
State-of-the-Art Polyurea Coatings: Synthesis Aspects, Structure–Properties Relationship, and Nanocomposites for Ballistic Protection Applications

 ,
,  ,
, 

Abstract
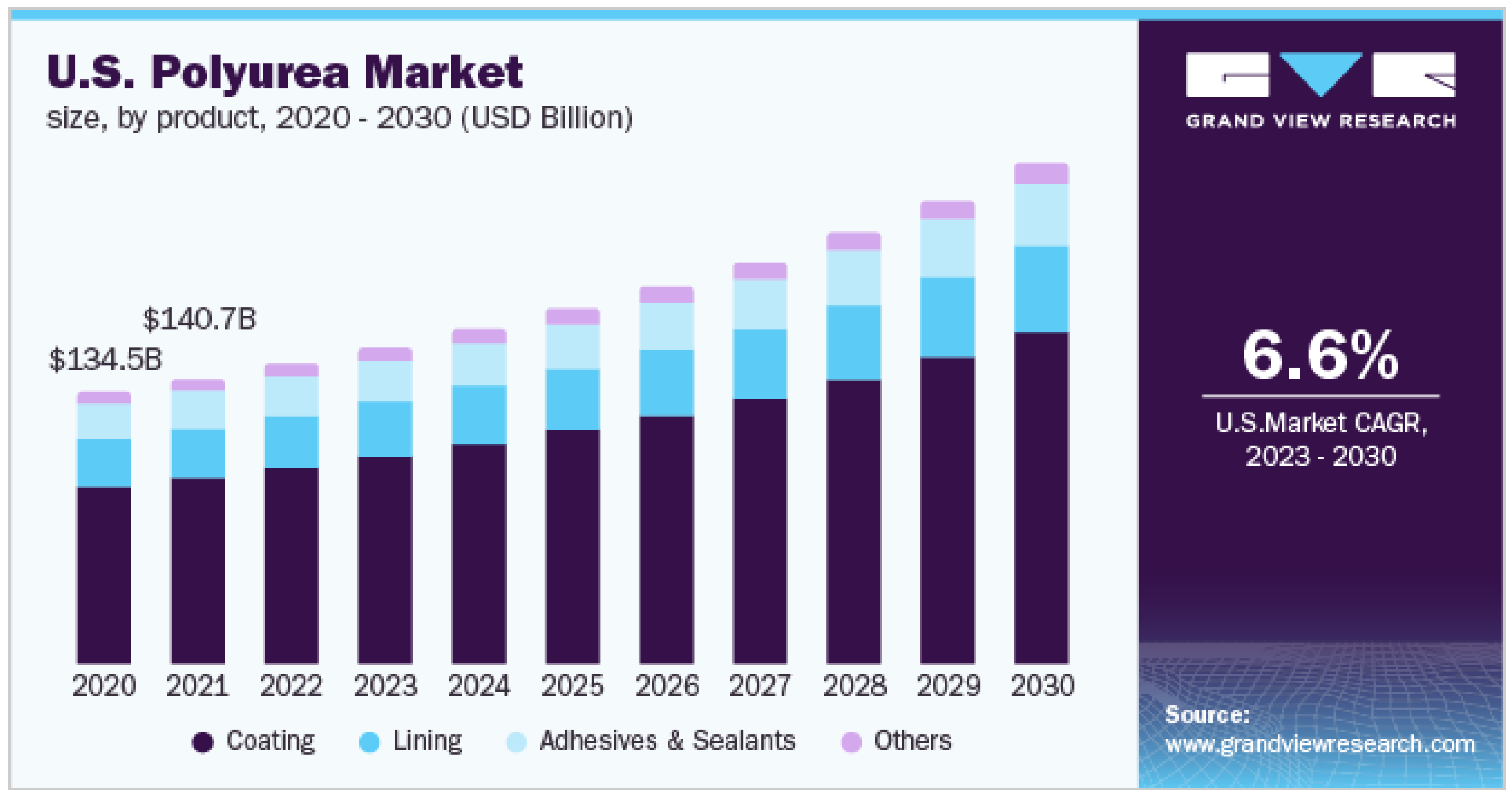
1. Introduction
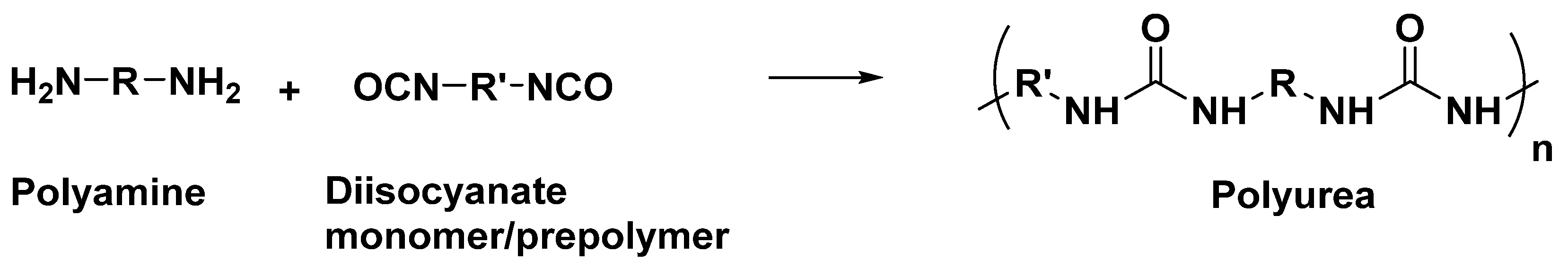
2. Polyurea Coatings for Ballistic Protection Applications
2.1. Overview
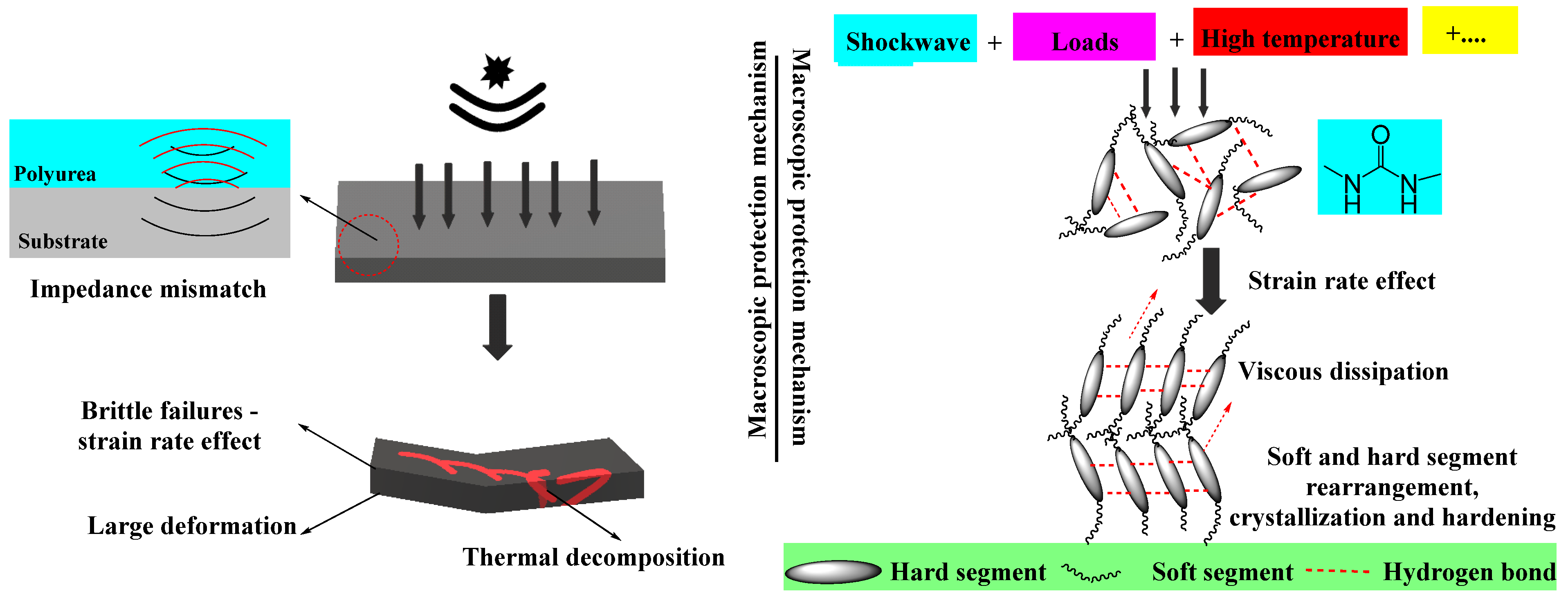
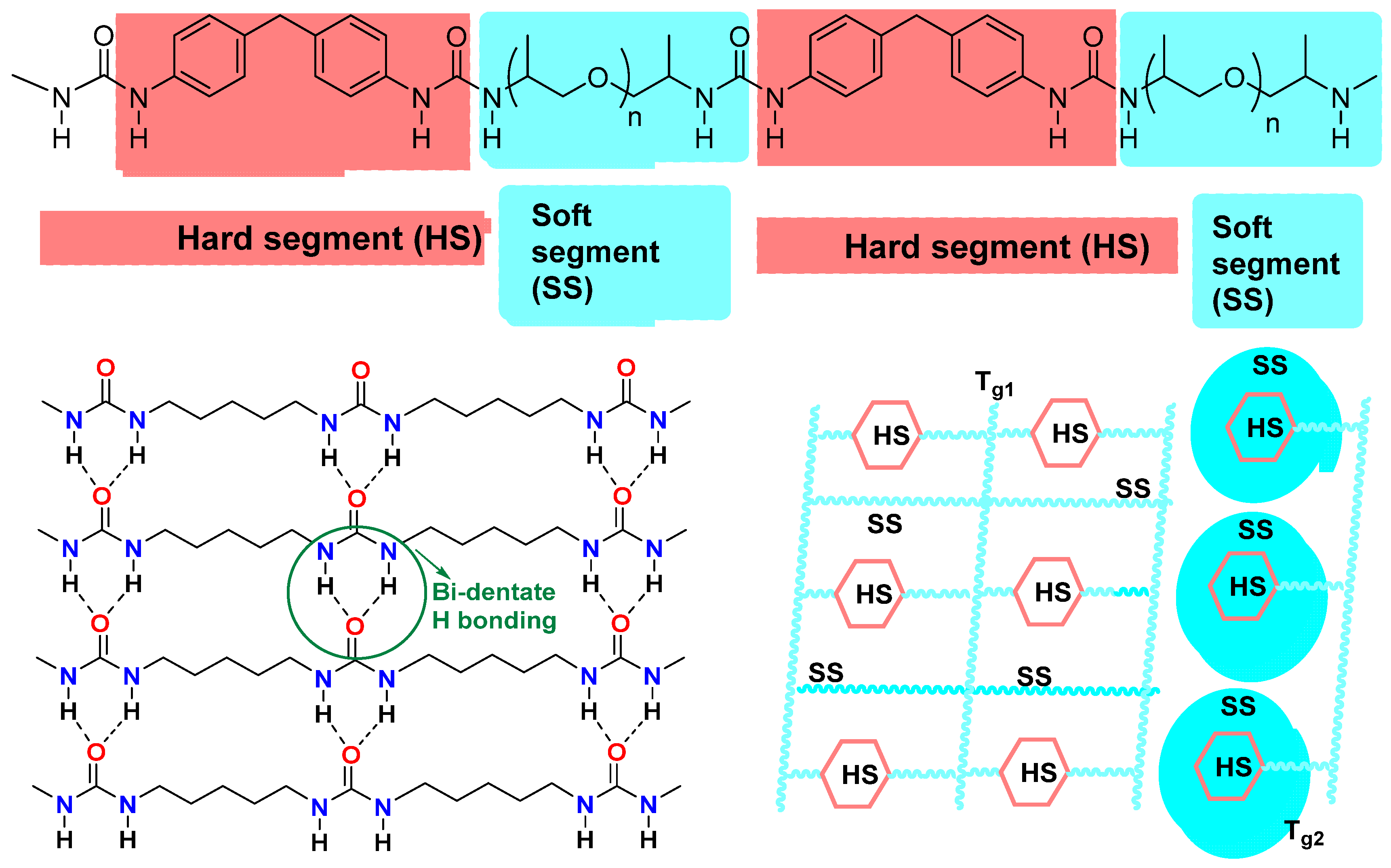
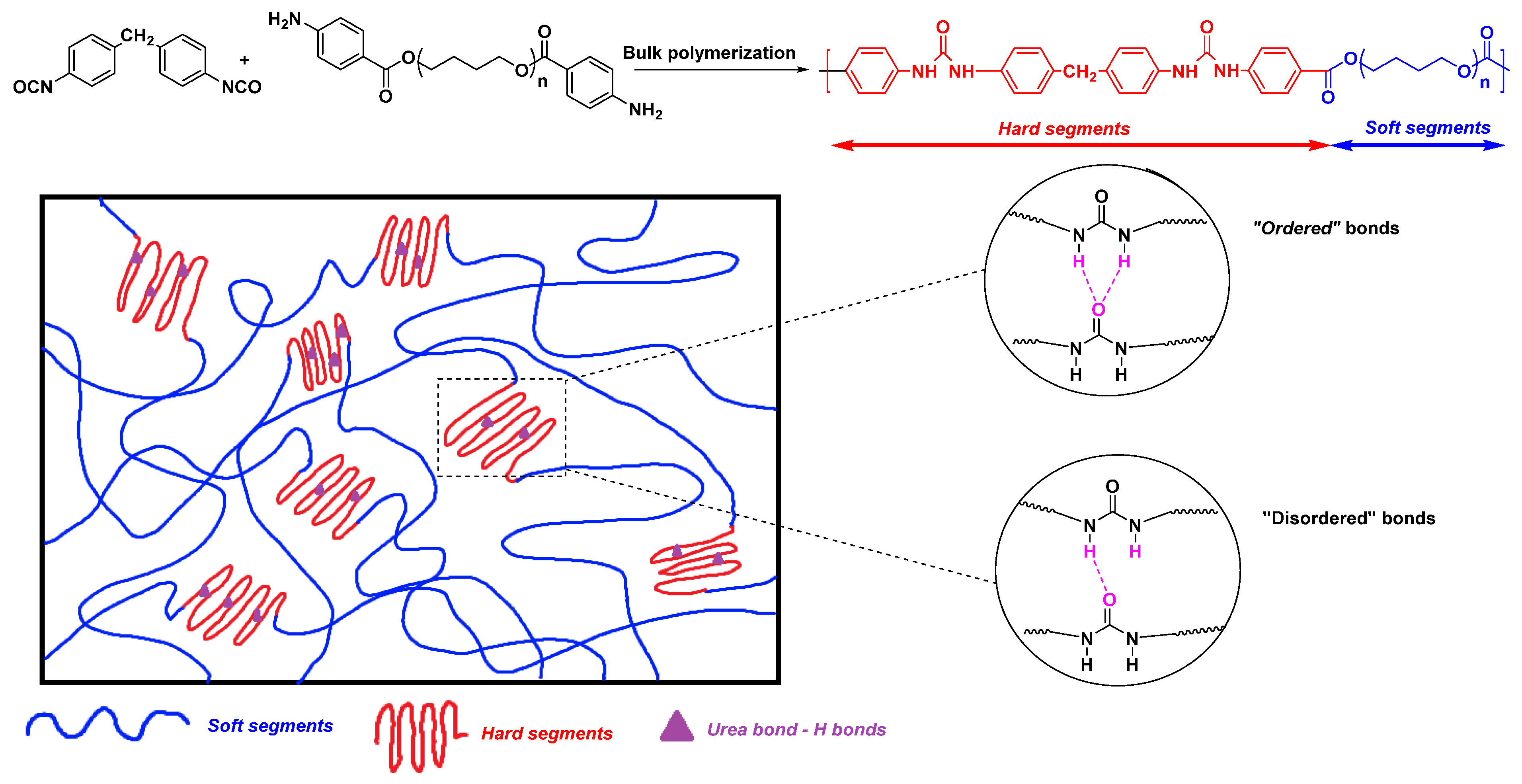
2.2. Structure–Properties Relationships
2.3. Specific Properties of Polyurea Coatings Employed for Ballistic Protection Applications
- The ratio between the hard and soft segments must be optimized to guarantee a high-strength material that can withstand high deformation and impede structural deterioration;
- They must present high thermal resistance while maintaining their mechanical properties;
- They must display a high elasticity modulus and long plastic stage when subjected to high-strain-rate loading and also a high loss modulus and storage modulus, thus dispersing all the energy during the deformation process.
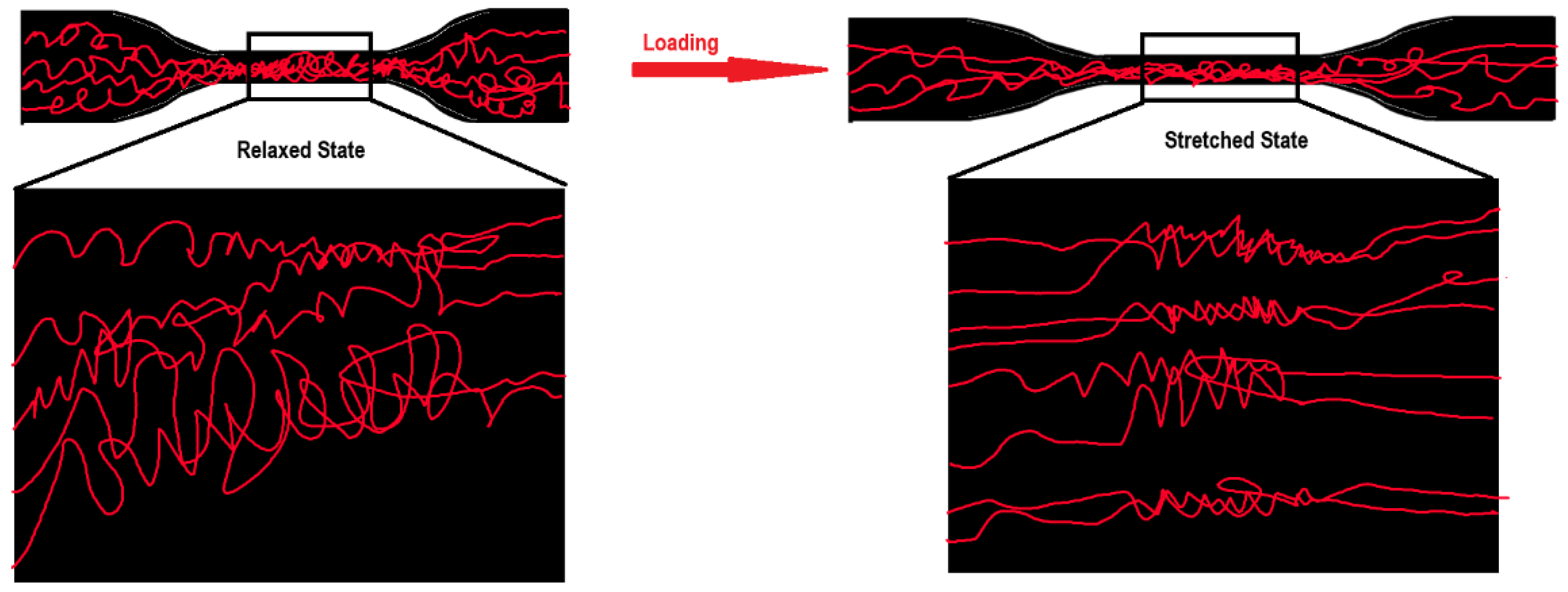
2.3.1. Mechanical Properties of Polyurea-Based Films Designed for Ballistic Protection Applications—Tensile Tests
2.3.2. Viscoelastic Properties of Polyurea-Based Films Designed for Ballistic Protection Applications—Dynamic Mechanical Analysis
2.3.3. Thermal Properties of Polyurea-Based Films Designed for Ballistic Protection Applications—Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA)
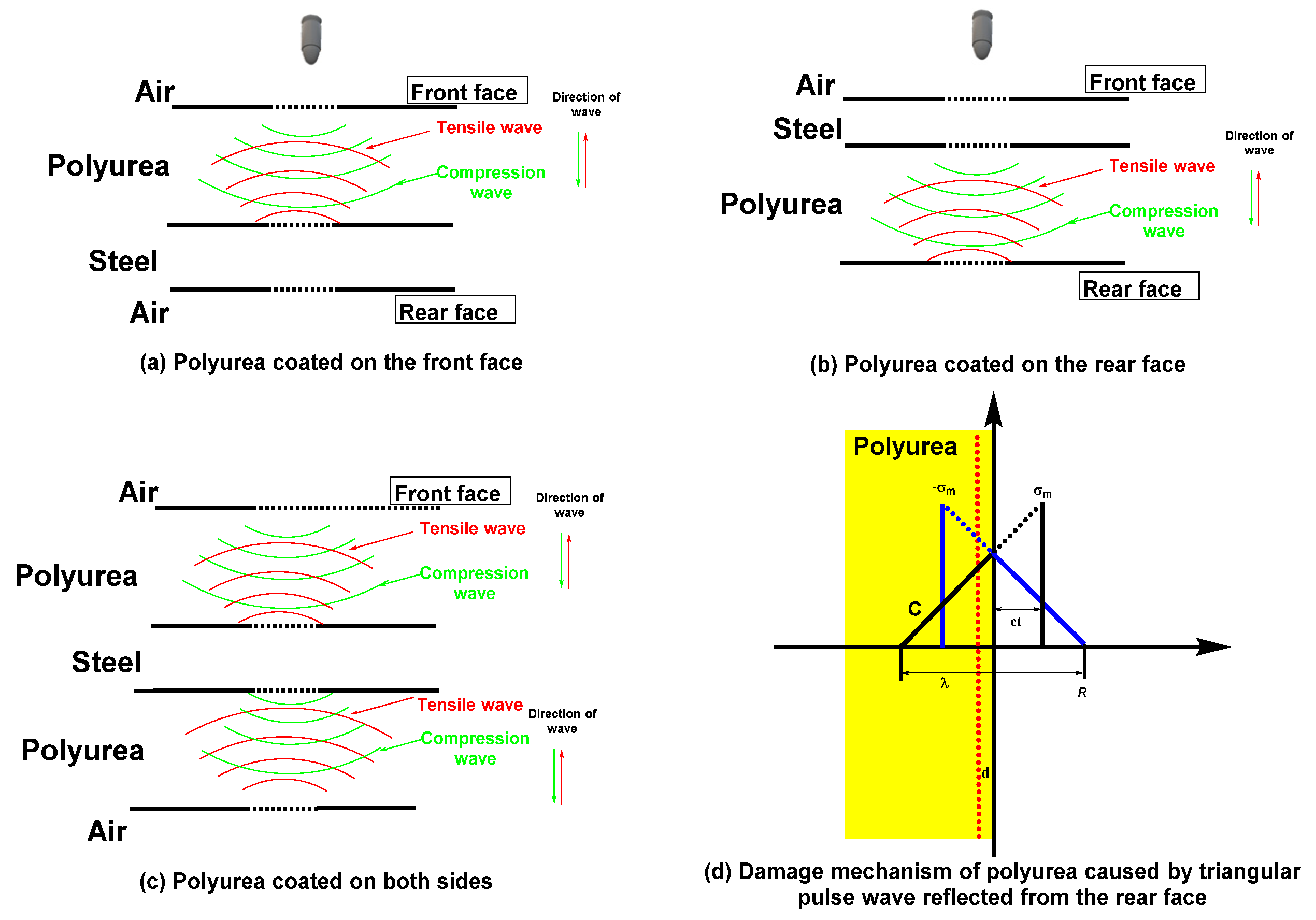
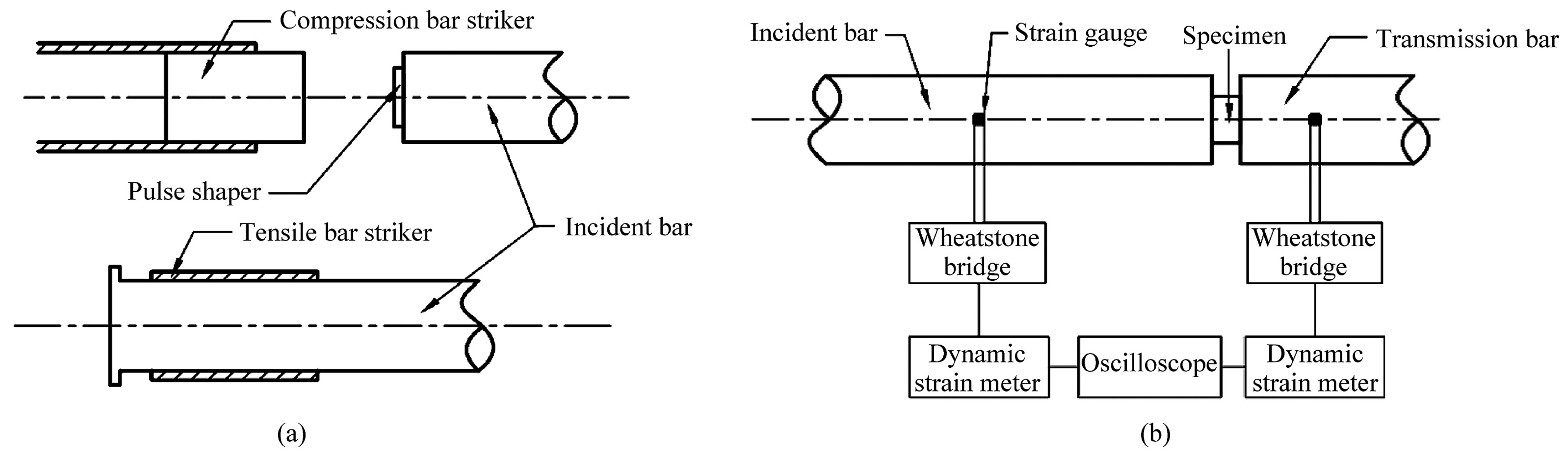
2.3.4. Impact Shielding Properties of Polyurea-Based Films Designed for Ballistic Protection Applications—Dynamic Regime Tests
3. Synthesis Principles for Tailoring the Final Properties of Polyurea
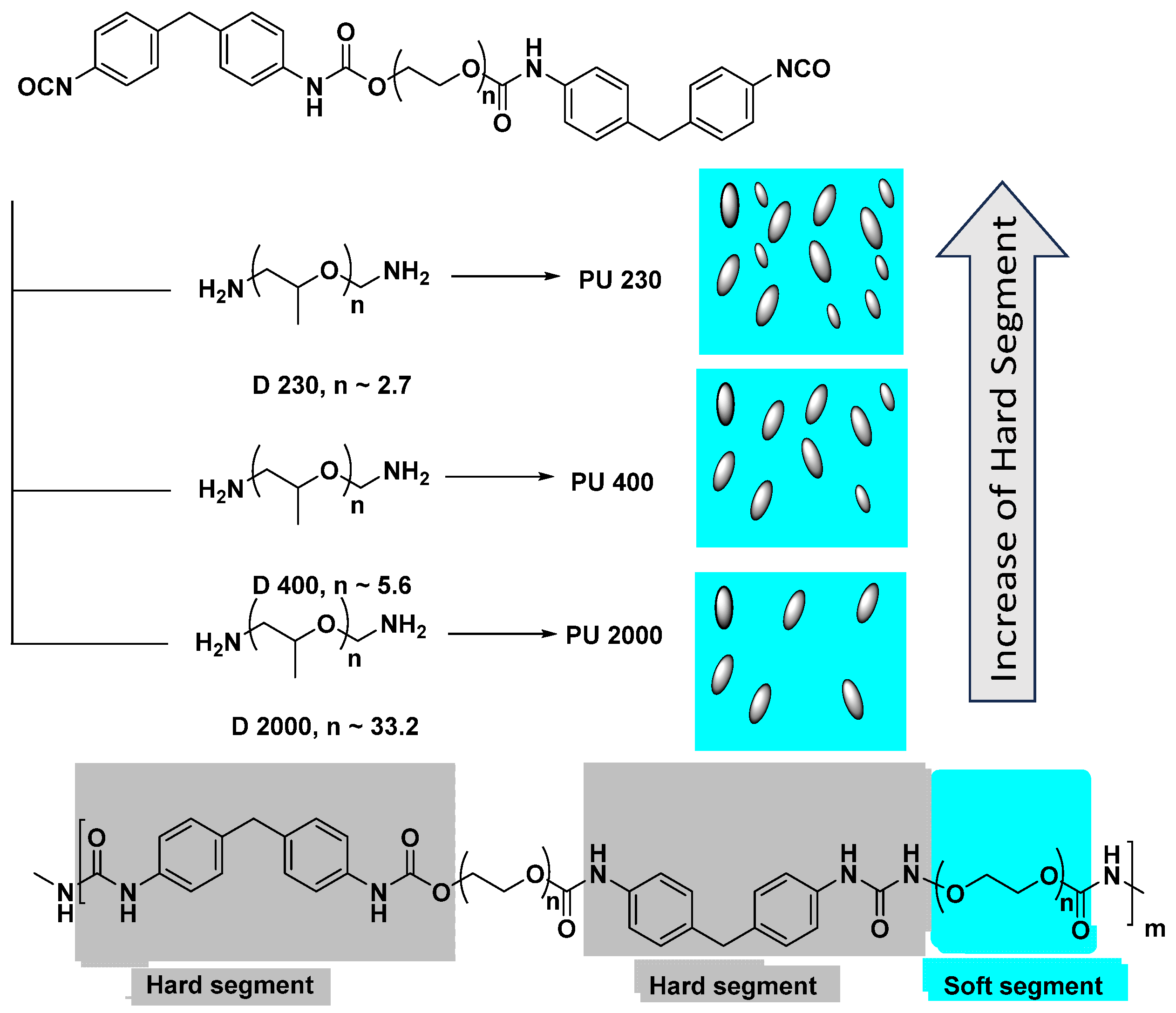
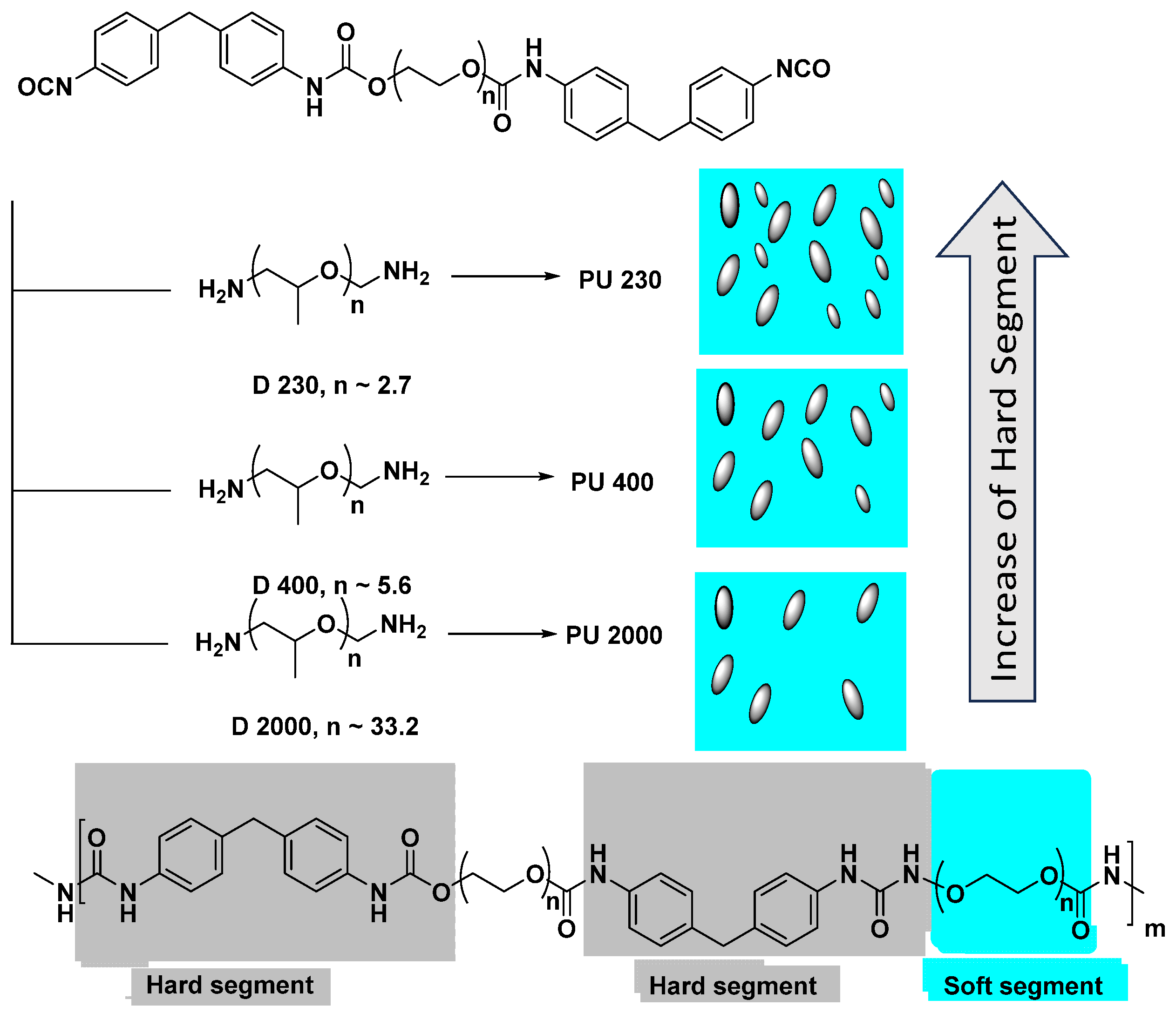
3.1. The Influence of Soft/Hard Segment Ratio on the Physical Properties of Polyurea
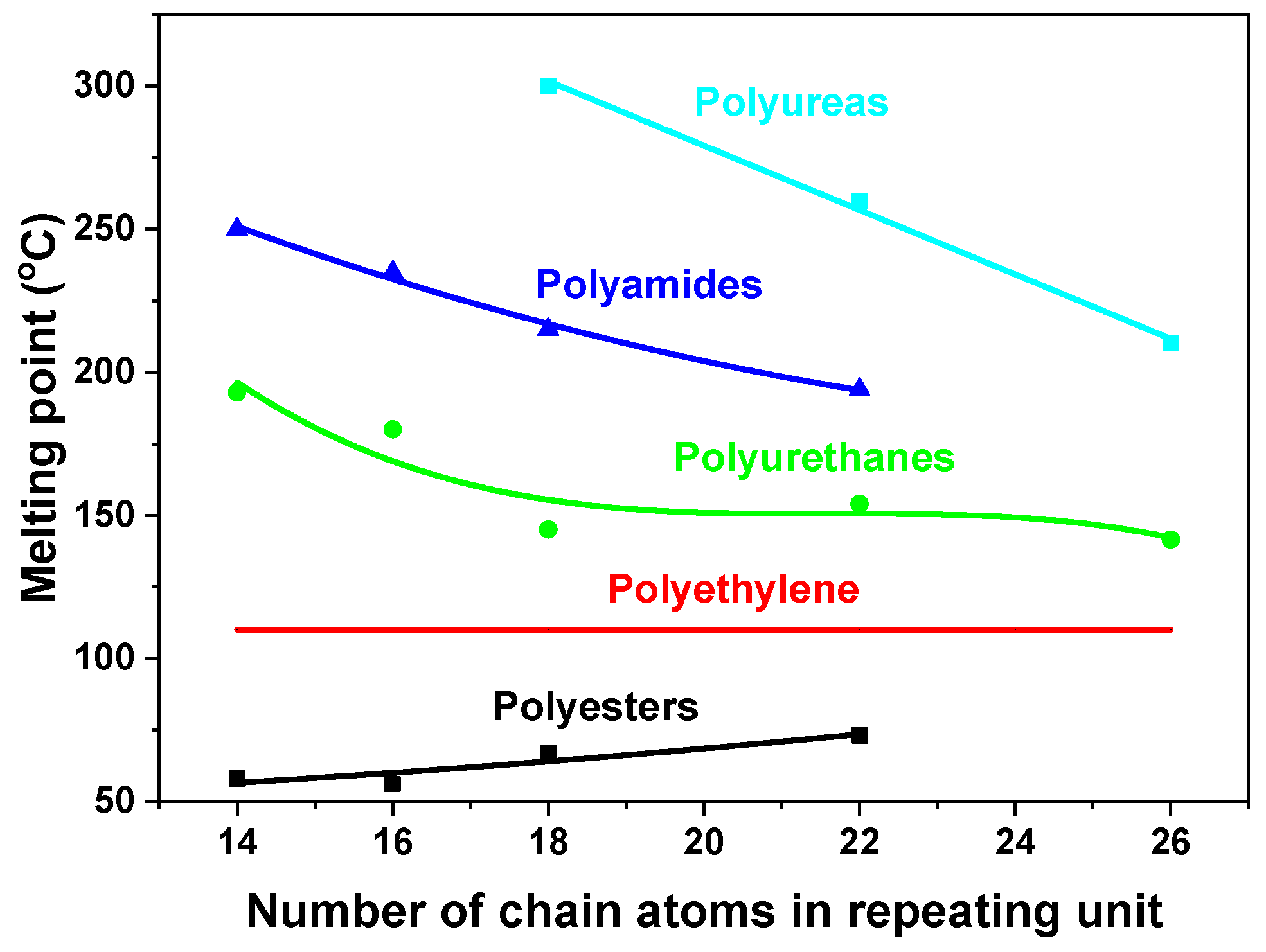
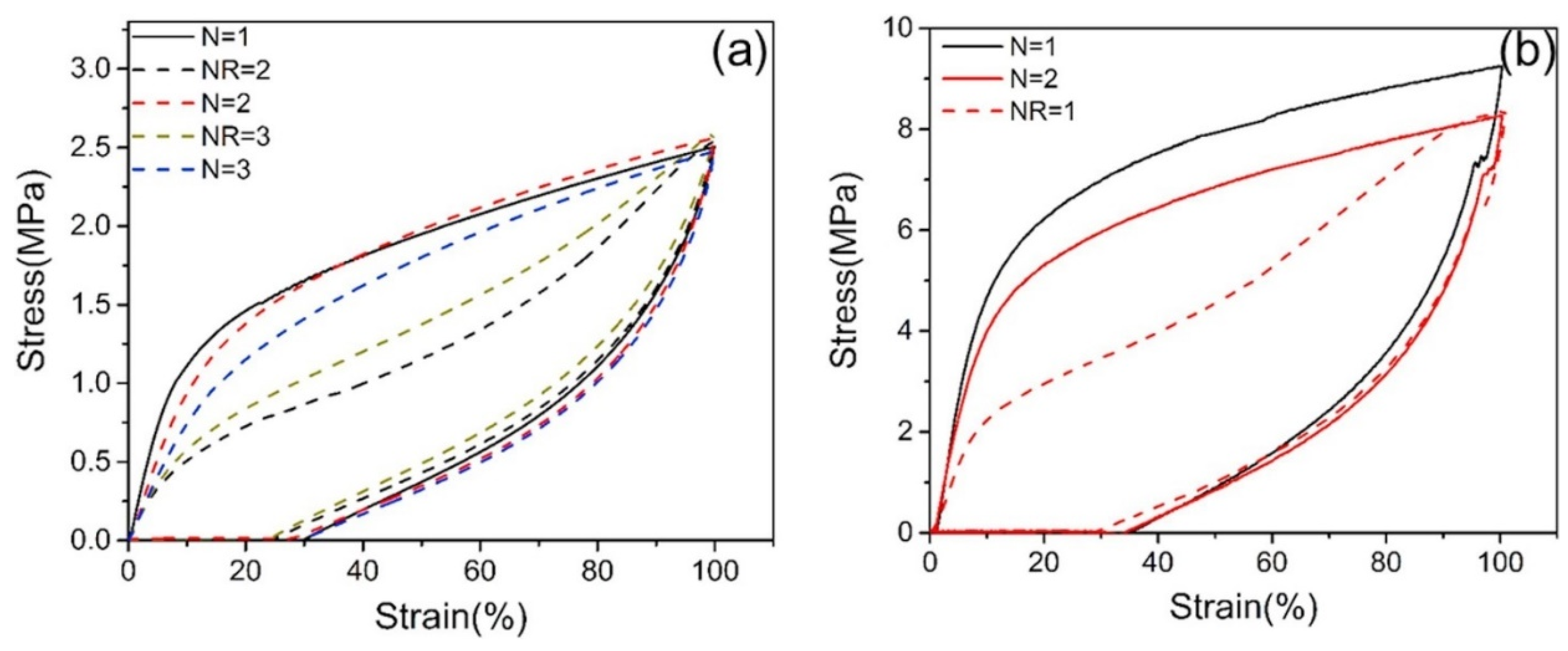
3.2. The Influence of the Chain Length on the Mechanical Properties of Polyurea Films
3.3. The Influence of the Diisocyanate Structure on the Mechanical Properties of Polyurea Films
3.4. The Influence of Crosslinking on the Mechanical Properties of Polyurea Films
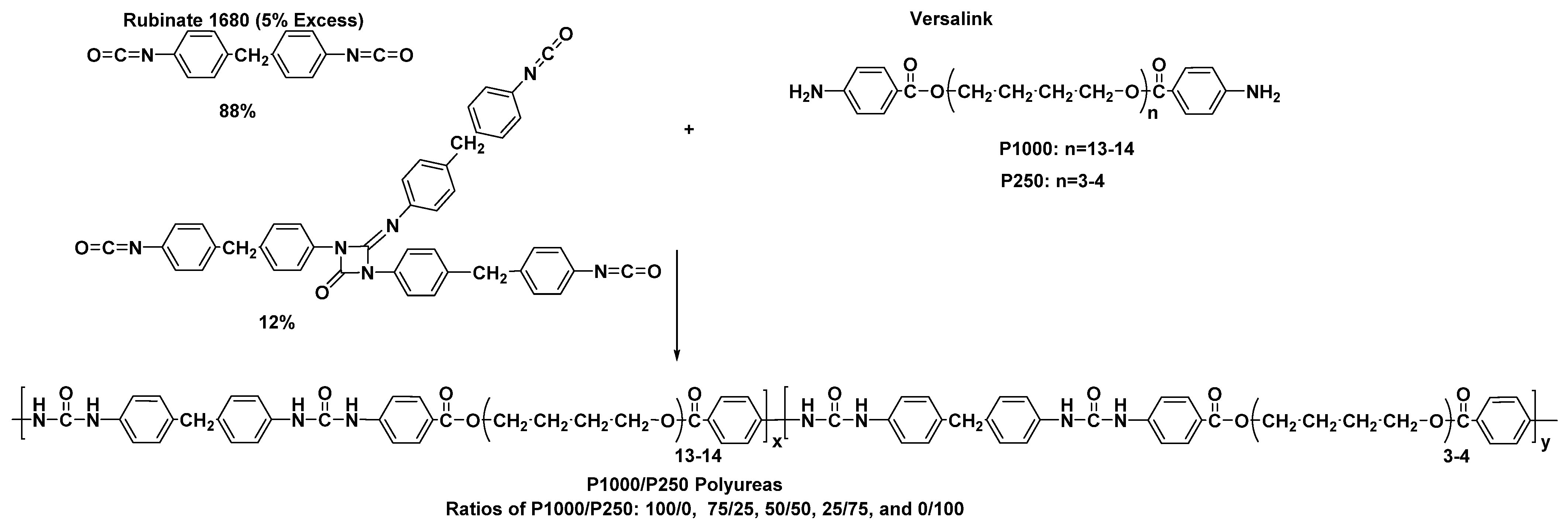
3.5. The Influence of the Chain Extender on the Mechanical Properties of Polyurea Films
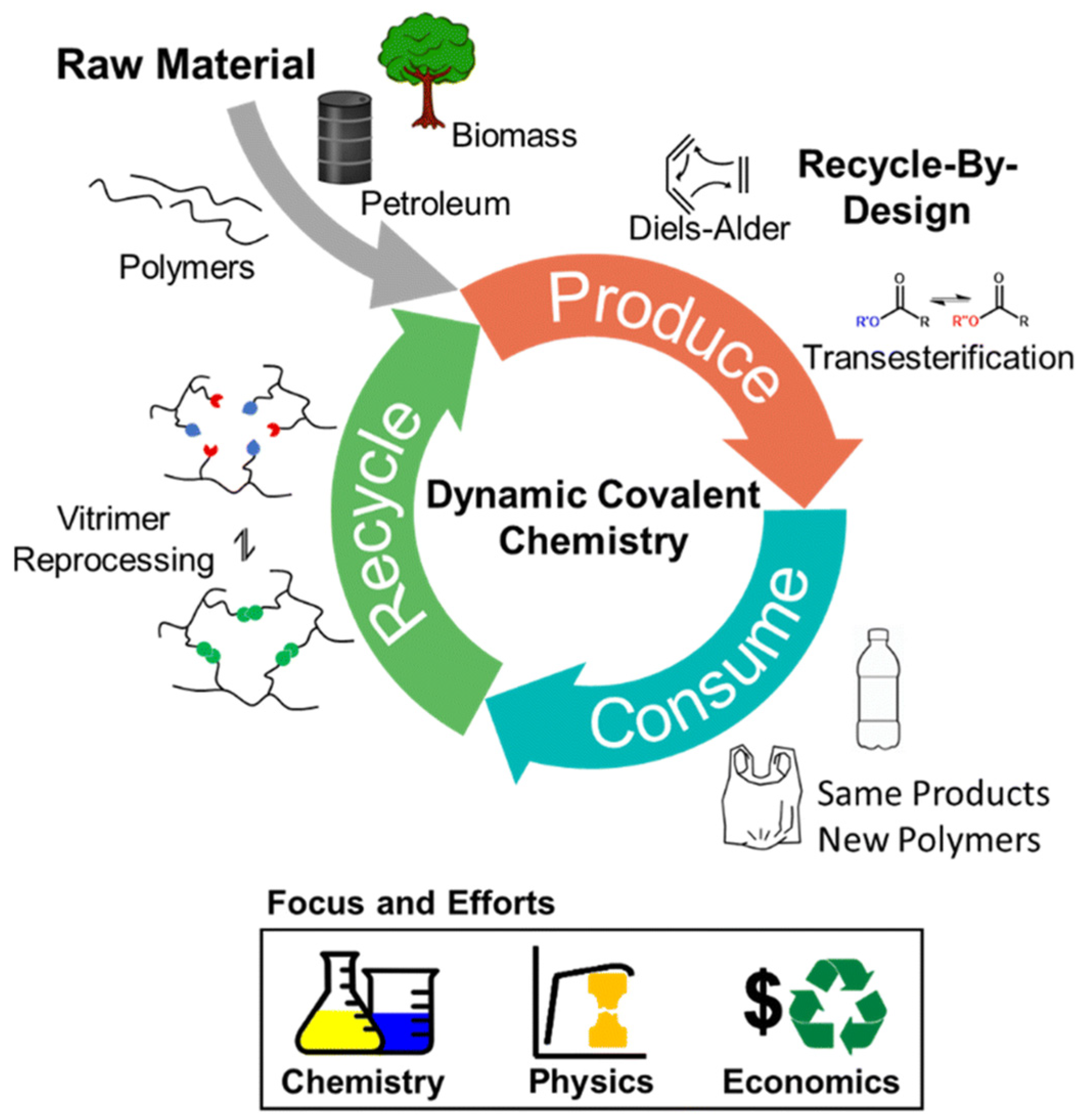
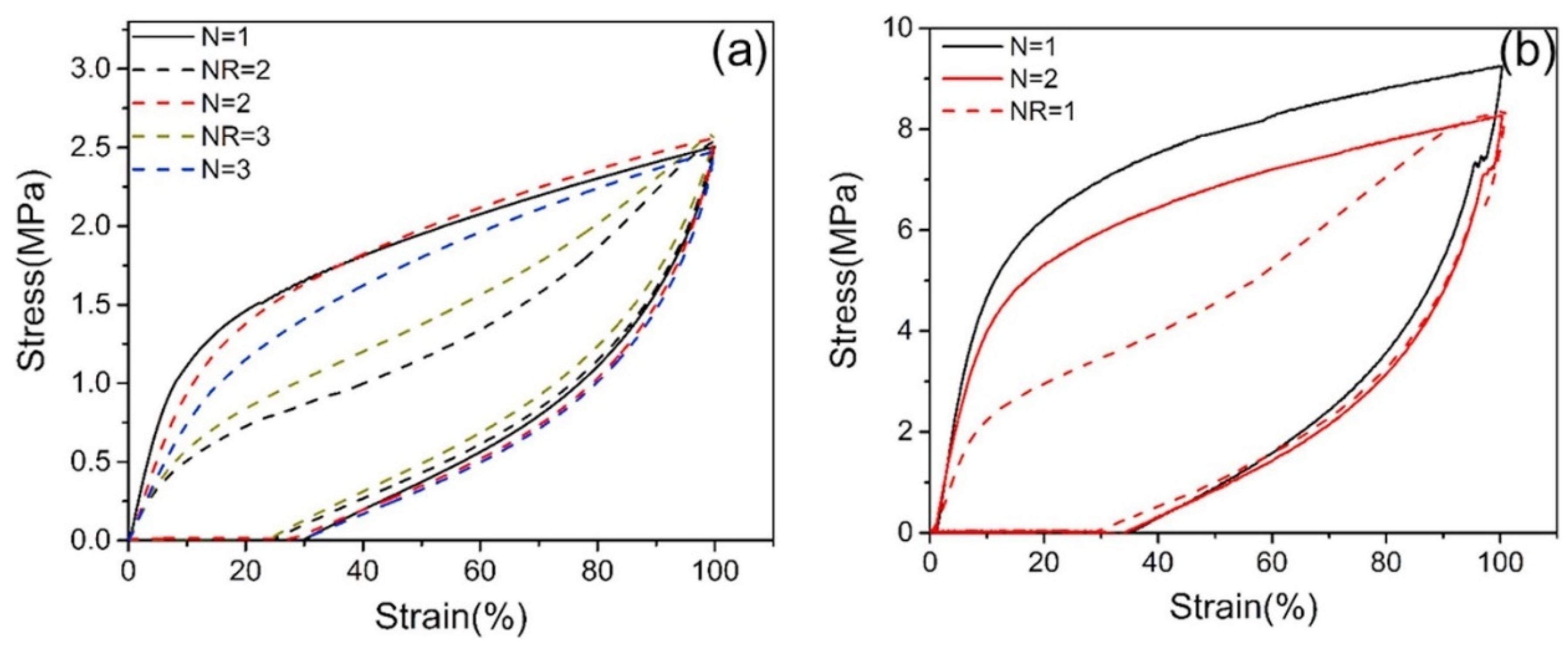
4. Self-Healing Polyureas
5. Polyurea-Based Nanocomposites
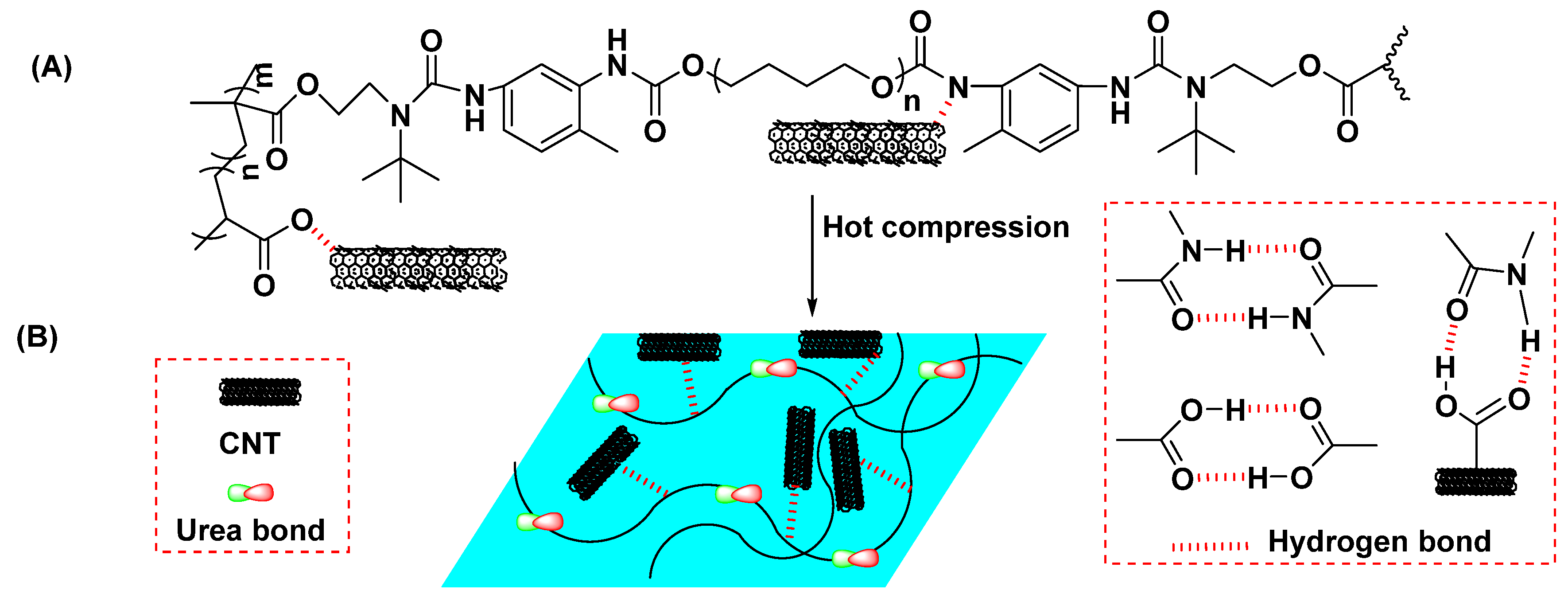
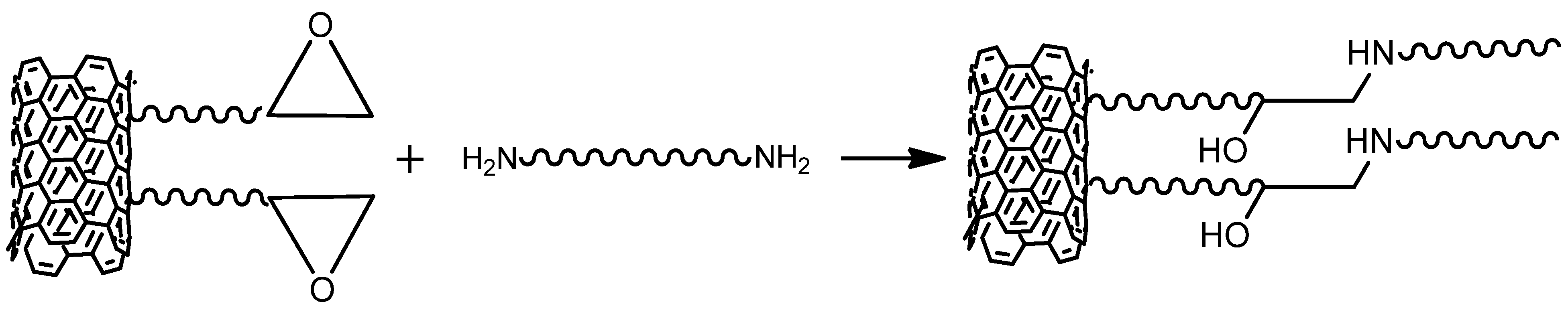
5.1. Polyurea Nanocomposites Based on Functionalized Nanofillers
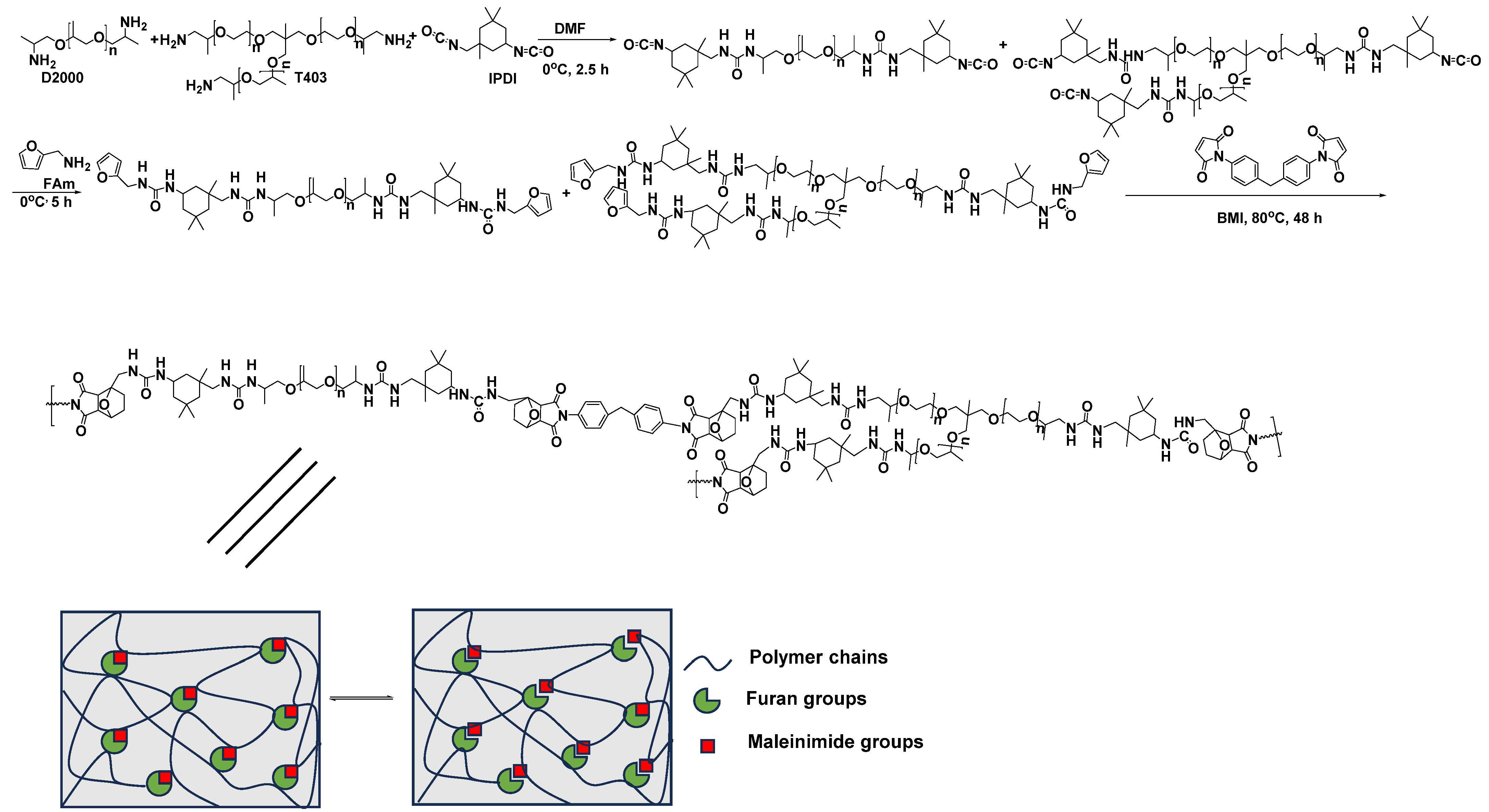
5.2. Self-Healing Nanocomposite Polyurea
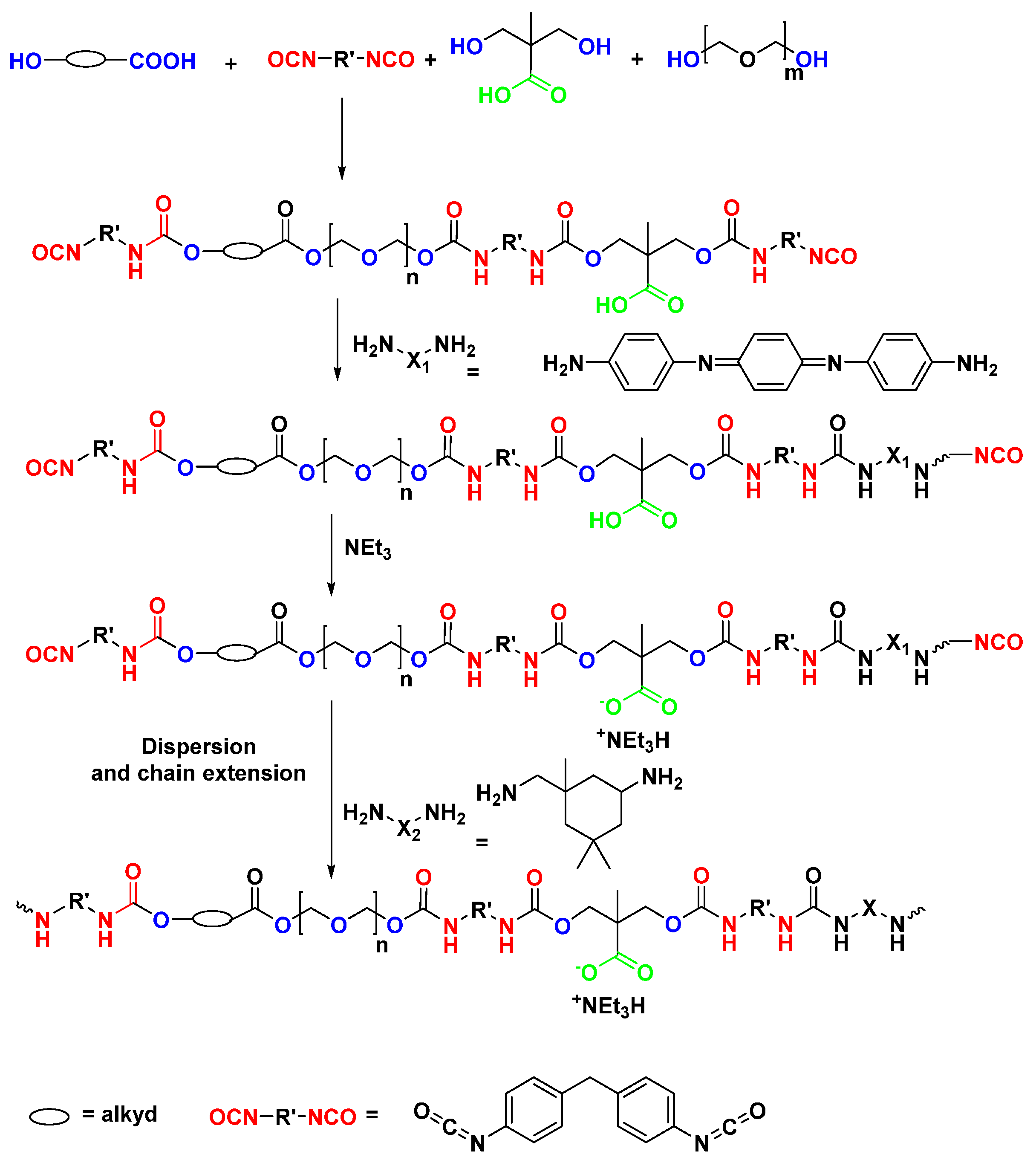
5.3. Hybrid Polyurea—Polyurethane Matrices from Renewable Sources
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Hill, R.; Walker, E.E. Polymer constitution and fiber properties. J. Polym. Sci. 1948, 3, 609–630. [Google Scholar] [CrossRef]
- Zhang, R.; Huang, W.; Lyu, P.; Yan, S.; Wang, X.; Ju, J. Polyurea for Blast and Impact Protection: A Review. Polymers 2022, 14, 2670. [Google Scholar] [CrossRef]
- Wolfgang, L.; Heinrich, R. Basic Polyureas. U.S. Patent 2,761,852, 4 September 1956. [Google Scholar]
- Wolfgang, L.; Heinrich, R. Process for the Production of Polyureas. U.S. Patent 4,028,310, 14 July 1958. [Google Scholar]
- Schlichter, M.E.; Barton, M.S. Polyurea/Polyurethane Edge Coating and Process for Making. U.S. Patent 5,534,295, 6 July 1996. [Google Scholar]
- Rice, D.M.; Dominguez, R.J. Reaction Injection Molded Elastomers Prepared from Amine Terminated Polyethers, Amine Terminated Chain Extender and Aromatic Polyisocyanate. U.S. Patent No. 4,433,067, 21 January 1984. [Google Scholar]
- Pires, R.F.; Bonifácio, V.D.B. Polyureas. In Kirk-Othmer Encyclopedia of Chemical Technology; Repository Universitas Perintis Indonesia: Makassar, Indonesia, 2017; pp. 1–12. [Google Scholar]
- Shojaei, B.; Najafi, M.; Yazdanbakhsh, A.; Abtahi, M.; Zhang, C. A review on the applications of polyurea in the construction industry. Polym. Adv. Technol. 2021, 32, 2797–2812. [Google Scholar] [CrossRef]
- Iqbal, N.; Tripathi, M.; Parthasarathy, S.; Kumar, D.; Roy, P.K. Polyurea spray coatings: Tailoring material properties through chemical crosslinking. Prog. Org. Coat. 2018, 123, 201–208. [Google Scholar] [CrossRef]
- Grujicic, M.; D’Entremont, B.P.; Pandurangan, B.; Runt, J.; Tarter, J.; Dillon, G. Concept-level analysis and design of polyurea for enhanced blast-mitigation performance. J. Mater. Eng. Perform. 2012, 21, 2024–2037. [Google Scholar] [CrossRef]
- Available online: https://www.grandviewresearch.com/industry-analysis/polyurea-market/methodology (accessed on 2 February 2023).
- Samiee, A.; Isaacs, J.; Nemat-Nasser, S. Ballistic Performance of Polyurea-Coated Armor Grade Ceramic Tiles; SPIE: Bellingham, WA, USA, 2010; Volume 7644. [Google Scholar]
- Si, P.; Liu, Y.; Yan, J.; Bai, F.; Huang, F. Ballistic Performance of Polyurea-Reinforced Ceramic/Metal Armor Subjected to Projectile Impact. Materials 2022, 15, 3918. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Z.; Zhao, P.; Zhang, L.; Jin, X.C.; Xu, Y. Experimental investigation on ballistic resistance of polyurea coated steel plates subjected to fragment impact. Thin Walled Struct. 2019, 144, 106342. [Google Scholar] [CrossRef]
- İşmal, Ö.E.; Paul, R. 17—Composite Textiles in High-Performance Apparel. In High-Performance Apparel; McLoughlin, J., Sabir, T., Eds.; Woodhead Publishing: Sawston, UK, 2018; pp. 377–420. [Google Scholar]
- Cernak, I.; Noble-Haeusslein, L.J. Traumatic Brain Injury: An Overview of Pathobiology with Emphasis on Military Populations. J. Cereb. Blood Flow Metab. 2010, 30, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Arman, B.; Reddy, A.S.; Arya, G. Viscoelastic Properties and Shock Response of Coarse-Grained Models of Multiblock versus Diblock Copolymers: Insights into Dissipative Properties of Polyurea. Macromolecules 2012, 45, 3247–3255. [Google Scholar] [CrossRef]
- Grujicic, M.; Snipes, J.S.; Ramaswami, S.; Yavari, R.; Runt, J.; Tarter, J.; Dillon, G. Coarse-grained Molecular-level Analysis of Polyurea Properties and Shock-mitigation Potential. J. Mater. Eng. Perform. 2013, 22, 1964–1981. [Google Scholar] [CrossRef]
- Bogoslovov, R.B.; Roland, C.M.; Gamache, R.M. Impact-induced glass transition in elastomeric coatings. Appl. Phys. Lett. 2007, 90, 221910. [Google Scholar] [CrossRef]
- Ćwik, T.K.; Iannucci, L.; Curtis, P.; Pope, D. Investigation of the ballistic performance of ultra high molecular weight polyethylene composite panels. Compos. Struct. 2016, 149, 197–212. [Google Scholar] [CrossRef]
- Iqbal, N.; Sharma, P.K.; Kumar, D.; Roy, P.K. Protective polyurea coatings for enhanced blast survivability of concrete. Constr. Build. Mater. 2018, 175, 682–690. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, P.; Jin, F.; He, H.; Zhou, Y.; Chen, H.; Zhou, J.; Wang, B.; Fan, H. Blast responses of polyurea-coated concrete arches. Arch. Civ. Mech. Eng. 2021, 21, 30. [Google Scholar] [CrossRef]
- Roland, C.M.; Fragiadakis, D.; Gamache, R.M. Elastomer–steel laminate armor. Compos. Struct. 2010, 92, 1059–1064. [Google Scholar] [CrossRef]
- Petre, R.; Zecheru, T.; Ginghina, R. Dynamic Tests on Polyurea-Based Hybrid Composites for Ballistic Protection. Mater. Plast. 2021, 58, 41–47. [Google Scholar] [CrossRef]
- Petre, R.; Zecheru, T.; Petrea, N.; Ginghina, R.; Sandu, S.; Muresan, M.; Matache, L.C.; Sava, A.C.; Neatu, F. Synthesis and Mechanical Properties of Polyurea-Based Hybrid Composites for Ballistic Individual Protection. Mater. Plast. 2018, 55, 315–319. [Google Scholar] [CrossRef]
- Jordan, J.L.; Casem, D.T.; Robinette, J. Hugoniot and dynamic strength in polyurea. J. Appl. Phys. 2022, 131, 165903. [Google Scholar] [CrossRef]
- Manav, M.; Ortiz, M. Molecular dynamics study of the shock response of polyurea. Polymer 2021, 212, 123109. [Google Scholar] [CrossRef]
- Sun, Y.-X.; Wang, X.; Ji, C.; Zhao, C.-X.; Liu, P.-L.; Meng, L.; Zhang, K.; Jiang, T. Experimental investigation on anti-penetration performance of polyurea-coated ASTM1045 steel plate subjected to projectile impact. Def. Technol. 2021, 17, 1496–1513. [Google Scholar] [CrossRef]
- Youssef, G.H. Dynamic Properties of Polyurea; University of California: Los Angeles, CA, USA, 2011. [Google Scholar]
- Giller, C.; Gamache, R.; Wahl, K.; Saab, A.; Roland, C. Coating/substrate interaction in elastomer-steel bilayer armor. J. Compos. Mater. 2016, 50, 2853–2859. [Google Scholar] [CrossRef]
- Yang, S.J.; Rosenbloom, S.I.; Fors, B.P.; Silberstein, M.N. Elucidating the impact of microstructure on mechanical properties of phase-segregated polyurea: Finite element modeling of molecular dynamics derived microstructures. Mech. Mater. 2024, 188, 104863. [Google Scholar] [CrossRef]
- Iqbal, N.; Tripathi, M.; Parthasarathy, S.; Kumar, D.; Roy, P.K. Polyurea coatings for enhanced blast-mitigation: A review. RSC Adv. 2016, 6, 109706–109717. [Google Scholar] [CrossRef]
- Heyden, S.; Ortiz, M.; Fortunelli, A. All-atom molecular dynamics simulations of multiphase segregated polyurea under quasistatic, adiabatic, uniaxial compression. Polymer 2016, 106, 100–108. [Google Scholar] [CrossRef]
- Toader, G.; Rusen, E.; Teodorescu, M.; Diacon, A.; Stanescu, P.O.; Rotariu, T.; Rotariu, A. Novel polyurea polymers with enhanced mechanical properties. J. Appl. Polym. Sci. 2016, 133, 43967. [Google Scholar] [CrossRef]
- He, Y.; Xie, D.; Zhang, X. The structure, microphase-separated morphology, and property of polyurethanes and polyureas. J. Mater. Sci. 2014, 49, 7339–7352. [Google Scholar] [CrossRef]
- Yildirim, E.; Yurtsever, M. The role of diisocyanate and soft segment on the intersegmental interactions in urethane and urea based segmented copolymers: A DFT study. Comput. Theor. Chem. 2014, 1035, 28–38. [Google Scholar] [CrossRef]
- Riehle, N.; Athanasopulu, K.; Kutuzova, L.; Götz, T.; Kandelbauer, A.; Tovar, G.E.M.; Lorenz, G. Influence of Hard Segment Content and Diisocyanate Structure on the Transparency and Mechanical Properties of Poly(dimethylsiloxane)-Based Urea Elastomers for Biomedical Applications. Polymers 2021, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.-X.; Gao, W.-C.; Ren, X.-M.; Ouyang, X.-Y.; Zhao, Y.; Zhao, H.; Wu, W.; Huang, C.-X.; Liu, Y.; Liu, X.-Y.; et al. A review of microphase separation of polyurethane: Characterization and applications. Polym. Test. 2022, 107, 107489. [Google Scholar] [CrossRef]
- He, Y.; Zhang, X.; Runt, J. The role of diisocyanate structure on microphase separation of solution polymerized polyureas. Polymer 2014, 55, 906–913. [Google Scholar] [CrossRef]
- Yilgor, I.; Yilgor, E. Structure-morphology-property behavior of segmented thermoplastic polyurethanes and polyureas prepared without chain extenders. Polym. Rev. 2007, 47, 487–510. [Google Scholar] [CrossRef]
- Chantawansri, T.L.; Sliozberg, Y.R.; Andzelm, J.W.; Hsieh, A.J. Coarse-grained modeling of model poly(urethane urea)s: Microstructure and interface aspects. Polymer 2012, 53, 4512–4524. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Xie, Z.; Xu, J.; Guo, B.-H. A multi-scale investigation on effects of hydrogen bonding on micro-structure and macro-properties in a polyurea. Polymer 2018, 145, 261–271. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Wang, Y.; Gu, J.; Ji, C.; Wu, G.; Cheng, L. High-hardness polyurea coated steel plates subjected to combined loadings of shock wave and fragments. Lat. Am. J. Solids Struct. 2022, 19, 1–24. [Google Scholar] [CrossRef]
- Zheng, T.; Zhang, Y.; Shi, J.; Xu, J.; Guo, B. Revealing the role of hydrogen bonding in polyurea with multiscale simulations. Mol. Simul. 2021, 47, 1258–1272. [Google Scholar] [CrossRef]
- Reinecker, M.; Soprunyuk, V.; Fally, M.; Sánchez-Ferrer, A.; Schranz, W. Two glass transitions of polyurea networks: Effect of the segmental molecular weight. Soft Matter 2014, 10, 5729–5738. [Google Scholar] [CrossRef]
- Sánchez-Ferrer, A.; Soprunyuk, V.; Engelhardt, M.; Stehle, R.; Gilg, H.A.; Schranz, W.; Richter, K. Polyurea Networks from Moisture-Cure, Reaction-Setting, Aliphatic Polyisocyanates with Tunable Mechanical and Thermal Properties. ACS Appl. Polym. Mater. 2021, 3, 4070–4078. [Google Scholar] [CrossRef]
- Fragiadakis, D.; Gamache, R.; Bogoslovov, R.B.; Roland, C.M. Segmental dynamics of polyurea: Effect of stoichiometry. Polymer 2010, 51, 178–184. [Google Scholar] [CrossRef]
- Tsagaropoulos, G.; Eisenburg, A. Direct observation of two glass transitions in silica-filled polymers. Implications to the morphology of random ionomers. Macromolecules 1995, 28, 396–398. [Google Scholar] [CrossRef]
- Holzworth, K.; Jia, Z.; Amirkhizi, A.V.; Qiao, J.; Nemat-Nasser, S. Effect of isocyanate content on thermal and mechanical properties of polyurea. Polymer 2013, 54, 3079–3085. [Google Scholar] [CrossRef]
- Chen, Z.S.; Yang, W.P.; Macosko, C.W. Polyurea Synthesis and Properties as a Function of Hard-Segment Content. Rubber Chem. Technol. 1988, 61, 86–99. [Google Scholar] [CrossRef]
- Castagna, A.M.; Pangon, A.; Choi, T.; Dillon, G.P.; Runt, J. The Role of Soft Segment Molecular Weight on Microphase Separation and Dynamics of Bulk Polymerized Polyureas. Macromolecules 2012, 45, 8438–8444. [Google Scholar] [CrossRef]
- Shen, Z.; Chen, J.; Li, G.; Situ, G.; Ma, X.; Sha, Y.; Zhao, D.; Gu, Q.; Zhang, M.; Luo, Y.; et al. Mechanically robust self-repairing polyurea elastomers: The roles of hard segment content and ordered/disordered hydrogen-bonding arrays. Eur. Polym. J. 2022, 181, 111657. [Google Scholar] [CrossRef]
- Humanitarian Demining, G.I.C.F. Explosive Weapon Effects; James Madison University: Harrisonburg, VA, USA, 2017. [Google Scholar]
- Wu, J.; Liu, Z.; Yu, J.; Xu, S. Experimental and numerical investigation of normal reinforced concrete panel strengthened with polyurea under near-field explosion. J. Build. Eng. 2022, 46, 103763. [Google Scholar] [CrossRef]
- Iqbal, N.; Kumar, D.; Roy, P.K. Emergence of time-dependent material properties in chain extended polyureas. J. Appl. Polym. Sci. 2018, 135, 46730. [Google Scholar] [CrossRef]
- Goswami, A.; Adhikary, S.D. Retrofitting materials for enhanced blast performance of Structures: Recent advancement and challenges ahead. Constr. Build. Mater. 2019, 204, 224–243. [Google Scholar] [CrossRef]
- Akulichev, A.G.; Tiwari, A.; Dorogin, L.; Echtermeyer, A.T.; Persson, B.N.J. Rubber adhesion below the glass transition temperature: Role of frozen-in elastic deformation. Europhys. Lett. 2017, 120, 36002. [Google Scholar] [CrossRef]
- Temizkan, E.; Eroğlu, G.; Ergün, A.; Deligöz, H. Preparation, characterization, and influence of polyurea coatings on their layered composite materials based on flexible rebonded polyurethane. Polym. Eng. Sci. 2021, 61, 1392–1404. [Google Scholar] [CrossRef]
- Iqbal, N.; Tripathi, M.; Parthasarathy, S.; Kumar, D.; Roy, P.K. Aromatic versus Aliphatic: Hydrogen Bonding Pattern in Chain-Extended High-Performance Polyurea. ChemistrySelect 2018, 3, 1976–1982. [Google Scholar] [CrossRef]
- Tripathi, M.; Parthasarathy, S.; Roy, P.K. Spray processable polyurea formulations: Effect of chain extender length on material properties of polyurea coatings. J. Appl. Polym. Sci. 2020, 137, 48573. [Google Scholar] [CrossRef]
- Tekalur, S.A.; Shukla, A.; Shivakumar, K. Blast resistance of polyurea based layered composite materials. Compos. Struct. 2008, 84, 271–281. [Google Scholar] [CrossRef]
- LeBlanc, J.; Shukla, A. Response of polyurea-coated flat composite plates to underwater explosive loading. J. Compos. Mater. 2015, 49, 965–980. [Google Scholar] [CrossRef]
- Pinto, M.; Shukla, A. Mitigation of pressure pulses from implosion of hollow composite cylinders. J. Compos. Mater. 2016, 50, 3709–3718. [Google Scholar] [CrossRef]
- Chen, C.; Wang, X.; Hou, H.; Cheng, Y.; Zhang, P.; Liu, J. Effect of strength matching on failure characteristics of polyurea coated thin metal plates under localized air blast loading: Experiment and numerical analysis. Thin Walled Struct. 2020, 154, 106819. [Google Scholar] [CrossRef]
- Hou, H.; Chen, C.; Cheng, Y.; Zhang, P.; Tian, X.; Liu, T.; Wang, J. Effect of structural configuration on air blast resistance of polyurea-coated composite steel plates: Experimental studies. Mater. Des. 2019, 182, 108049. [Google Scholar] [CrossRef]
- Shi, S.; Liao, Y.; Peng, X.; Liang, C.; Sun, J. Behavior of polyurea-woven glass fiber mesh composite reinforced RC slabs under contact explosion. Int. J. Impact Eng. 2019, 132, 103335. [Google Scholar] [CrossRef]
- Wu, G.; Wang, X.; Ji, C.; Liu, Q.; Gao, Z.; Zhang, K.; Zhao, C. Experimental and numerical simulation study on polyurea-coated fuel tank subjected to combined action of blast shock waves and fragments. Thin Walled Struct. 2021, 169, 108436. [Google Scholar] [CrossRef]
- Wu, G.; Wang, X.; Ji, C.; Gao, Z.; Jiang, T.; Zhao, C.; Liu, Y. Anti-blast properties of 6063-T5 aluminum alloy circular tubes coated with polyurea elastomer: Experiments and numerical simulations. Thin Walled Struct. 2021, 164, 107842. [Google Scholar] [CrossRef]
- Lyu, P.; Fang, Z.; Wang, X.; Huang, W.; Zhang, R.; Sang, Y.; Sun, P. Explosion Test and Numerical Simulation of Coated Reinforced Concrete Slab Based on BLAST Mitigation Polyurea Coating Performance. Materials 2022, 15, 2607. [Google Scholar] [CrossRef]
- Rosenbloom, S.I.; Yang, S.J.; Tsakeredes, N.J.; Fors, B.P.; Silberstein, M.N. Microstructural evolution of polyurea under hydrostatic pressure. Polymer 2021, 227, 123845. [Google Scholar] [CrossRef]
- Leite, F.; Mota, C.; Bessa, J.; Cunha, F.; Fangueiro, R.; Gomes, G.; Mingote, J. Advanced Coatings of Polyureas for Building Blast Protection: Physical, Chemical, Thermal and Mechanical Characterization. Appl. Sci. 2022, 12, 10879. [Google Scholar] [CrossRef]
- Auckloo, S.A.B.; Palaniandy, K.; Hung, Y.M.; Lazzara, G.; Chai, S.-P.; Pasbakhsh, P. Nonporous, Strong, Stretchable, and Transparent Electrospun Aromatic Polyurea Nanocomposites as Potential Anticorrosion Coating Films. Nanomaterials 2021, 11, 2998. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.; Fragiadakis, D.; Roland, C.M.; Runt, J. Microstructure and Segmental Dynamics of Polyurea under Uniaxial Deformation. Macromolecules 2012, 45, 3581–3589. [Google Scholar] [CrossRef]
- Iqbal, N.; Tripathi, M.; Parthasarathy, S.; Kumar, D.; Roy, P.K. Tuning the properties of segmented polyurea by regulating soft-segment length. J. Appl. Polym. Sci. 2018, 135, 46284. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Yu, R.-P.; Ren, J.-W.; Zhang, Q.-C.; Zhao, Z.-Y.; Ni, C.-Y.; Han, B.; Lu, T.J. Enhanced vibration and damping characteristics of novel corrugated sandwich panels with polyurea-metal laminate face sheets. Compos. Struct. 2020, 251, 112591. [Google Scholar] [CrossRef]
- Hagen, R.; Salmén, L.; Lavebratt, H.; Stenberg, B. Comparison of dynamic mechanical measurements and Tg determinations with two different instruments. Polym. Test. 1994, 13, 113–128. [Google Scholar] [CrossRef]
- Weibo, H. Spray Polyurea Elastomer Technology and Its Application in Heavy-Duty Anti-Corrosion. Corros. Sci. Technol. Prot. 2003, 15, 56–58. [Google Scholar]
- Zhang, L.; Ji, C.; Wang, X.; Wang, Y.; Wu, G.; Zhu, H.; Han, Z. Strengthening and converse strengthening effects of polyurea layer on polyurea–steel composite structure subjected to combined actions of blast and fragments. Thin Walled Struct. 2022, 178, 109527. [Google Scholar] [CrossRef]
- Luo, J.; Wang, T.; Sim, C.; Li, Y. Mini-Review of Self-Healing Mechanism and Formulation Optimization of Polyurea Coating. Polymers 2022, 14, 2808. [Google Scholar] [CrossRef]
- Toader, G.; Diacon, A.; Rusen, E.; Rizea, F.; Teodorescu, M.; Stanescu, P.O.; Damian, C.; Rotariu, A.; Trana, E.; Bucur, F.; et al. A Facile Synthesis Route of Hybrid Polyurea-Polyurethane-MWCNTs Nanocomposite Coatings for Ballistic Protection and Experimental Testing in Dynamic Regime. Polymers 2021, 13, 16. [Google Scholar] [CrossRef]
- Yong, A.X.H.; Sims, G.D.; Gnaniah, S.J.P.; Ogin, S.L.; Smith, P.A. Heating rate effects on thermal analysis measurement of Tg in composite materials. Adv. Manuf. Polym. Compos. Sci. 2017, 3, 43–51. [Google Scholar] [CrossRef]
- Raghu, S.; Staub, F.W. Obtaining the surface temperature distribution in a shock wave-boundary layer interaction region using a liquid crystal technique. Exp. Therm. Fluid Sci. 1994, 9, 283–288. [Google Scholar] [CrossRef]
- Won-in, K.; Boonruang, C.; Dararutana, P. Characterization of polyurea elastomer used for blast mitigation. In AIP Conference Proceedings; AIP Publishing: College Park, MA, USA, 2020; Volume 2279. [Google Scholar] [CrossRef]
- Weinberg, K.; Khosravani, M.R.; Thimm, B.; Reppel, T.; Bogunia, L.; Aghayan, S.; Nötzel, R. Hopkinson bar experiments as a method to determine impact properties of brittle and ductile materials. GAMM Mitteilungen 2018, 41, e201800008. [Google Scholar] [CrossRef]
- Wang, H.; Deng, X.; Wu, H.; Pi, A.; Li, J.; Huang, F. Investigating the dynamic mechanical behaviors of polyurea through experimentation and modeling. Def. Technol. 2019, 15, 875–884. [Google Scholar] [CrossRef]
- Pangon, A.; Dillon, G.P.; Runt, J. Influence of mixed soft segments on microphase separation of polyurea elastomers. Polymer 2014, 55, 1837–1844. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Lee, S.; Park, J.; Shin, E.-J. One-Shot Synthesis of Thermoplastic Polyurethane Based on Bio-Polyol (Polytrimethylene Ether Glycol) and Characterization of Micro-Phase Separation. Polymers 2022, 14, 4269. [Google Scholar] [CrossRef]
- Marcano, A.; Fatyeyeva, K.; Koun, M.; Dubuis, P.; Grimme, M.; Marais, S. Recent developments in the field of barrier and permeability properties of segmented polyurethane elastomers. Rev. Chem. Eng. 2019, 35, 445–474. [Google Scholar] [CrossRef]
- Hummel, R.E.; Dubroca, T. Differential Reflectance Spectroscopy in Analysis of Surfaces. In Encyclopedia of Analytical Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 1–25. [Google Scholar]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nutt, S.R.; Sheran, K.; Wudl, F. A Thermally Re-mendable Cross-Linked Polymeric Material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, Y.; Wu, Q.; Moore, J.S. Architecture-Controlled Ring-Opening Polymerization for Dynamic Covalent Poly(disulfide)s. J. Am. Chem. Soc. 2019, 141, 17075–17080. [Google Scholar] [CrossRef]
- Zhang, Y.; Ying, H.; Hart, K.R.; Wu, Y.; Hsu, A.J.; Coppola, A.M.; Kim, T.A.; Yang, K.; Sottos, N.R.; White, S.R.; et al. Malleable and Recyclable Poly(urea-urethane) Thermosets bearing Hindered Urea Bonds. Adv. Mater. 2016, 28, 7646–7651. [Google Scholar] [CrossRef]
- Fu, D.; Pu, W.; Escorihuela, J.; Wang, X.; Wang, Z.; Chen, S.; Sun, S.; Wang, S.; Zuilhof, H.; Xia, H. Acylsemicarbazide Moieties with Dynamic Reversibility and Multiple Hydrogen Bonding for Transparent, High Modulus, and Malleable Polymers. Macromolecules 2020, 53, 7914–7924. [Google Scholar] [CrossRef]
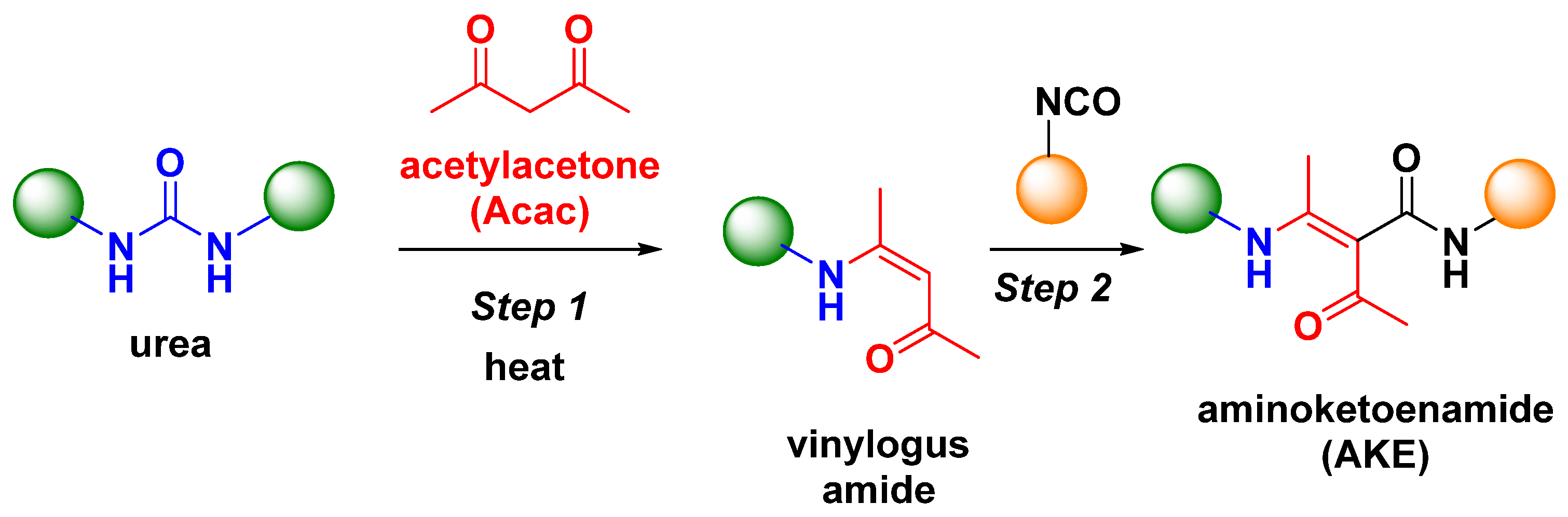
- Ma, Y.; Jiang, X.; Yin, J.; Weder, C.; Berrocal, J.A.; Shi, Z. Chemical Upcycling of Conventional Polyureas into Dynamic Covalent Poly(aminoketoenamide)s. Angew. Chem. Int. Ed. 2023, 62, e202212870. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Wang, Y.; Fu, X.; Jiang, L.; Wang, Y.; Yuan, A.; Xu, H.; Lei, J. Recyclable and Reprocessable Thermosetting Polyurea with High Performance Based on Diels-Alder Dynamic Covalent Crosslinking. Macromol. Res. 2021, 29, 562–568. [Google Scholar] [CrossRef]
- Das, S.; Cox, D.F.; Wilkes, G.L.; Klinedinst, D.B.; Yilgor, I.; Yilgor, E.; Beyer, F.L. Effect of Symmetry and H-bond Strength of Hard Segments on the Structure-Property Relationships of Segmented, Nonchain Extended Polyurethanes and Polyureas. J. Macromol. Sci. Part B 2007, 46, 853–875. [Google Scholar] [CrossRef]
- Sami, S.; Yildirim, E.; Yurtsever, M.; Yurtsever, E.; Yilgor, E.; Yilgor, I.; Wilkes, G.L. Understanding the influence of hydrogen bonding and diisocyanate symmetry on the morphology and properties of segmented polyurethanes and polyureas: Computational and experimental study. Polymer 2014, 55, 4563–4576. [Google Scholar] [CrossRef]
- Sheth, J.P.; Klinedinst, D.B.; Wilkes, G.L.; Yilgor, I.; Yilgor, E. Role of chain symmetry and hydrogen bonding in segmented copolymers with monodisperse hard segments. Polymer 2005, 46, 7317–7322. [Google Scholar] [CrossRef]
- Adhikari, R.; Gunatillake, P.A.; Meijs, G.F.; McCarthy, S.J. The effect of diisocyanate isomer composition on properties and morphology of polyurethanes based on 4,4′-dicyclohexyl methane diisocyanate and mixed macrodiols (PDMS–PHMO). J. Appl. Polym. Sci. 1999, 73, 573–582. [Google Scholar] [CrossRef]
- Saralegi, A.; Etxeberria, A.; Fernández-d’Arlas, B.; Mondragon, I.; Eceiza, A.; Corcuera, M.A. Effect of H12MDI isomer composition on mechanical and physico-chemical properties of polyurethanes based on amorphous and semicrystalline soft segments. Polym. Bull. 2013, 70, 2193–2210. [Google Scholar] [CrossRef]
- Joseph, M.D.; Savina, M.R.; Harris, R.F. Effects on properties of varying the cis/trans isomer distribution in polyurethane elastomers made with 1,4-cyclohexane diisocyanate. J. Appl. Polym. Sci. 1992, 44, 1125–1133. [Google Scholar] [CrossRef]
- Prisacariu, C.; Buckley, C.P.; Caraculacu, A.A. Mechanical response of dibenzyl-based polyurethanes with diol chain extension. Polymer 2005, 46, 3884–3894. [Google Scholar] [CrossRef]
- Prisacariu, C.; Scortanu, E. Effect of Increasing the Hard Segment Percentage on the Mechanical Response of Selected Polyurethane Films. High Perform. Polym. 2010, 22, 876–887. [Google Scholar] [CrossRef]
- Toader, G.; Moldovan, A.E.; Diacon, A.; Dirloman, F.M.; Rusen, E.; Podaru, A.; Rotariu, T.; Ginghina, R.E.; Hoza, O.E. Effect of Aromatic Chain Extenders on Polyurea and Polyurethane Coatings Designed for Defense Applications. Polymers 2023, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Huang, D. Preparation of polymer nanoparticles, and the effect of nanoconfinement on glass transition, structural relaxation and crystallization. Front. Chem. China 2011, 6, 332–340. [Google Scholar] [CrossRef]
- Wang, W.; Lu, W.; Goodwin, A.; Wang, H.; Yin, P.; Kang, N.-G.; Hong, K.; Mays, J.W. Recent advances in thermoplastic elastomers from living polymerizations: Macromolecular architectures and supramolecular chemistry. Prog. Polym. Sci. 2019, 95, 1–31. [Google Scholar] [CrossRef]
- Petrović, Z.S.; Ferguson, J. Polyurethane elastomers. Prog. Polym. Sci. 1991, 16, 695–836. [Google Scholar] [CrossRef]
- Hsu, J.-M.; Yang, D.-L.; Huang, S.K. Study on thermal transitions of toluene diisocyanate-based polyurethane elastomers with poly (tetramethylene oxide) as the soft segment by TSC/RMA, DSC and DMA thermal analyzers. J. Polym. Res. 1999, 6, 67–78. [Google Scholar] [CrossRef]
- Bae, J.Y.; Chung, D.J.; An, J.H.; Shin, D.H. Effect of the structure of chain extenders on the dynamic mechanical behaviour of polyurethane. J. Mater. Sci. 1999, 34, 2523–2527. [Google Scholar] [CrossRef]
- Sonnenschein, M.F. Polyurethanes: Science, Technology, Markets, and Trends; Wiley: Hoboken, NJ, USA, 2020. [Google Scholar]
- Yuan, Z.; Yan, J.; Gao, F.; Cheng, J.; Zhang, J. High-performance, fluorescent, UV-shielding, triboelectric, super-flexible polyurea elastomers via strong π–π stacking of pyrene and hydrogen bonding strategies. J. Mater. Chem. C 2023, 11, 8971–8981. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Chang, F.-C.; Chen, J.-K.; Wang, T.-Y.; Lee, D.-J. High-efficiency self-healing materials based on supramolecular polymer networks. RSC Adv. 2015, 5, 101148–101154. [Google Scholar] [CrossRef]
- Li, B.; Cao, P.-F.; Saito, T.; Sokolov, A.P. Intrinsically Self-Healing Polymers: From Mechanistic Insight to Current Challenges. Chem. Rev. 2023, 123, 701–735. [Google Scholar] [CrossRef]
- Hao, Y.; Zhu, G.; Li, B. Mechanically Robust and Self-Healable Polyureas Based on Multiple Dynamic Bonds. Ind. Eng. Chem. Res. 2023, 62, 10444–10453. [Google Scholar] [CrossRef]
- Zhang, Z.; Qian, L.; Cheng, J.; Xie, Q.; Ma, C.; Zhang, G. Room-Temperature Self-Healing Polyurea with High Puncture and Impact Resistances. Chem. Mater. 2023, 35, 1806–1817. [Google Scholar] [CrossRef]
- Hao, Y.; Zhu, G.; Li, B.; Ren, T. A high-performance self-healing polyurea material based on exchangeable aromatic disulfide. J. Appl. Polym. Sci. 2022, 139, e52992. [Google Scholar] [CrossRef]
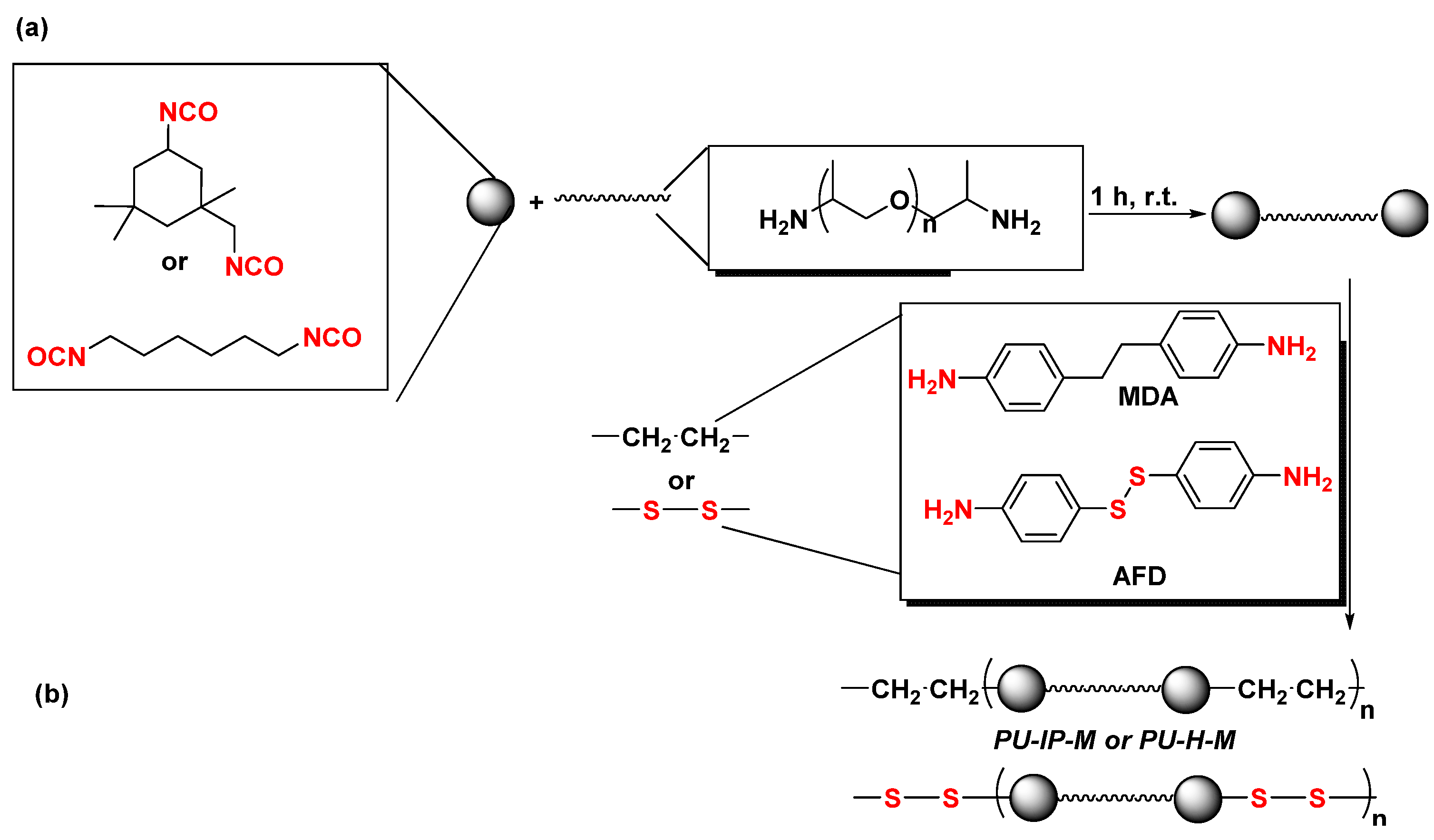
- Li, T.; Xie, Z.; Xu, J.; Weng, Y.; Guo, B.-H. Design of a self-healing cross-linked polyurea with dynamic cross-links based on disulfide bonds and hydrogen bonding. Eur. Polym. J. 2018, 107, 249–257. [Google Scholar] [CrossRef]
- Xiang, Z.; Chu, C.; Xie, H.; Xiang, T.; Zhou, S. Multifunctional Thermoplastic Polyurea Based on the Synergy of Dynamic Disulfide Bonds and Hydrogen Bond Cross-Links. ACS Appl. Mater. Interfaces 2021, 13, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zheng, T.; Han, J.; Liu, Z.; Guo, Z.-X.; Zhuang, Z.; Xu, J.; Guo, B.-H. Effects of Diisocyanate Structure and Disulfide Chain Extender on Hard Segmental Packing and Self-Healing Property of Polyurea Elastomers. Polymers 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed]
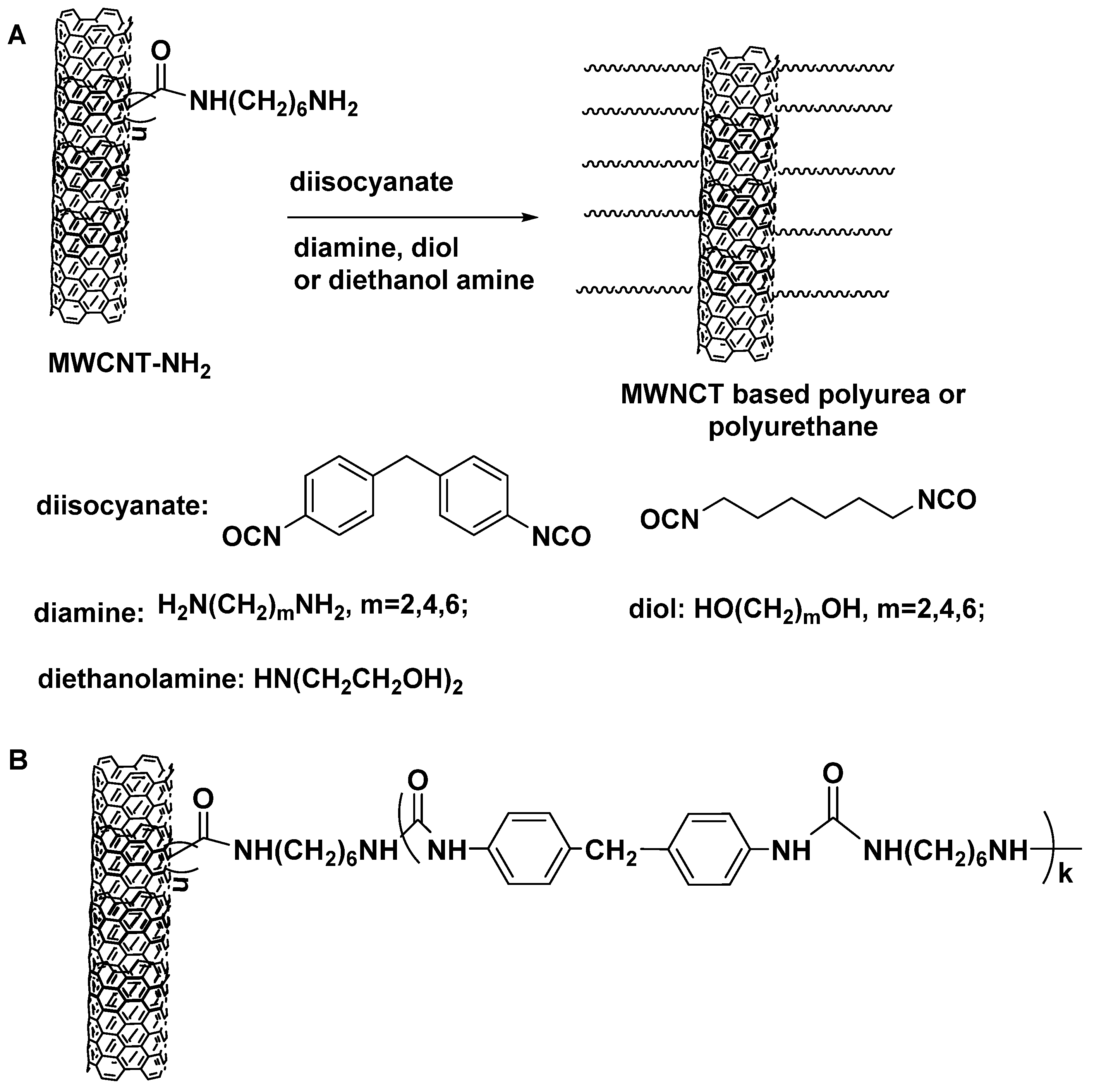
- Toader, G.; Rusen, E.; Teodorescu, M.; Diacon, A.; Stanescu, P.O.; Damian, C.; Rotariu, T.; Rotariu, A. New polyurea MWCNTs nanocomposite films with enhanced mechanical properties. J. Appl. Polym. Sci. 2017, 134, 7. [Google Scholar] [CrossRef]
- Chin, D.C.H.; Palaniandy, K.; Hia, I.L.; Pasbakhsh, P. High performance aliphatic polyurea films reinforced using nonfunctionalized multiwalled carbon nanotubes. Polym. Compos. 2020, 41, 1036–1044. [Google Scholar] [CrossRef]
- Gao, C.; Jin, Y.Z.; Kong, H.; Whitby, R.L.D.; Acquah, S.F.A.; Chen, G.Y.; Qian, H.; Hartschuh, A.; Silva, S.R.P.; Henley, S.; et al. Polyurea-Functionalized Multiwalled Carbon Nanotubes: Synthesis, Morphology, and Raman Spectroscopy. J. Phys. Chem. B 2005, 109, 11925–11932. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, Y.; Guo, A.; Xue, T.; Li, X.; Yu, C.; Zhang, F. Remotely fast response healing crosslinked polyurea nanocomposites with recyclability via two-step method. Compos. Sci. Technol. 2022, 224, 109462. [Google Scholar] [CrossRef]
- Jagtap, S.B.; Patil, V.D.; Suresh, K.; Ram, F.; Mohan, M.S.; Rajput, S.S.; Patil, S.; Shukla, P.G.; Shanmuganathan, K. Functionalized carbon nanotube reinforced polymer nanocomposite microcapsules with enhanced stiffness. Colloids Surf. A Physicochem. Eng. Asp. 2018, 550, 82–89. [Google Scholar] [CrossRef]
- Cai, D.; Song, M. High mechanical performance polyurea/organoclay nanocomposites. Compos. Sci. Technol. 2014, 103, 44–48. [Google Scholar] [CrossRef]
- Lee, J.-H.; Choi, E.-K.; Song, J.-Y.; Kim, S.-Y.; Oh, S.-K. Applicability of Composite Polyurea Method Considering the Required Performance in Underground Parking Lot Upper Slab. J. Korean Recycl. Constr. Resour. Inst. 2019, 7, 243–254. [Google Scholar] [CrossRef]
- Irshidat, M.; Al-Ostaz, A.; Cheng, A.H.-D. Correlating Micromorphology and Nanomorphology to High Strain Rate Performance of Nanoparticle Reinforced Polymeric Materials. J. Nanomechanics Micromechanics 2012, 2, 55–64. [Google Scholar] [CrossRef]
- Fukao, K. Dynamics in thin polymer films by dielectric spectroscopy. Eur. Phys. J. E 2003, 12, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Elmahdy, M.M.; Chrissopoulou, K.; Afratis, A.; Floudas, G.; Anastasiadis, S.H. Effect of Confinement on Polymer Segmental Motion and Ion Mobility in PEO/Layered Silicate Nanocomposites. Macromolecules 2006, 39, 5170–5173. [Google Scholar] [CrossRef]
- Roland, C.M.; Fragiadakis, D.; Gamache, R.M.; Casalini, R. Factors influencing the ballistic impact resistance of elastomer-coated metal substrates. Philos. Mag. 2013, 93, 468–477. [Google Scholar] [CrossRef]
- Dewapriya, M.A.N.; Miller, R.E. Molecular dynamics study of the penetration resistance of multilayer polymer/ceramic nanocomposites under supersonic projectile impacts. Extrem. Mech. Lett. 2021, 44, 101238. [Google Scholar] [CrossRef]
- Xue, Q.; He, Y.; Zhang, X.; Zhang, X.; Cai, M.; Guo, C.F.; Yang, C. Strong Interfaces Enable Efficient Load Transfer for Strong, Tough, and Impact-Resistant Hydrogel Composites. ACS Appl. Mater. Interfaces 2022, 14, 33797–33805. [Google Scholar] [CrossRef]
- Fan, T.; Xue, S.-S.; Zhu, W.-B.; Zhang, Y.-Y.; Li, Y.-Q.; Chen, Z.-K.; Huang, P.; Fu, S.-Y. Multifunctional Polyurethane Composite Foam with Outstanding Anti-impact Capacity for Soft Body Armors. ACS Appl. Mater. Interfaces 2022, 14, 13778–13789. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, Y.; Lv, Z.; Wang, J.; Zhang, W.; Wu, Y. Research on impact resistance of ceramic matrix composites. Compos. Struct. 2021, 268, 113977. [Google Scholar] [CrossRef]
- Huang, W.; Shishehbor, M.; Guarín-Zapata, N.; Kirchhofer, N.D.; Li, J.; Cruz, L.; Wang, T.; Bhowmick, S.; Stauffer, D.; Manimunda, P.; et al. A natural impact-resistant bicontinuous composite nanoparticle coating. Nat. Mater. 2020, 19, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qin, B.; Xu, J.-F.; Zhang, X. Multi-recyclable Shape Memory Supramolecular Polyurea with Long Cycle Life and Superior Stability. ACS Mater. Lett. 2021, 3, 331–336. [Google Scholar] [CrossRef]
- Garcia, S.J. Effect of polymer architecture on the intrinsic self-healing character of polymers. Eur. Polym. J. 2014, 53, 118–125. [Google Scholar] [CrossRef]
- Dry, C. Passive Tuneable Fibers and Matrices. Int. J. Mod. Phys. B 1992, 6, 2763–2771. [Google Scholar] [CrossRef]
- de Gennes, P.G. Reptation of a Polymer Chain in the Presence of Fixed Obstacles. J. Chem. Phys. 2003, 55, 572–579. [Google Scholar] [CrossRef]
- Wool, R.P.; O’Connor, K.M. A theory crack healing in polymers. J. Appl. Phys. 1981, 52, 5953–5963. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, P.; Yu, Y.; Liu, J.; Su, X.; Kuan, H.-C.; Wang, B.; Zhang, L.; Zhang, Y.; Losic, D.; et al. Polyaspartic polyurea/graphene nanocomposites for multifunctionality: Self-healing, mechanical resilience, electrical and thermal conductivities, and resistance to corrosion and impact. Thin Walled Struct. 2023, 189, 110853. [Google Scholar] [CrossRef]
- Qian, Y.; Zhou, Y.; Li, L.; Liu, W.; Yang, D.; Qiu, X. Facile preparation of active lignin capsules for developing self-healing and UV-blocking polyurea coatings. Prog. Org. Coat. 2020, 138, 105354. [Google Scholar] [CrossRef]
- Zeng, Q.; Xue, S.; Li, J.; Jiang, W.; Ding, Y.; Shen, L. Preparation of bio-based air-drying water-borne polyurea coatings with excellent coating properties and anticorrosive performance. Prog. Org. Coat. 2022, 171, 107040. [Google Scholar] [CrossRef]
- Niesiobędzka, J.; Datta, J. Challenges and recent advances in bio-based isocyanate production. Green Chem. 2023, 25, 2482–2504. [Google Scholar] [CrossRef]
- Vencorex. Available online: https://www.vencorex.com/product/tolonate-x-flo-100/ (accessed on 15 July 2023).
- Covestro. Available online: https://solutions.covestro.com/en/products/desmodur/desmodur-eco-n-7300_84603813-18871289?SelectedCountry=PL (accessed on 20 October 2023).
- Mitsui Chemicals Inc. Available online: https://jp.mitsuichemicals.com/en/service/product/stabio.htm (accessed on 25 October 2023).
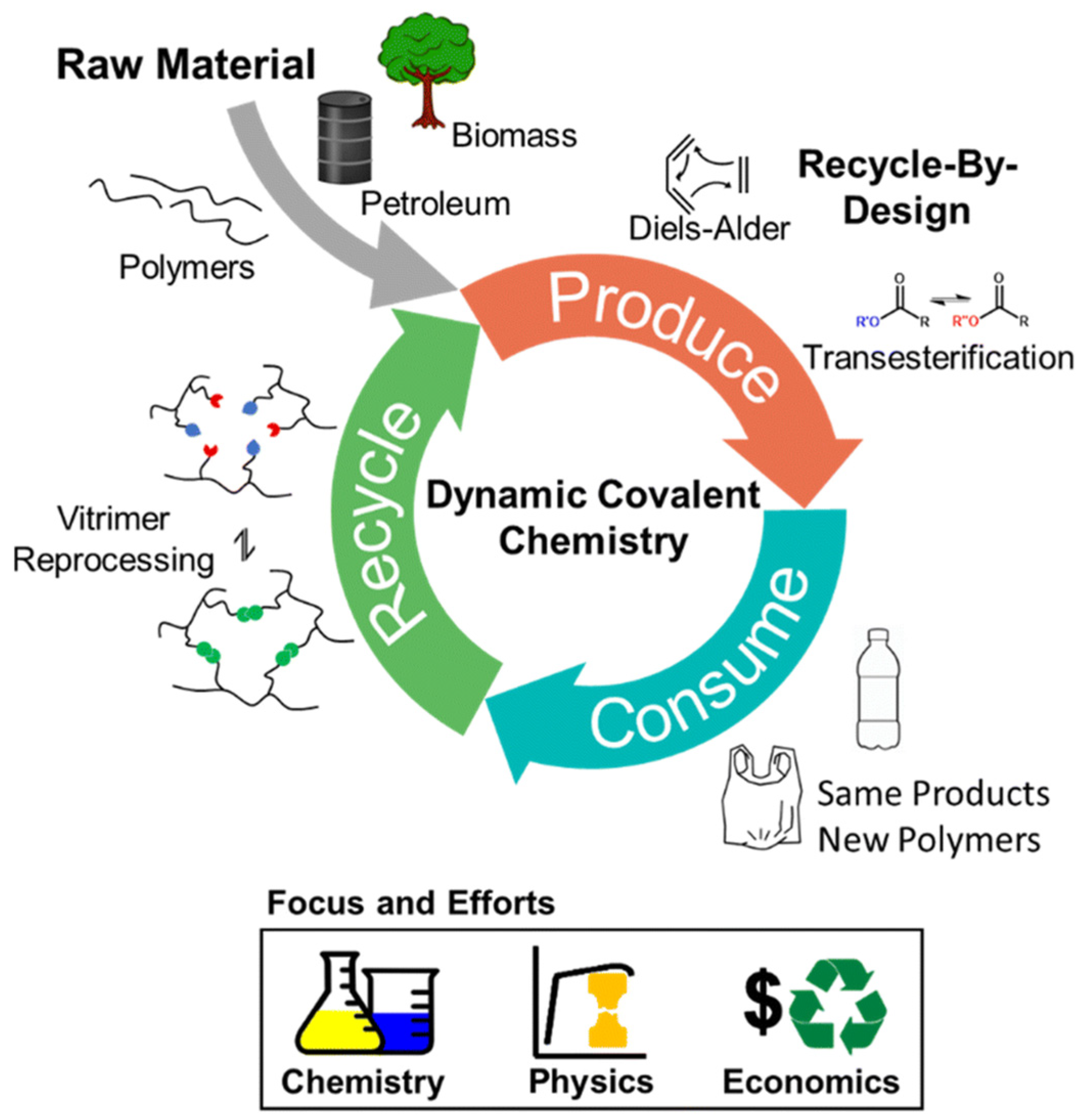
- Yan, T.; Balzer, A.H.; Herbert, K.M.; Epps, T.H.; Korley, L.T.J. Circularity in polymers: Addressing performance and sustainability challenges using dynamic covalent chemistries. Chem. Sci. 2023, 14, 5243–5265. [Google Scholar] [CrossRef]







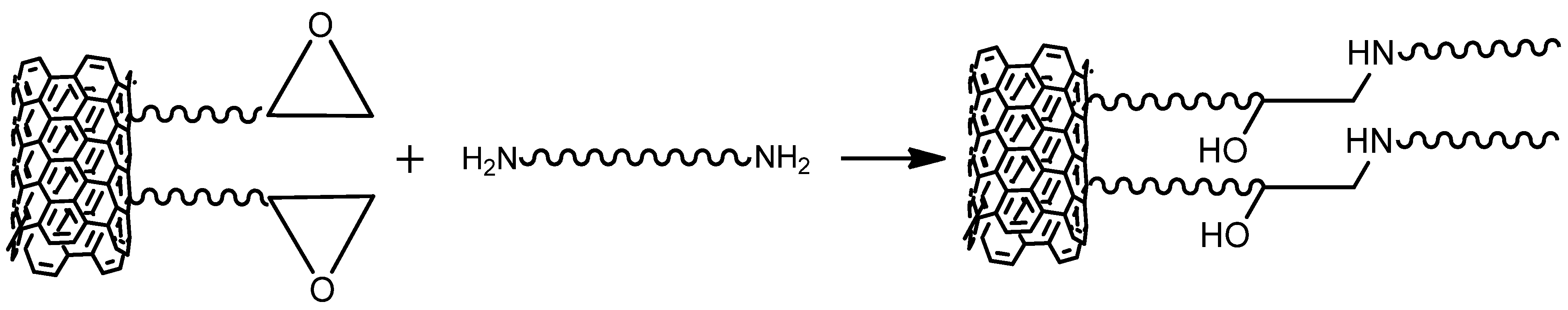
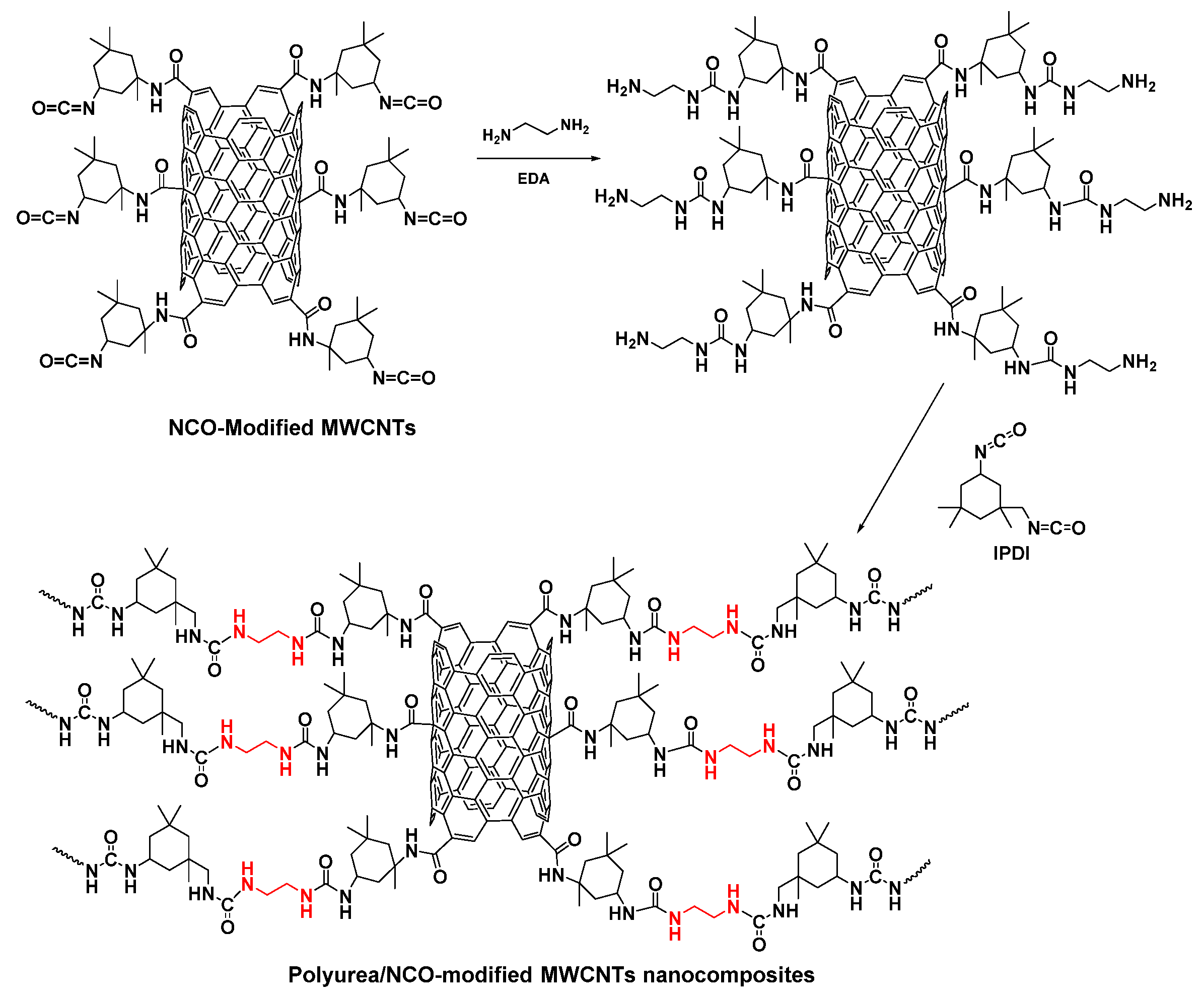
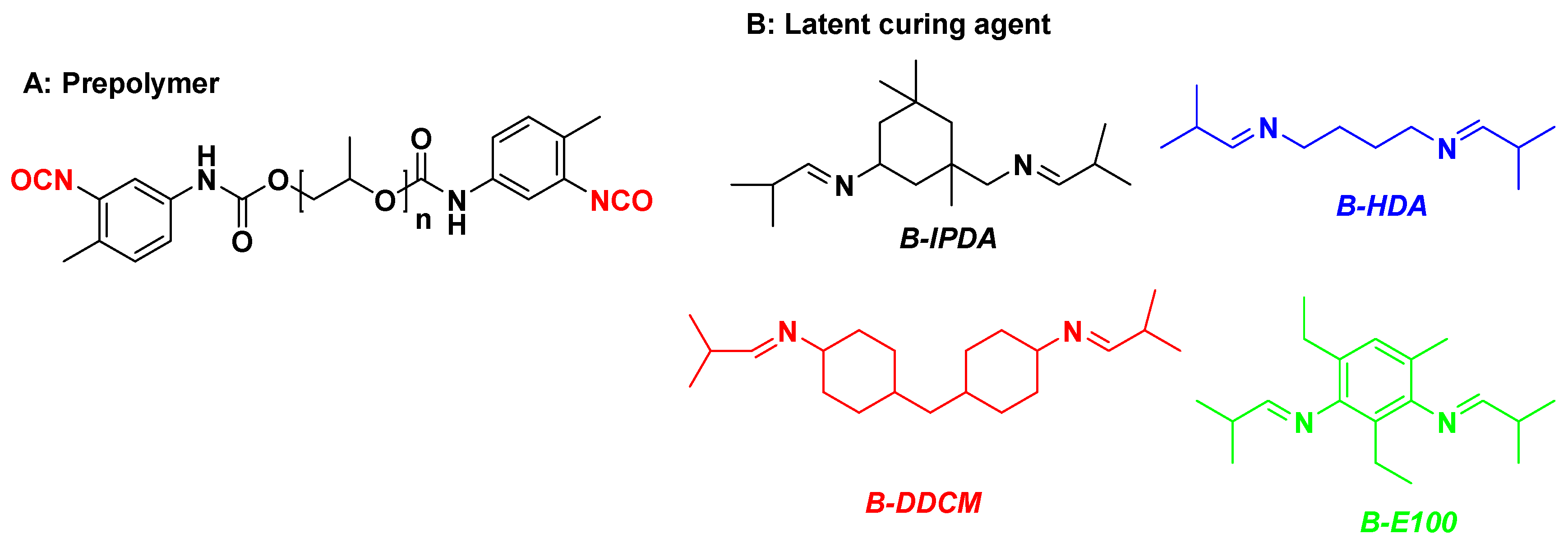

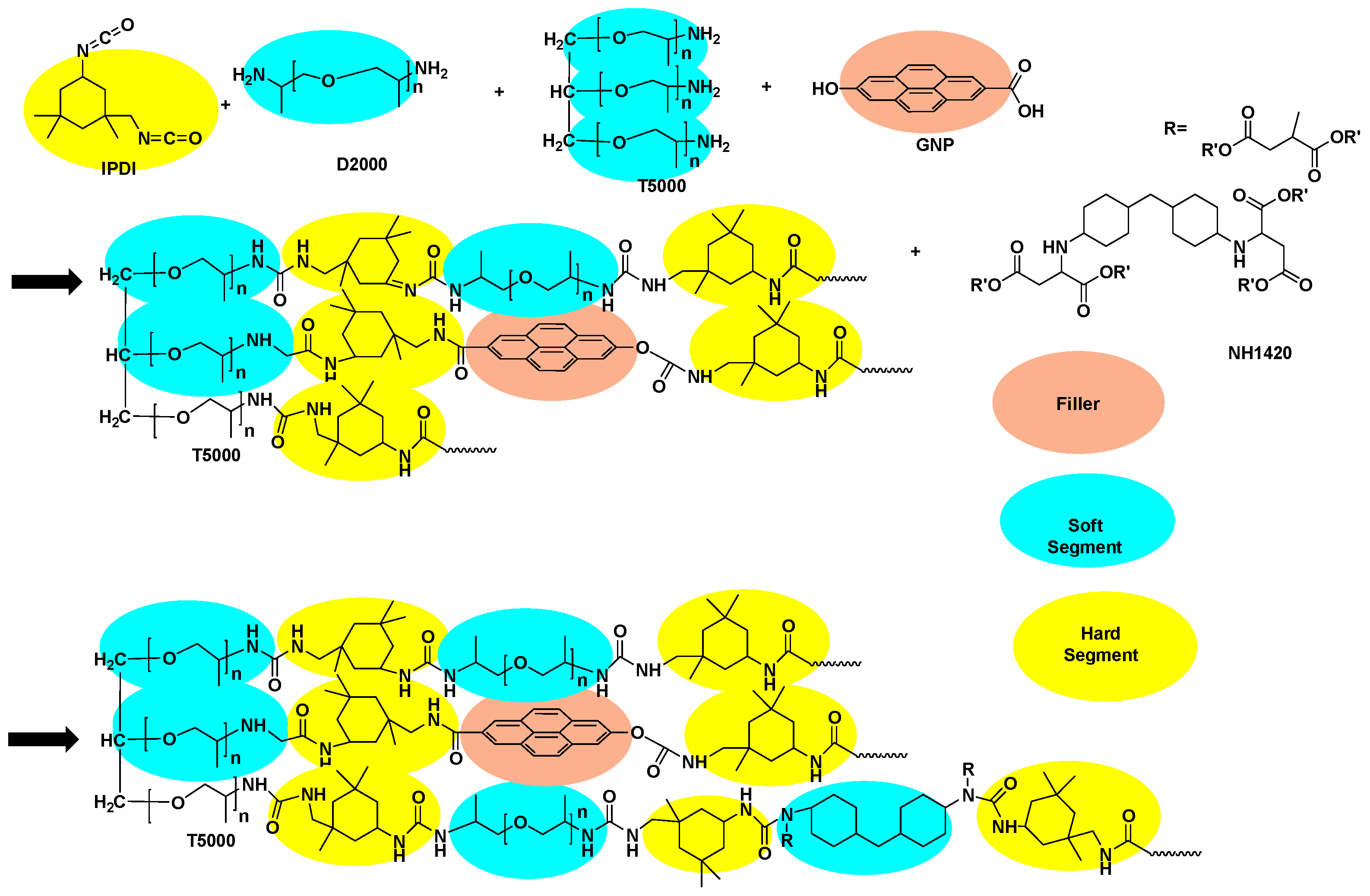


| Parameter | Value Range |
|---|---|
| Tensile strength, MPa | 7.5–27.5 |
| Shore hardness | A20–D65 |
| Elongation, % | 20–1000 |
| Modulus, MPa | 3.4–13.8 |
| Water absorption, % | 5–16 |
| Abrasion resistance, mg weight loss | 6–70 |
| Impact resistance, in kg | 25–90 |
| Isocyanate Type | Hard Segment Content (wt. %) | Tensile Strength (MPa) | Ultimate Elongation (%) |
|---|---|---|---|
| Isophorone diisocyanate (IPDI) | 16.8 | 7.15 | 1070 |
| Bis(4-isocyanatocyclohexyl)methane (HMDI) | 19.3 | 19.6 | 925 |
| 2,6- and 2,4-toluene diisocyanate (TDI) | 13.7 | 12.3 | 760 |
| p-diphenylmethane diisocyanate (MDI) | 18.5 | 16.9 | 670 |
| 1,3-phenylene diisocyanate (MPDI) | 12.7 | 5.4 | 700 |
| 1,4-phenylene diisocyanate (PPDI) | 12.7 | 19.5 | 540 |
| 1,6-hexamethylene diisocyanate (HDI) | 13.2 | 24.7 | 760 |
| 1,4-cyclohexyl diisocyanate (CHDI) | 13.1 | 30.0 | 980 |
| No. | Brand | Year | Mechanical Properties Details | Reference | |||
|---|---|---|---|---|---|---|---|
| Tensile Strength (MPa) | Elongation (%) | Tear Strength (kN/m) | Shore Hardness | ||||
| 1 | EP JS | 2008 | 20.34 | 350 | 87.5 | [61] | |
| 2 | Dragon Shield-BC | 2014 | 11.75 | 50 | - | - | [62] |
| 3 | HM-VK | 2016 | 10 | 500 | - | - | [63] |
| 4 | Link-XS350 | 2019 | 22.39 | 163 | - | 60 ± 1 HD | [64,65] |
| 5 | SWD562 | 2019 | 16.5 | 160 | - | - | [66] |
| 6 | SPUA 306 | 2019 | 24 | 400 | 85 | 85–95 HA | [14] |
| 7 | SPUA 307 | 2019 | 25 | 45 | 81 | 65–75 HD | [14] |
| 8 | AMMT-53 | 2020 | 25 | 50 | 90 | 70 HD | [28,67] |
| 9 | AMMT-55 | 2020 | 35 | 300 | 115 | 65 HD | [28,67] |
| 10 | AP103 | 2021 | 16 | 450 | - | - | [68] |
| 11 | - | 2021 | 18 | 350 | 45 | 90–96 HA | [22] |
| 12 | Qtech T26 | 2022 | 25.4 | 451.88 | 75.5 | - | [69] |
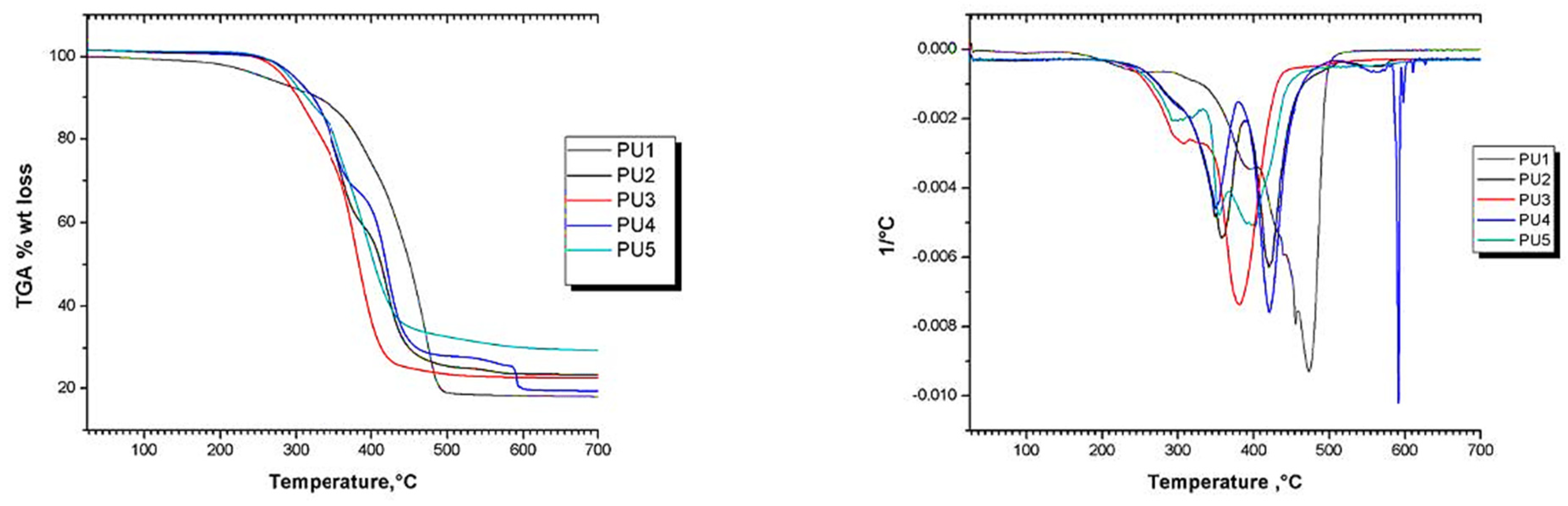
| Sample | Decomposition Temperature, °C | ||
|---|---|---|---|
| Tinitial | T50% | Tend | |
| PU-1 | 141 | 452 | 517 |
| PU-2 | 155 | 387 | 642 |
| PU-3 | 176 | 374 | 446 |
| PU-4 | 164 | 409 | 700 |
| PU-5 | 177 | 390 | 700 |
| Pressure (Bar) | Aluminum Plate (Uncoated) | Aluminum Plate (Coated with PU) | Aluminum Plate (Coated with PU-NC2) | Aluminum Plate (Coated with PU-NC3) | Aluminum Plate (Coated with PU-NC4) |
|---|---|---|---|---|---|
| Maximum Force (kN) | Maximum Force (kN) | Maximum Force (kN) | Maximum Force (kN) | Maximum Force (kN) | |
| 0.2 | 2.0 | 2.0 | 2.1 | - | - |
| 0.3 | 2.6 | 3.0 | 2.8 | 2.3 | 3.0 |
| 0.4 | 2.5 | 3.6 | 3.8 | 3.8 | 4.0 |
| 0.5 | 1.8 | 4.1 | 4.3 | 5.0 | 4.4 |
| 0.6 | - | 3.6 | 3.9 | 4.1 | 4.8 |
| 0.7 | - | - | - | - | 4.0 |
| Component | Role | Equivalent Number neq | Viscosity η30 °C (MPa s−1) | Density ρ (g cm−3) | Structure |
|---|---|---|---|---|---|
| SUPRASEC 2054 | Major monomer | 3.6 | 880 | 1.09 | 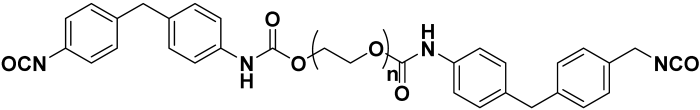 |
| Propylene carbonate | Diluent | — | 2 | 1.20 |  |
| Ethylene diamine | Chain extender | 33.3 | 1.5 × 10−3 | 0.90 | 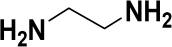 |
| JEFFAMINE D-230 | Chain extender | 8.1–8.7 | 7 | 0.93 | 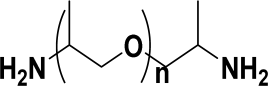 n ≈ 2.5 n ≈ 2.5 |
| JEFFAMINE D-400 | Chain extender | 4.1–4.7 | 21 | 0.96 | n ≈ 6.1 |
| JEFFAMINE D-2000 | Major monomer | 0.9–1.0 | 182 | 0.98 | n ≈ 33 |
| DETDA | Chain extender | 11.2 | 106 | 1.02 | 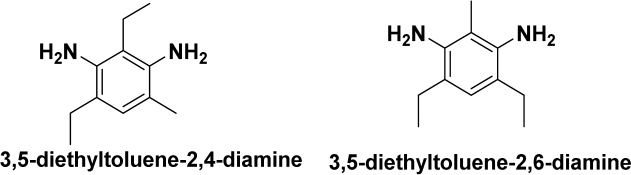 |
| Sample Code | Amine (Part per Unit Volume) | HS (%) | Contribution of Aromatic Chain Extender (%) | ||||
|---|---|---|---|---|---|---|---|
| Chain Extender | D-2000 | ||||||
| EDA | D-230 | D-400 | DEDTA | ||||
| PUAr-76 | — | — | — | 21.22 | 78.78 | 60.86 | 76 |
| PU230Ar-63 | — | 5.48 | — | 17.52 | 77.00 | 61.65 | 63 |
| PU230Ar-36 | — | 16.41 | — | 11.05 | 72.54 | 63.72 | 36 |
| PU230Ar-10 | — | 27.35 | — | 2.68 | 69.97 | 64.84 | 10 |
| PU230Ar-0 | — | 31.30 | — | — | 68.70 | 65.43 | — |
| PU400Ar-62 | — | — | 10.94 | 17.25 | 71.81 | 64.25 | 62 |
| PU400Ar-47 | — | — | 21.88 | 13.28 | 64.84 | 66.65 | 47 |
| PU400Ar-33 | — | — | 32.82 | 9.3 | 57.88 | 71.06 | 33 |
| PU400Ar-19 | — | — | 43.76 | 5.53 | 50.91 | 74.49 | 19 |
| PU400Ar-0 | — | — | 58.42 | — | 41.58 | 79.1 | — |
| PUEDAr-32 a | 5.65 | — | — | 9.85 | 84.50 | 61.74 | 32 |
| PUEDAr-0 a | 8.65 | — | — | — | 91.35 | 54 | — |
| Coating | Tg (°C) | Peak G″ (Mpa) | V-50 a |
|---|---|---|---|
| Neat | −68.7 | 62 | 1.3 |
| 5% Nanoclay | −68.7 | 67 | 1.29 |
| 2% POSS | −63.9 | 76 | 1.34 |
| 1% MWCNT | −63.2 | 62 | 1.30 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toader, G.; Diacon, A.; Axinte, S.M.; Mocanu, A.; Rusen, E. State-of-the-Art Polyurea Coatings: Synthesis Aspects, Structure–Properties Relationship, and Nanocomposites for Ballistic Protection Applications. Polymers 2024, 16, 454. https://doi.org/10.3390/polym16040454
Toader G, Diacon A, Axinte SM, Mocanu A, Rusen E. State-of-the-Art Polyurea Coatings: Synthesis Aspects, Structure–Properties Relationship, and Nanocomposites for Ballistic Protection Applications. Polymers. 2024; 16(4):454. https://doi.org/10.3390/polym16040454
Chicago/Turabian StyleToader, Gabriela, Aurel Diacon, Sorin Mircea Axinte, Alexandra Mocanu, and Edina Rusen. 2024. "State-of-the-Art Polyurea Coatings: Synthesis Aspects, Structure–Properties Relationship, and Nanocomposites for Ballistic Protection Applications" Polymers 16, no. 4: 454. https://doi.org/10.3390/polym16040454
APA StyleToader, G., Diacon, A., Axinte, S. M., Mocanu, A., & Rusen, E. (2024). State-of-the-Art Polyurea Coatings: Synthesis Aspects, Structure–Properties Relationship, and Nanocomposites for Ballistic Protection Applications. Polymers, 16(4), 454. https://doi.org/10.3390/polym16040454