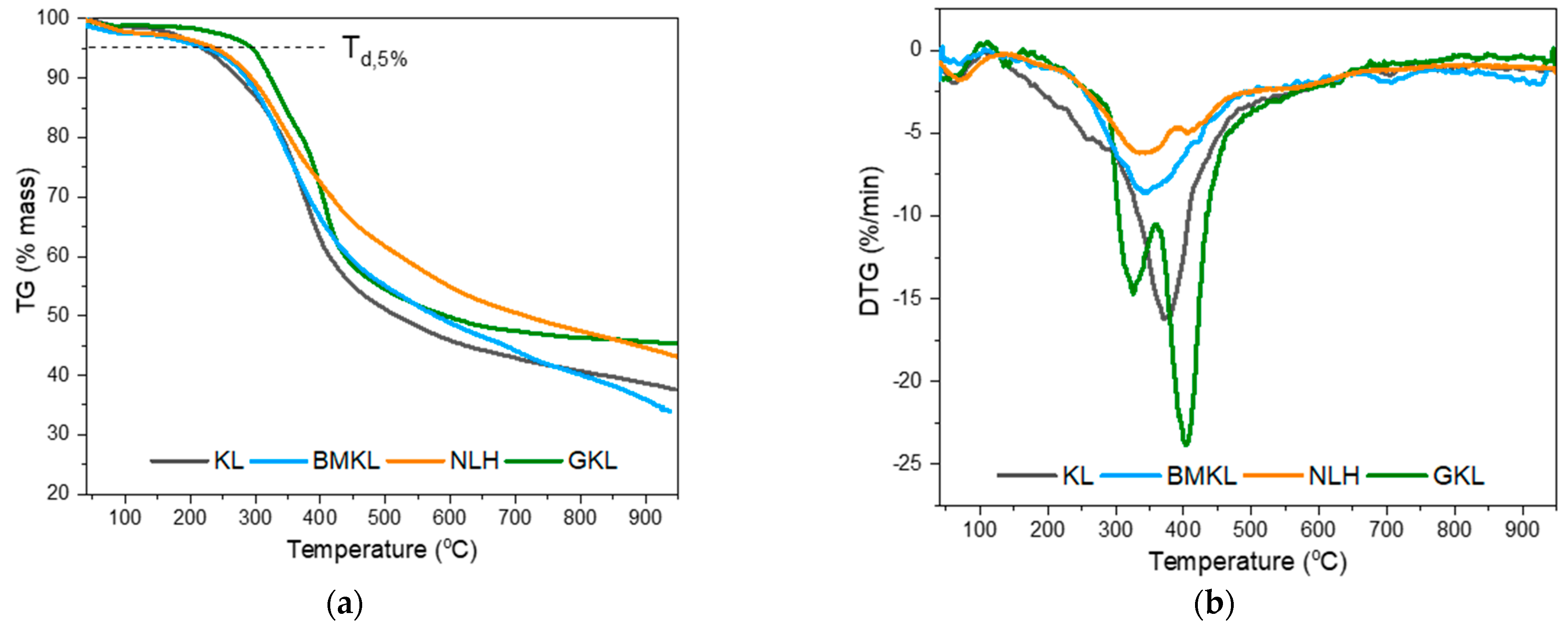
| Lignin | Tg (°C) a | Td,5% (°C) b | DTGmax (°C) b | Residue at 800 °C (%) b |
|---|---|---|---|---|
| KL | 143 | 219 | 372 | 40.7 |
| BMKL | 148 | 220 | 343 | 40.1 |
| NLH | 116 | 233 | 344 | 47.2 |
| GKL | 103 | 294 | 325 & 403 | 46.4 |
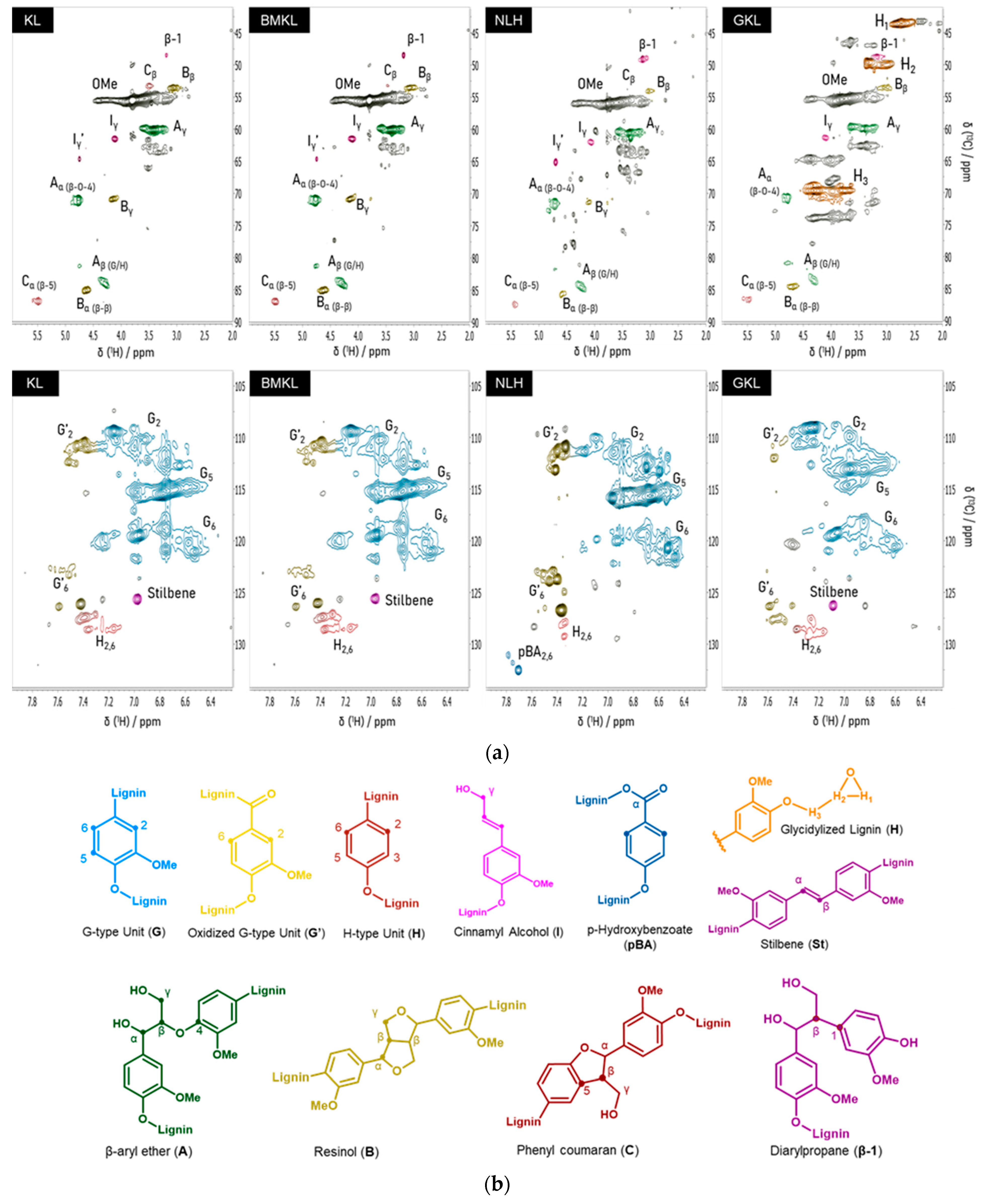
| KL | BMKL | NLH | GKL | ||
|---|---|---|---|---|---|
| Lignin interunit bonds—Abundance (per 100 Ar a) | Chemical shifts (δC/δH) | ||||
| Arylglycerol-β-aryl ethers (β-O-4) | 22.8 | 23.7 | 18.3 | 5.7 | 71.1/4.8 |
| Pinoresinols (β-β) | 10.9 | 12.3 | 4.8 | 4.1 | 84.8/4.6 |
| Phenyl coumaran (β-5) | 6.9 | 7.6 | 4.5 | 2.7 | 86.9/5.5 |
| Diarylpropane (β-1) | 1.4 | 3.9 | 3.9 | 3.4 | 48.7/3.1 |
| Lignin aromatic units—Abundance (%) | |||||
| G/H ratio | 91.2/8.8 | 91.7/8.3 | 92.3/7.7 | 96.5/3.5 | |
| Lignin functional groups—Abundance (mmol/gr) b | Chemical shifts (δ31p) | ||||
| Aliphatic OH | 2.25 | 2.30 | 2.47 | 2.43 | 145–150 |
| Condensed OH | 1.59 | 1.49 | 1.05 | 0.04 | 140.3–144.5 |
| Non-condensed OH | 2.02 | 2.13 | 2.58 | 0 | 137.2–140.3 |
| Guaiacyl OH | 1.81 | 1.83 | 2.31 | 0 | 138.4–140.3 |
| p-Hydroxyphenyl OH | 0.21 | 0.30 | 0.27 | 0 | 137.2–138.4 |
| Total PhOH | 3.61 | 3.62 | 3.63 | 0.04 | 137.2–144.3 |
| -COOH | 0.45 | 0.46 | 0.94 | 0 | 134–136 |
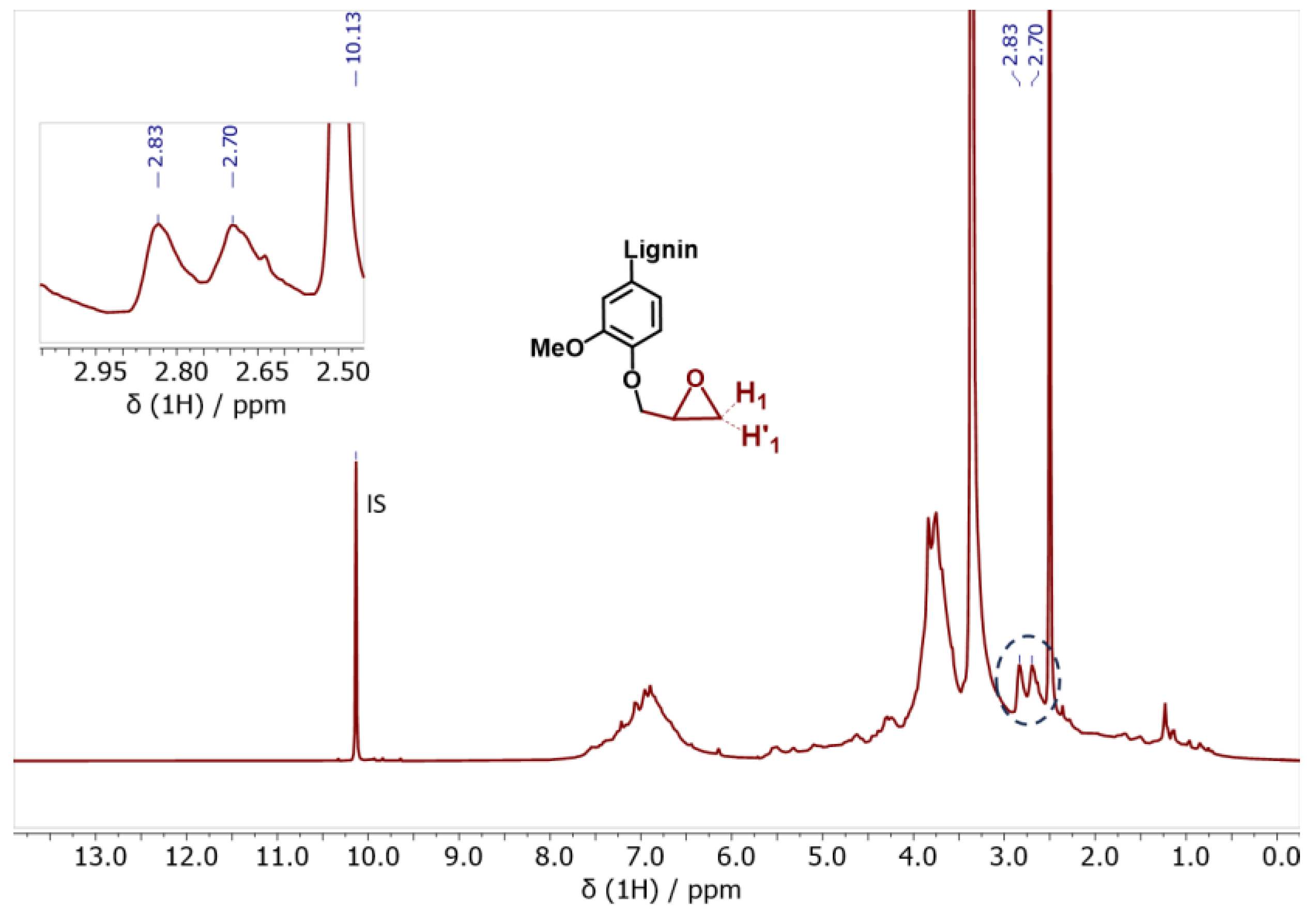
| Epoxy content c | - | - | - | 3.80 | - |
| Tensile Strength | DMA | |||||
|---|---|---|---|---|---|---|
| KL Content | D-230 Replacement | Ultimate Strength | Strain at Break | Young Modulus, E | Storage Modulus E′ at the Glassy State 1 | Tg 2 |
| (wt.%) | (wt.%) | (MPa) | (%) | (MPa) | (MPa) | (°C) |
| 0 | 0 | 65.6 ± 3.6 | 6.1 ± 0.5 | 1329 ± 22 | 1580 ± 74 | 81.5 ± 0.6 |
| 3 | 5 | 81.6 ± 5.6 | 6.0 ± 0.6 | 1686 ± 15 | 1500 ± 62 | 86.5 ± 1.7 |
| 6 | 10 | 55.6 ± 4.7 | 3.7 ± 0.4 | 1545 ± 118 | 2082 ± 130 | 86.1 ± 1.1 |
| 9 | 14 | 45.2 ± 0.9 | 2.8 ± 0.1 | 1688 ± 46 | 2748 ± 210 | 86.3 ± 2.4 |
| 45 | 100 | Sample too brittle for tensile and DMA measurement | ||||
| Tensile Strength | DMA | ||||
|---|---|---|---|---|---|
| KL Content | Ultimate Strength | Strain at Break | Young’s Modulus, E | Storage Modulus E′ at the Glassy State 1 | Tg 2 |
| (wt.%) | (MPa) | (%) | (MPa) | (MPa) | (°C) |
| Pristine | 65.6 ± 3.6 | 6.1 ± 0.5 | 1329 ± 22 | 1580 ± 74 | 81.5 ± 0.6 |
| 1 wt.% KL | 82.0 ± 5.3 | 6.2 ± 0.5 | 1644 ± 53 | 2512 ± 180 | 74.8 ± 0.9 |
| 3 wt.% KL | 89.1 ± 5.7 | 6.3 ± 0.4 | 1714 ± 38 | 2621 ± 322 | 78.0 ± 1.6 |
| 6 wt.% KL | 75.0 ± 1.4 | 5.4 ± 0.2 | 1613 ± 14 | 1411 ± 152 | 77.1 ± 1.2 |
| 9 wt.% KL | 66.3 ± 1.2 | 4.5 ± 0.2 | 1656 ± 36 | 1561 ± 181 | 79.1 ± 2.2 |
| 16.6 wt.% KL | 60.2 ± 1.8 | 4.2 ± 0.1 | 1620 ± 26 | 1515 ± 71 | 92.8 ± 1.5 |
| 23 wt.% KL | 58.3 ± 0.8 | 4.1 ± 0.3 | 1550 ±69 | - | - |
| 30 wt.% KL | 58.2 ± 4.1 | 3.9 ± 0.4 | 1598 ± 39 | 2691 ± 69 | 60.9 ± 2.1 |
| 44.5 wt.% KL | 13.6 ± 10.5 | 1.4 ± 2.1 | 800 ± 287 | - | - |
| 3 wt.% BMKL | 68.4 ± 1.4 | 6.1 ± 0.3 | 1400 ± 55 | 1847 ± 95 | 78.9 ± 0.7 |
| 6 wt.% BMKL | 71.6 ± 4.3 | 5.9 ± 0.4 | 1329 ± 13 | 2513 ± 185 | 87.7 ± 2.1 |
| 9 wt.% BMKL | 54.0 ± 1.7 | 3.8 ± 0.2 | 1536 ± 57 | 1341 ± 72 | 82.5 ± 1.5 |
| 16.6 wt.% BMKL | 56.1 ± 6.3 | 3.8 ± 0.4 | 1569 ± 91 | - | - |
| 3 wt.% NLH | 68.2 ± 1.1 | 7.6 ± 0.1 | 1379 ± 23 | 1928 ± 88 | 84.8 ± 1.2 |
| 6 wt.% NLH | 72.0 ± 4.3 | 5.0 ± 0.2 | 1520 ± 41 | 2001 ± 90 | 91.8 ± 1.6 |
| 9 wt.% NLH | 80.5 ± 6.1 | 7.4 ± 0.6 | 1361 ± 19 | 2229 ± 209 | 93.6 ± 1.1 |
| 16.6 wt.% NLH | 48.7 ± 4.6 | 3.4 ± 0.2 | 1498 ± 53 | 3709 ± 422 | 90.7 ± 0.9 |
| 30 wt.% NLH | 64.2 ± 5.6 | 4.3 ± 0.5 | 1543 ± 34 | 1662 ± 110 | 88.2 ± 1.9 |
| Tensile Strength | DMA | ||||
|---|---|---|---|---|---|
| KL Content | Ultimate Strength | Strain at Break | Young’s Modulus, E | Storage Modulus at the Glassy State 1 | Tg 2 |
| (wt.%) | (MPa) | (%) | (MPa) | (MPa) | (°C) |
| Pristine | 65.6 ± 3.6 | 6.1 ± 0.5 | 1329 ± 22 | 1580 ± 74 | 81.5 ± 0.6 |
| GL4/3GKL | 74.4 ± 2.2 | 7.5 ± 0.1 | 1362 ± 54 | 1516 ± 95 | 90.1 ± 1.7 |
| GL12/9GKL | 80.8 ± 1.5 | 6.8 ± 0.3 | 1401 ± 17 | 1390 ± 45 | 95.1 ± 2.3 |
| GL21/16.6GKL | 80.4 ± 1.1 | 6.7 ± 0.3 | 1415 ± 5 | 1095 ± 130 | 94.1 ± 1.4 |
| GL38/30GKL | 64.1 ± 3.3 | 5.7 ± 0.4 | 1241 ± 69 | 1397 ± 320 | 74.3 & 98.9 |
| GL50/40GKL | 41.3 ± 32 | 4.3 ± 0.4 | 1052 ± 10 | 1109 ± 51 | 70.5 & 109 |
| Sample | Td,5% (°C) | DTGmax (°C) | Char700 (%) | |
|---|---|---|---|---|
| Effect of the kraft content | Pristine | 348 | 376 | 9.9 |
| 3 wt.% KL | 348 | 374 | 11.8 | |
| 9 wt.% KL | 348 | 374 | 14.8 | |
| 44.5 wt.% KL | 301 | 358 | 29.1 | |
| Effects of the lignin type andcontent | 3 wt.% BMKL | 348 | 378 | 14.6 |
| 9 wt.% BMKL | 341 | 372 | 12.6 | |
| 3 wt.% NLH | 338 | 375 | 12.4 | |
| 9 wt.% NLH | 337 | 365 | 11.9 | |
| 3 wt.% GKL | 353 | 376 | 12.0 | |
| 9 wt.% GKL | 341 | 371 | 12.9 | |
| Effect of replacement by lignin | GL14/9KL | 341 | 379 | 13.6 |
| GL12/9GKL | 336 | 368 | 13.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pappa, C.P.; Cailotto, S.; Gigli, M.; Crestini, C.; Triantafyllidis, K.S. Kraft (Nano)Lignin as Reactive Additive in Epoxy Polymer Bio-Composites. Polymers 2024, 16, 553. https://doi.org/10.3390/polym16040553
Pappa CP, Cailotto S, Gigli M, Crestini C, Triantafyllidis KS. Kraft (Nano)Lignin as Reactive Additive in Epoxy Polymer Bio-Composites. Polymers. 2024; 16(4):553. https://doi.org/10.3390/polym16040553
Chicago/Turabian StylePappa, Christina P., Simone Cailotto, Matteo Gigli, Claudia Crestini, and Konstantinos S. Triantafyllidis. 2024. "Kraft (Nano)Lignin as Reactive Additive in Epoxy Polymer Bio-Composites" Polymers 16, no. 4: 553. https://doi.org/10.3390/polym16040553
APA StylePappa, C. P., Cailotto, S., Gigli, M., Crestini, C., & Triantafyllidis, K. S. (2024). Kraft (Nano)Lignin as Reactive Additive in Epoxy Polymer Bio-Composites. Polymers, 16(4), 553. https://doi.org/10.3390/polym16040553