
Synthesis and Characterization of DOPO-Containing Poly(2,6-dimethyl-1,4-phenylene oxide)s by Oxidative Coupling Polymerization


Abstract
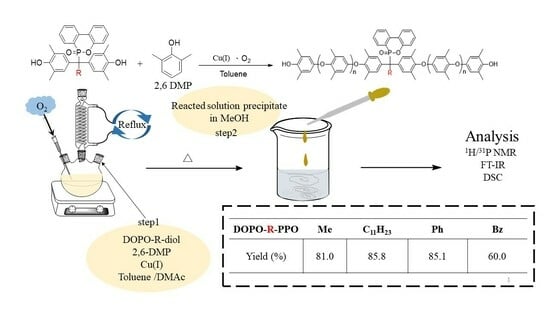
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
2.2.1. Synthesis of 3,5-Dimethyl-4-hydroxylacetophenone (DMP−Keto−Me)
2.2.2. Synthesis of 3,5-Dimethyl-4-hydroxylbenzophenone (DMP−Keto−Ph)
2.2.3. Synthesis of 1-(4-Hydroxy-3,5-dimethylphenyl) dodecan-1-one (DMP−Keto−C11)
2.2.4. Synthesis of Benzyl 3,5-Dimethyl-4-hydroxyl ketone (DMP−Keto−Bz)
2.2.5. Synthesis of 6-(1,1-bis(4-Hydroxy-3,5-dimethylphenyl) ethyl) dibenzo[c,e][1,2] oxa phosphinine 6-oxide) (DOPO−Me−diol)
2.2.6. Synthesis of 6-(1,1-bis(4-Hydroxy-3,5-dimethylphenyl) dodecyl) dibenzo[c,e][1,2] oxaphosphinine 6-oxide (DOPO−C11−diol)
2.2.7. Synthesis of 6-(bis(4-Hydroxy-3,5- dimethylphenyl)(phenyl)methyl)dibenzo[c,e][1,2] oxaphosphinine 6-oxide (DOPO−Ph−diol)
2.2.8. Synthesis of 6-(1,1-bis(4-Hydroxy-3,5-dimethylphenyl)-2- phnylethyl) dibenzo[c,e][1,2]oxaphosphinine 6-oxide (DOPO−Bz−diol)
2.2.9. Synthesis of DOPO−Me−Incorporated Poly(2,6-dimethyl-1,4-phenylene oxide) (DOPO−Me−PPO)
2.2.10. Synthesis of DOPO−C11−Incorporated Poly(2,6-dimethyl-1,4-phenylene oxide) (DOPO−C11−PPO)
2.2.11. Synthesis of DOPO−Ph−Incorporated Poly(2,6-dimethyl-1,4-phenylene oxide) (DOPO−Ph−PPO)
2.2.12. Synthesis of DOPO−Bz−Incorporated Poly(2,6-dimethyl-1,4-henylene oxide) (DOPO−Bz−PPO)
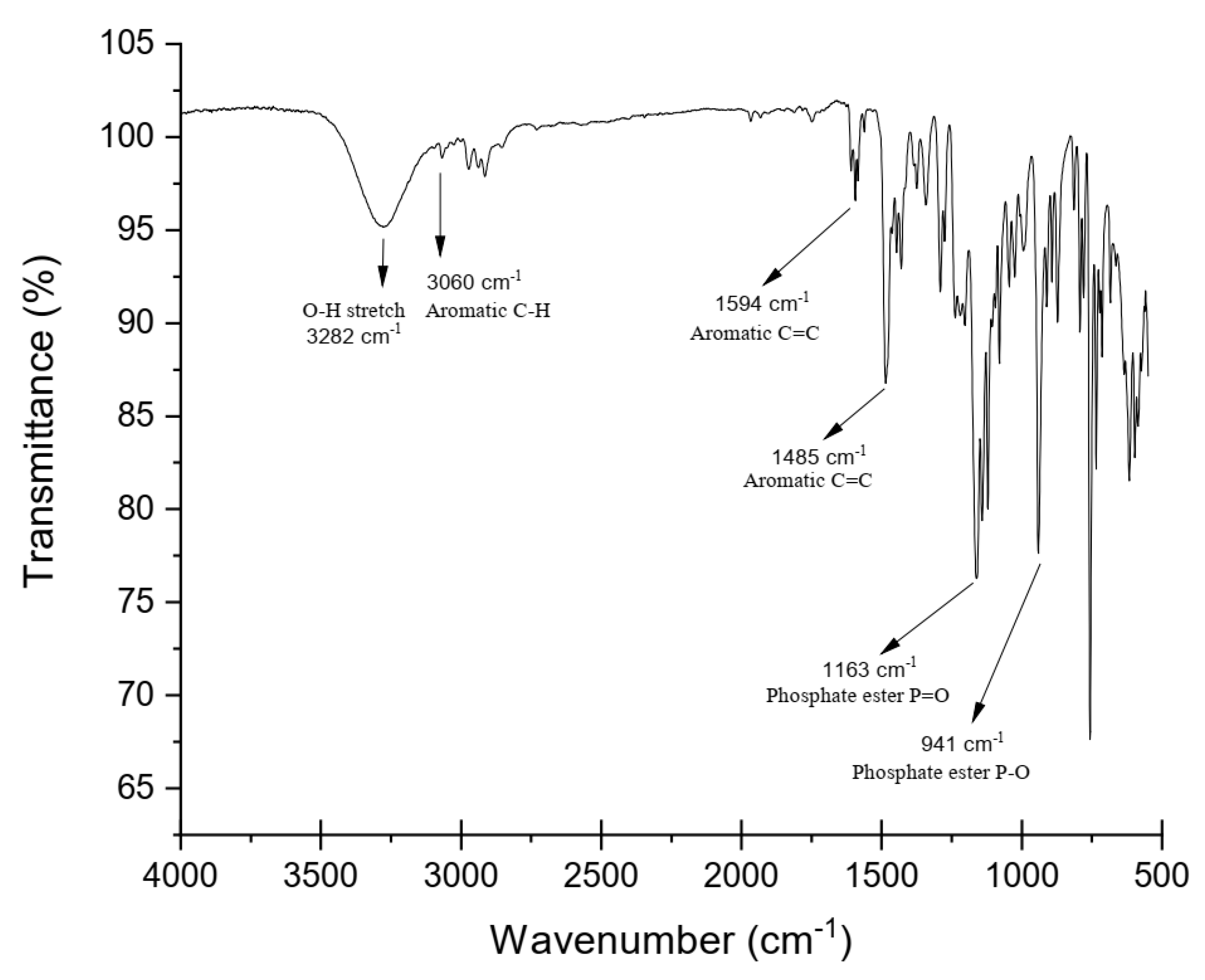
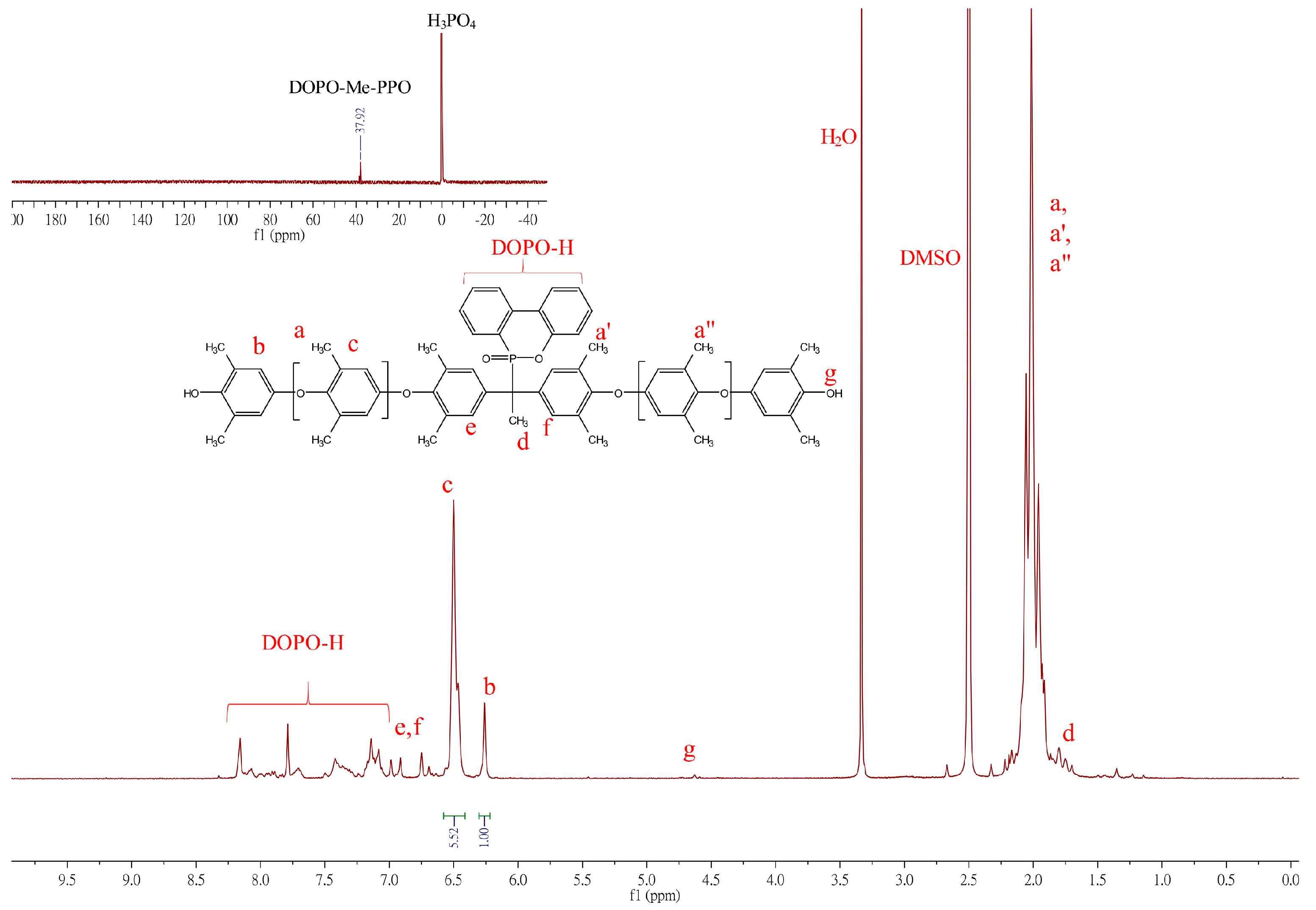
2.3. Equipment and Characterization
3. Results and Discussions
3.1. Preparation of DOPO-Incorporated Polyphenylene Oxides
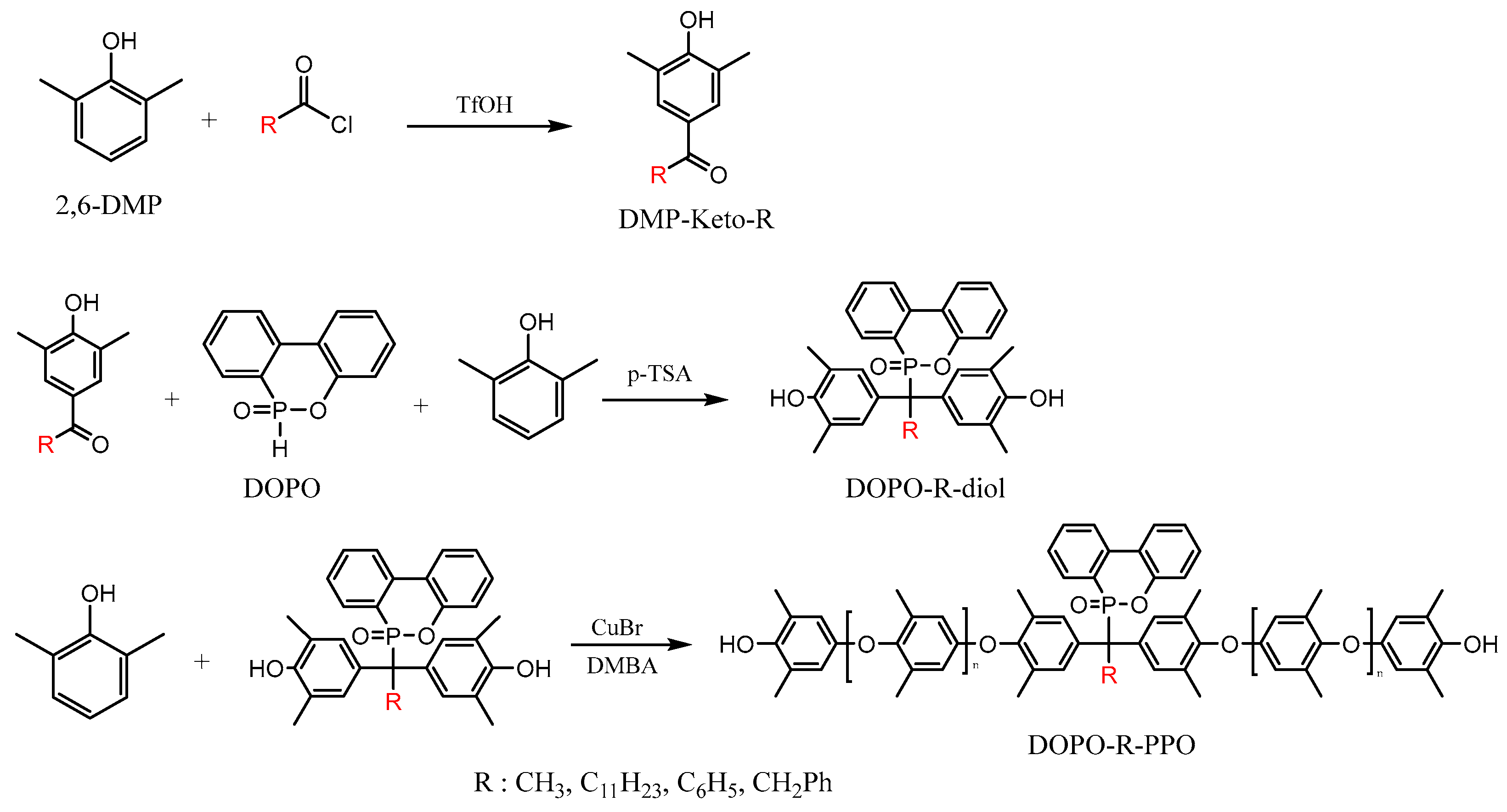
3.1.1. Synthesis of DMP−Keto−R
3.1.2. Synthesis of DOPO−R−diol
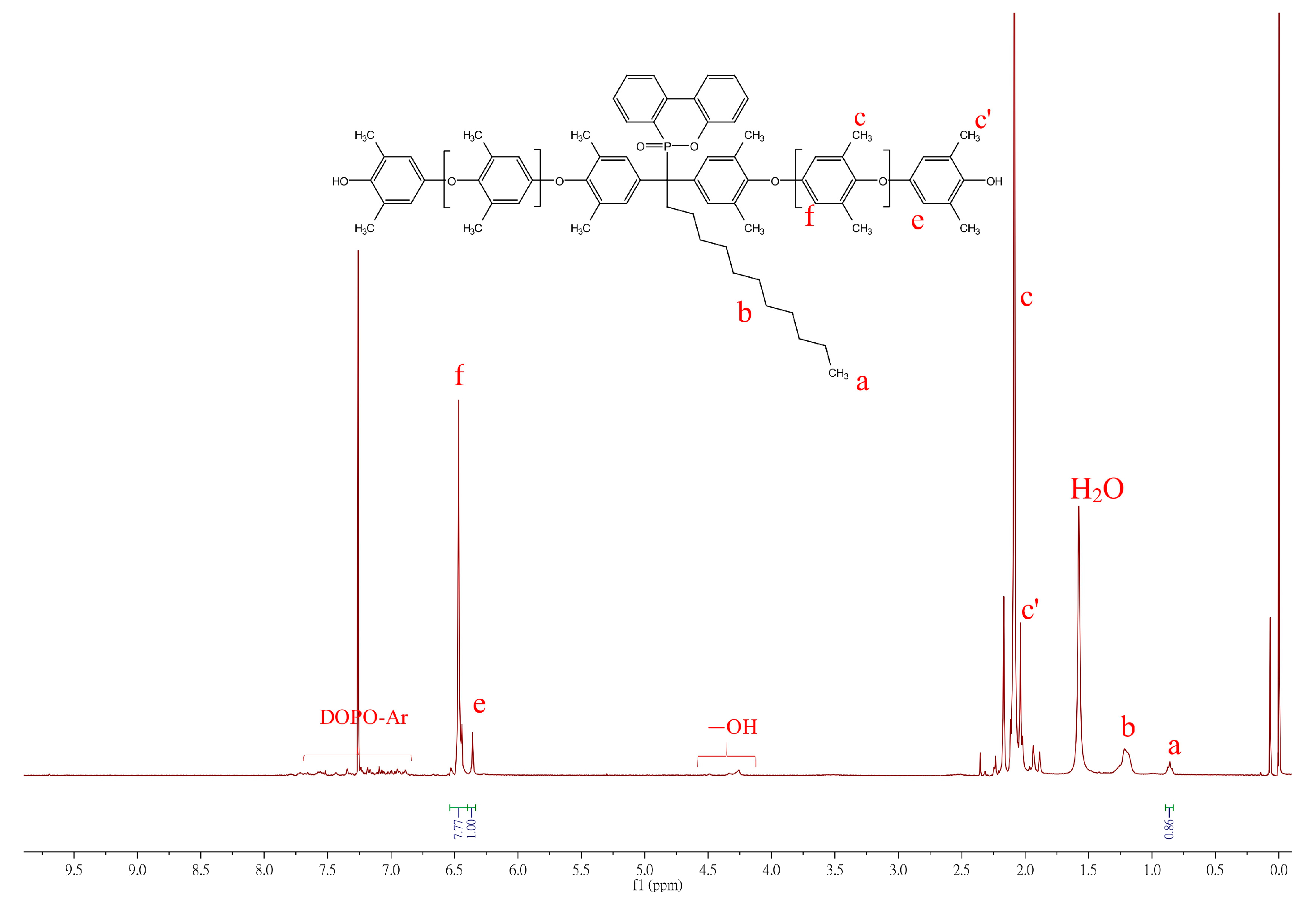
3.1.3. Synthesis of DOPO−R−PPO
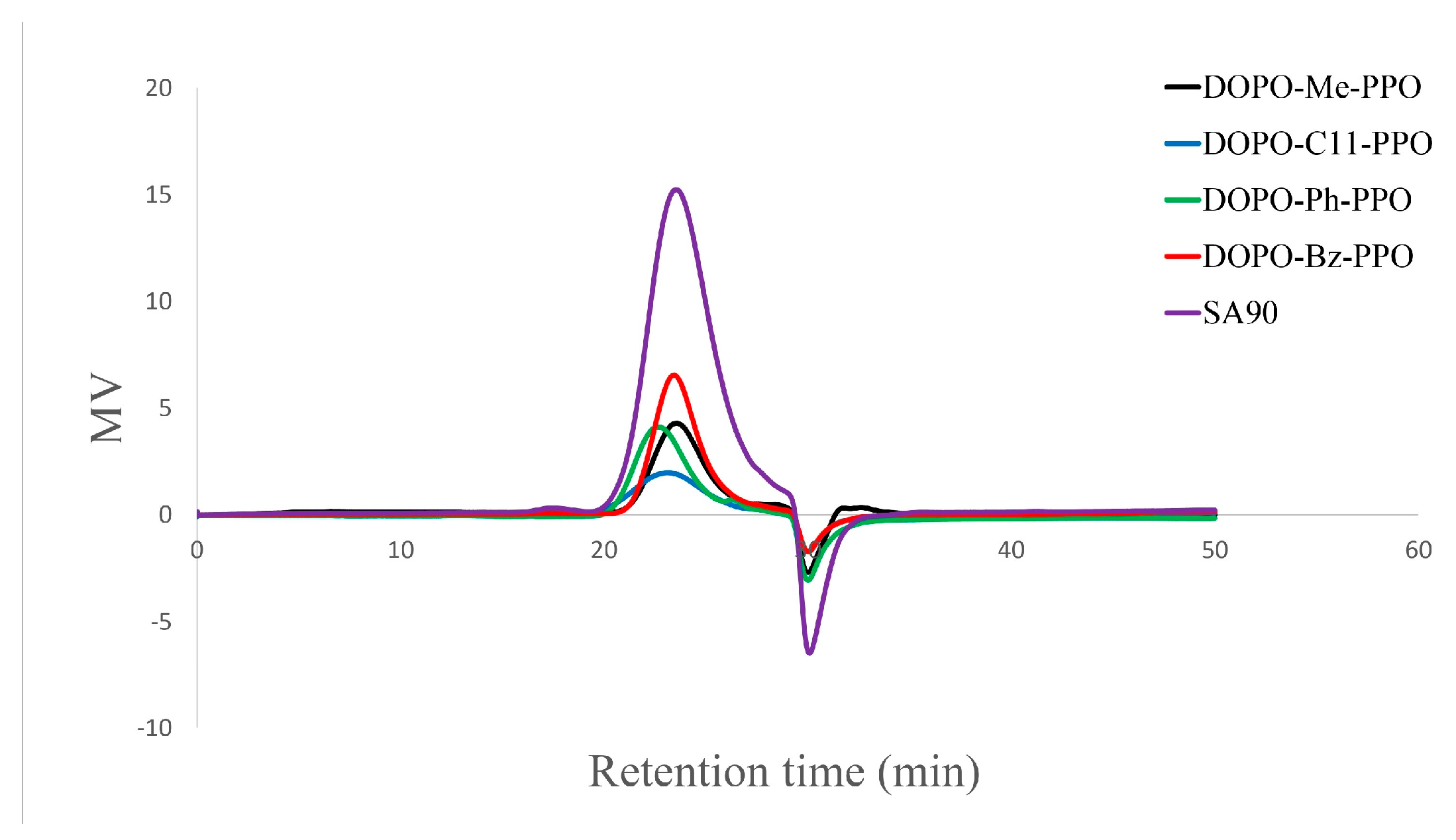
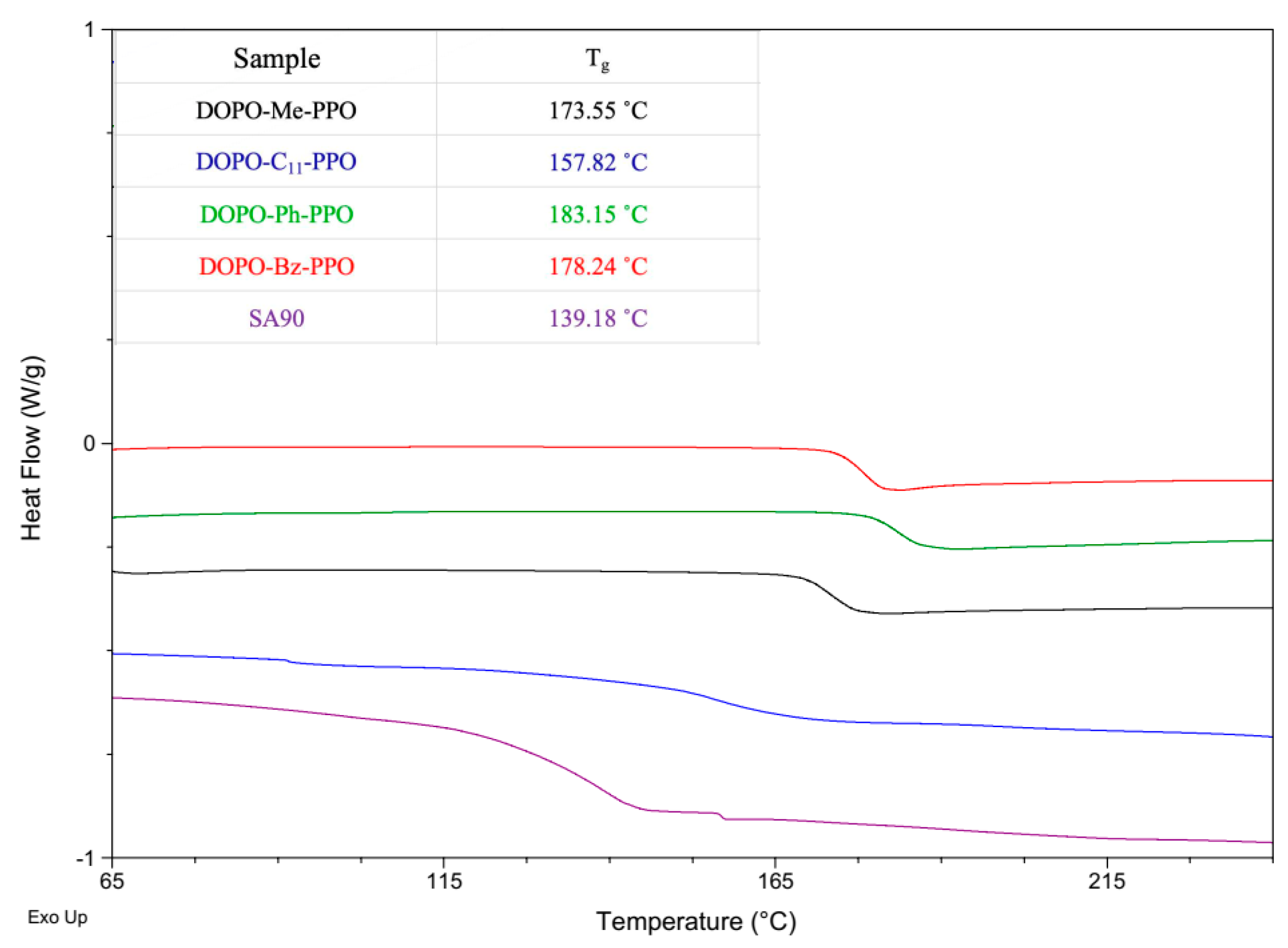
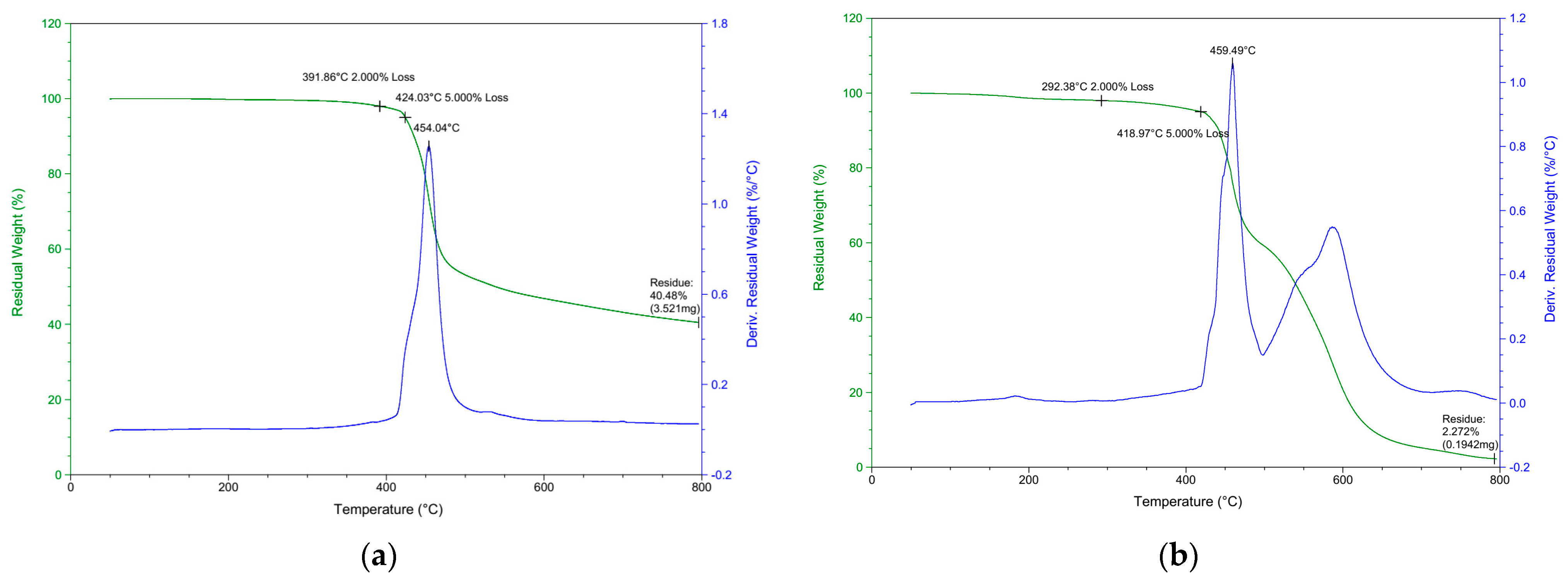
3.2. Thermal Properties of DOPO−R−PPO
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aycock, D.; Abolins, V.; White, D. Encyclopedia of Polymer Science and Engineering, 2nd ed.; Wiley-Interscience: New York, NY, USA, 1986; Volume 13, pp. 1–30. [Google Scholar]
- Lu, S.Y.; Hamerton, I. Recent developments in the chemistry of halogen-free flame retardant polymers. Prog. Polym. Sci. 2002, 27, 1661–1712. [Google Scholar] [CrossRef]
- Rakotomalala, M.; Wagner, S.; Doring, M. Recent developments in halogen free flame retardants for epoxy resins for electrical and electronic applications. Materials 2010, 3, 4300–4327. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Valle, R.C.S.C.; Valle, J.A.B.; Maestá, F.B.; Meng, X.; Arias, M.J.L. Recent Progress of DOPO-Containing Compounds as Flame Retardants for Versatile Polymeric Materials: Review. World J. Text. Eng. Technol. 2020, 6, 89–103. [Google Scholar]
- He, X.; Zhang, W.; Yang, R. The characterization of DOPO/MMT nanocompound and its effect on flame retardancy of epoxy resin. Compos. Part A Appl. Sci. Manuf. 2017, 98, 124–135. [Google Scholar] [CrossRef]
- Zhang, W.; He, X.; Song, T.; Jiao, Q.; Yang, R. Comparison of intumescence mechanism and blowing-out effect in flame-retarded epoxy resins. Polym. Degrad. Stab. 2015, 112, 43–51. [Google Scholar] [CrossRef]
- Varganici, C.; Rosu, L.; Bifulco, A.; Rosu, D.; Mustata, F.; Gaan, S. Recent advances in flame retardant epoxy systems from reactive DOPO–based phosphorus additives. Polym. Degrad. Stab. 2022, 202, 110020. [Google Scholar] [CrossRef]
- Qian, X.; Liu, Q.; Zhang, L.; Li, H.; Liu, J.; Yan, S. Synthesis of reactive DOPO-based flame retardant and its application in rigid polyisocyanurate-polyurethane foam. Polym. Degrad. Stab. 2022, 197, 109852. [Google Scholar] [CrossRef]
- König, A.; Kroke, E. Methyl-DOPO—A new flame retardant for flexible polyurethane foam. Polym. Adv. Technol. 2011, 22, 5–13. [Google Scholar] [CrossRef]
- Gaan, S.; Liang, S.; Mispreuve, H.; Perler, H.; Naescher, R.; Neisius, M. Flame retardant flexible polyurethane foams from novel DOPOphosphonamidate additives. Polym. Degrad. Stab. 2015, 113, 180–188. [Google Scholar] [CrossRef]
- Wang, H.; Wang, S.; Du, X.; Wang, H.; Cheng, X.; Du, Z. Synthesis of a novel flame retardant based on DOPO derivatives and its application in waterborne polyurethane. RSC Adv. 2019, 9, 7411–7419. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Q.; Zhao, X.; Jin, Z. Synthesis of reactive DOPO-based flame retardant and its application in polyurethane elastomers. Polym. Degrad. Stab. 2021, 183, 109440. [Google Scholar] [CrossRef]
- Ma, S.; Xiao, Y.; Zhou, F.; Schartel, B.; Chan, Y.Y.; Korobeinichev, O.P.; Trubachev, S.A.; Hu, W.; Ma, C.; Hu, Y. Effects of novel phosphorus-nitrogen-containing DOPO derivative salts on mechanical properties, thermal stability and flame retardancy of flexible polyurethane foam. Polym. Degrad. Stab. 2020, 177, 109160. [Google Scholar] [CrossRef]
- Zhang, W.; Li, X.; Guo, X.; Yang, R. Mechanical and thermal properties and flame retardancy of phosphoruscontaining polyhedral oligomeric silsesquioxane (DOPO-POSS)/polycarbonate composites. Polym. Degrad. Stab. 2010, 95, 2541–2546. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, L.; Zhao, B.; Luo, Y.; Wang, D.Y.; Wang, Y.Z. A novel efficient halogen-free flame retardant system for polycarbonate. Polym. Degrad. Stab. 2011, 96, 320–327. [Google Scholar] [CrossRef]
- Ni, P.; Fang, Y.; Qian, L.; Qiu, Y. Flame-retardant behavior of a phosphorus/silicon compound on polycarbonate. J. Appl. Polym. Sci. 2018, 135, 45815–45823. [Google Scholar] [CrossRef]
- Liu, C.; Yao, Q. Design and synthesis of efficient phosphorus flame retardant for polycarbonate. Ind. Eng. Chem. Res. 2017, 56, 8789–8796. [Google Scholar] [CrossRef]
- Wang, Q.Z.; Liu, C.; Xu, Y.J.; Liu, Y.; Zhu, P.; Wang, Y.Z. Highly efficient flame retardation of polyester fabrics via novel DOPO-modified sol-gel coatings. Polymer 2021, 226, 123761–123769. [Google Scholar] [CrossRef]
- Haubold, T.S.; Hartwig, A.; Koschek, K. Synthesis and application studies of DOPO-based organophosphorous derivatives to modify the thermal behavior of polybenzoxazine. Polymers 2022, 14, 606. [Google Scholar] [CrossRef]
- Vasiljevic, J.; Colovi’c, M.; Korošin, N.C.; Šobak, M.; Štirn, Z.; Jerman, I. Effect of different flame-retardant bridged DOPO derivatives on properties of in situ produced fiber-forming polyamide 6. Polymers 2020, 12, 675. [Google Scholar] [CrossRef]
- Hwang, H.J.; Hsu, S.W.; Wang, C.S. Synthesis and Physical Properties of Low-Molecular-Weight Redistributed Poly (2,6-dimethyl-1,4-phenylene oxide) for Epoxy Resin. J. Vinyl Addit. Technol. 2009, 15, 54–59. [Google Scholar] [CrossRef]
- Lin, C.H.; Tsai, Y.J.; Shih, Y.S.; Chang, H.C. Catalyst-free synthesis of phosphinated poly(2,6-dimethyl-1,4-phenylene oxide) with high-Tg and low-dielectric characteristic. Polym. Degrad. Stab. 2014, 99, 105–110. [Google Scholar] [CrossRef]
- Reddy, K.S.K.; Chen, Y.C.; Lin, Y.A.; Shih, Y.L.; Wang, M.W.; Lin, C.H. Synthesis of high-Tg, flame-retardant, and low-dissipation telechelic oligo (2,6-dimethylphenylene ether) resins for high-frequency communications. ACS Appl. Polym. Mater. 2022, 4, 3225–3235. [Google Scholar] [CrossRef]
- Lin, Y.C.; Feng, H.C.; Yang, M.S.; Yu, H.A.; Huang, C.C.; Liang, M. Synthesis, characterization and thermal properties of functionalized poly(2,6-dimethyl-1,4-phenylene oxide)s containing ethylenic, aldehydic, hydroxyl and acrylate. Polym. Int. 2012, 61, 719–726. [Google Scholar] [CrossRef]
- Su, C.T.; Lin, K.U.; Lee, T.J.; Liang, M. Preparation, characterization and curing properties of epoxy-terminated poly (2.6-dimethyl-1,4 phenylene oxide)s. Eur. Polym. J. 2010, 46, 1488–1497. [Google Scholar] [CrossRef]
- Lee, T.J.; Fang, Y.D.; Yuan, W.G.; Wei, K.M.; Liang, M. Synthesis, structures and thermal properties of new class epoxide-terminated telechelic poly(2,6-dimethyl-1,4-phenylene oxide)s. Polymer 2007, 48, 734–742. [Google Scholar] [CrossRef]
- Huang, C.C.; Yang, M.S.; Liang, M. Synthesis of new thermosetting poly(2,6-dimethyl-1,4-phenylene oxide) containing epoxide pendant groups. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 5875–5886. [Google Scholar] [CrossRef]
- Murashige, R.; Hayashi, Y.; Ohmori, S.; Torii, A.; Aizu, Y.; Muto, Y.; Murai, Y.; Oda, Y.; Hashimoto, M. Comparisons of O-acylation and Friedele-Crafts acylation of phenols and acyl chlorides and Fries rearrangement of phenyl esters in trifluoromethanesulfonic acid: Effective synthesis of optically active homotyrosines. Tetrahedron 2011, 67, 641–649. [Google Scholar] [CrossRef]
- Naeimi, H.; Moradi, L. Efficient and mild synthesis of ortho-hydroxyaryl ketones catalyzed by zinc chloride under solvent-free condition and microwave irradiation. Catal. Commun. 2006, 7, 1067–1071. [Google Scholar] [CrossRef]
- Liu, Y.L. Phosphorous-Containing Epoxy Resins from a Novel Synthesis Route. J. Appl. Polym. Sci. 2002, 83, 1697–1701. [Google Scholar] [CrossRef]
- Lin, C.H.; Wang, M.W.; Chang, S.L.; Wei, T.P.; Ding, S.H.; Su, W.C. Facile, one-pot synthesis of phosphinate-substituted bisphenol A and its alkaline-stable diglycidyl ether derivative. Polym. Degrad. Stab. 2010, 95, 1167–1176. [Google Scholar] [CrossRef]
- White, D.M.; Nye, S.A. 13C NMR study of poly(2,6-dimethyl-1,4-phenylene oxide)s. Sites of amine incorporation. Macromolecules 1990, 23, 1318–1329. [Google Scholar] [CrossRef]
- Hay, A.S. Polymerization by oxidative coupling: Discovery and commercialization of PPO and Noryl resins. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 505–517. [Google Scholar] [CrossRef]
- Wu, C.S.; Liu, Y.L.; Chiu, Y.C.; Chiu, Y.S. Thermal stability of epoxy resins containing flame retardant. components: An evaluation with thermogravimetric analysis. Polym. Degrad. Stab. 2002, 78, 41–48. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | DMP−Keto−R | Acid:Monomer | Temp | Time | Yield |
|---|---|---|---|---|---|
| R | Mole Ratio | °C | h | % | |
| 1 | Me | 2:1 | 25 | 1.5 | 73.5 |
| 2 | Me | 2:1 | 50 | 1.5 | 82.6 |
| 3 | Me | 2:1 | 50 | 2 | 85.3 |
| 4 | C11 | 2:1 | 25 | 2 | 60.6 |
| 5 | C11 | 2:1 | 50 | 2 | 90.8 |
| 6 | Ph | 2:1 | 25 | 2 | 50.5 |
| 7 | Ph | 2:1 | 50 | 2 | 63.3 |
| 8 | Ph | 2:1 | 50 | 4 | 83.4 |
| 9 | Bz | 2:1 | 25 | 1 | 44.1 |
| 10 | Bz | 2:1 | 50 | 1 | 68.5 |
| 11 | Bz | 2:1 | 50 | 2 | 76.2 |
| Entry | R | Precipitated Solvent | Acid | Time | Tm | Yield |
|---|---|---|---|---|---|---|
| h | °C | % | ||||
| 12 | Me | EtOH | p-TSA | 17 | 285 | 94 |
| 13 | C11 | Hexane | p-TSA | 18 | 156 | 75 |
| 14 | Ph | Ether | p-TSA | 12 | 277 | 73 |
| 15 | Bz | Hexane | p-TSA | 16 | 209 | 75 |
| DOPO−R−PPO | Toluene:DMAc | Cu:Monomer | Cu:Base | Conc. | Temp | Time | Mn | Mw | Đ | DOPO-R:2,6-DMP | Mn | Tg |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vol. | mol | mol | M | °C | h | GPC | GPC | mol | H-NMR | °C | ||
| Me | 4:1 | 0.5:100 | 1:30 | 2 | 30 | 6 | 2288 | 3157 | 1.37 | 1:16.3 | 2472 | 173.6 |
| C11 | 1:0 | 1.5:100 | 1:15 | 0.5 | 50 | 8 | 2822 | 4970 | 1.76 | 1:15.3 | 2493 | 157.8 |
| Ph | 4:1 | 0.5:100 | 1:75 | 2 | 50 | 2.5 | 3561 | 5191 | 1.45 | 1:19.0 | 2858 | 183.2 |
| Bz | 4:1 | 0.5:100 | 1:75 | 2 | 50 | 2.5 | 2386 | 3243 | 1.35 | 1:13.9 | 2245 | 178.2 |
| SA-90 | - | - | - | - | - | - | 2288 | 3582 | 1.56 | - | - | 139.2 |
| Sample Name | Td2% (°C) | Td5% (°C) | Tdmax (°C) | Char % | Char % * | Char % | |||
|---|---|---|---|---|---|---|---|---|---|
| N2 | Air | N2 | Air | N2 | Air | N2 | Air | Air | |
| DOPO−Me−PPO | 391.9 | 292.4 | 424.0 | 418.9 | 454.0 | 459.5 | 40.48 | 20.72 | 2.72 |
| DOPO−C11−PPO | 256.8 | 239.3 | 346.6 | 407.5 | 459.4 | 461.0 | 34.21 | 14.2 | 4.94 |
| DOPO−Ph−PPO | 323.7 | 356.1 | 394.5 | 404.8 | 454.3 | 556.1 | 28.66 | 15.21 | 2.47 |
| DOPO−Bz−PPO | 357.4 | 261.3 | 409.4 | 413.9 | 452.9 | 453.6 | 33.62 | 14.13 | 2.68 |
| SA-90 | 302.2 | 291.3 | 364.0 | 407.7 | 449.8 | 463.3 | 27.75 | 2.71 | 0.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-H.; Chang, C.; Huang, Y.-C.; You, J.-X.; Liang, M. Synthesis and Characterization of DOPO-Containing Poly(2,6-dimethyl-1,4-phenylene oxide)s by Oxidative Coupling Polymerization. Polymers 2024, 16, 303. https://doi.org/10.3390/polym16020303
Lu C-H, Chang C, Huang Y-C, You J-X, Liang M. Synthesis and Characterization of DOPO-Containing Poly(2,6-dimethyl-1,4-phenylene oxide)s by Oxidative Coupling Polymerization. Polymers. 2024; 16(2):303. https://doi.org/10.3390/polym16020303
Chicago/Turabian StyleLu, Cheng-Hao, Chi Chang, Yu-Chen Huang, Jun-Xiang You, and Mong Liang. 2024. "Synthesis and Characterization of DOPO-Containing Poly(2,6-dimethyl-1,4-phenylene oxide)s by Oxidative Coupling Polymerization" Polymers 16, no. 2: 303. https://doi.org/10.3390/polym16020303
APA StyleLu, C.-H., Chang, C., Huang, Y.-C., You, J.-X., & Liang, M. (2024). Synthesis and Characterization of DOPO-Containing Poly(2,6-dimethyl-1,4-phenylene oxide)s by Oxidative Coupling Polymerization. Polymers, 16(2), 303. https://doi.org/10.3390/polym16020303