
Characterization of Agarose Gels in Solvent and Non-Solvent Media
 , ,
, , 
Abstract

1. Introduction
2. Materials and Methods
2.1. Agarose Samples Preparation
2.2. NMR Experiments
2.3. Water Regain and Swelling Degree
2.4. Differential Scanning Calorimetry
2.5. Rheology
3. Results and Discussion
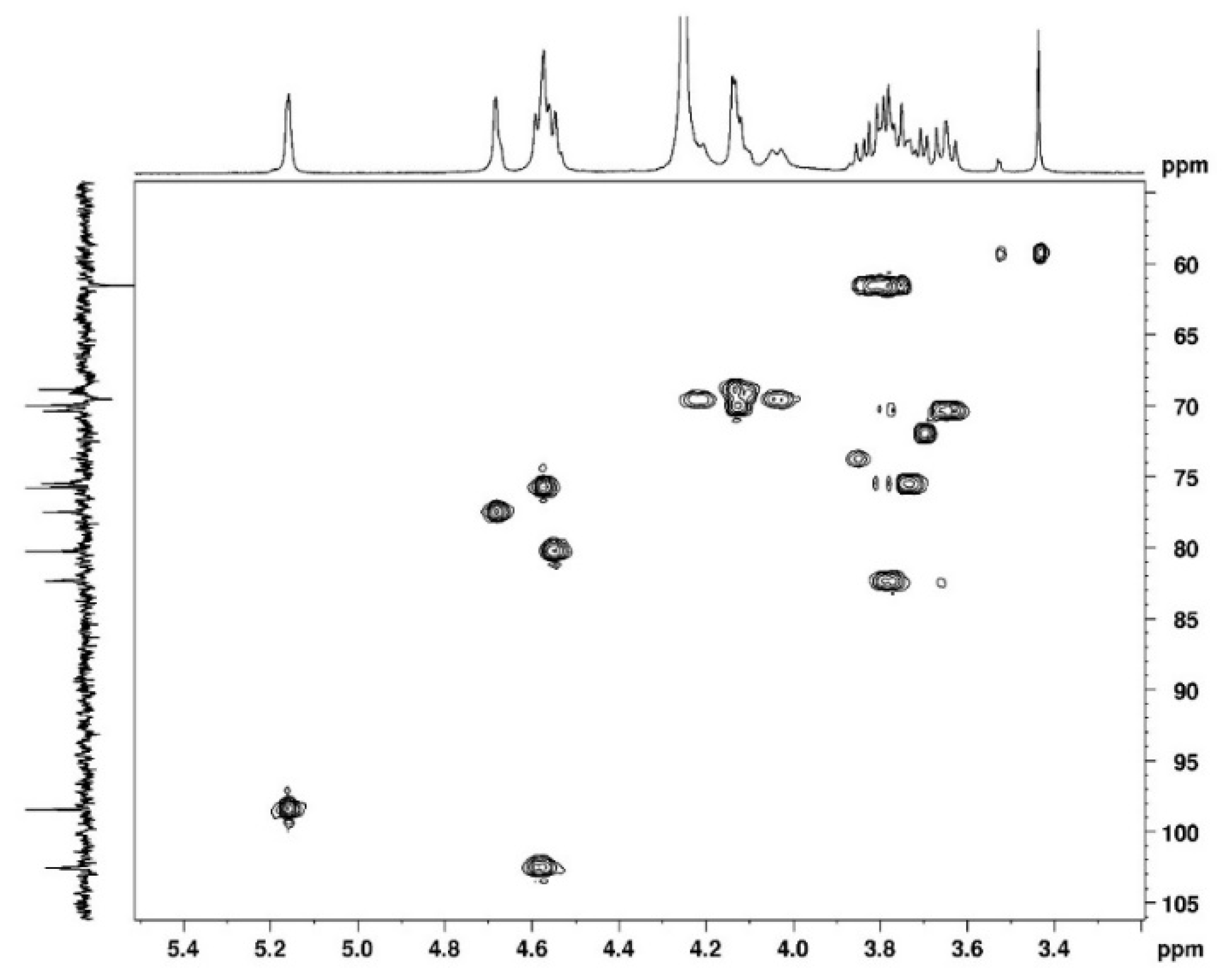
3.1. NMR Study
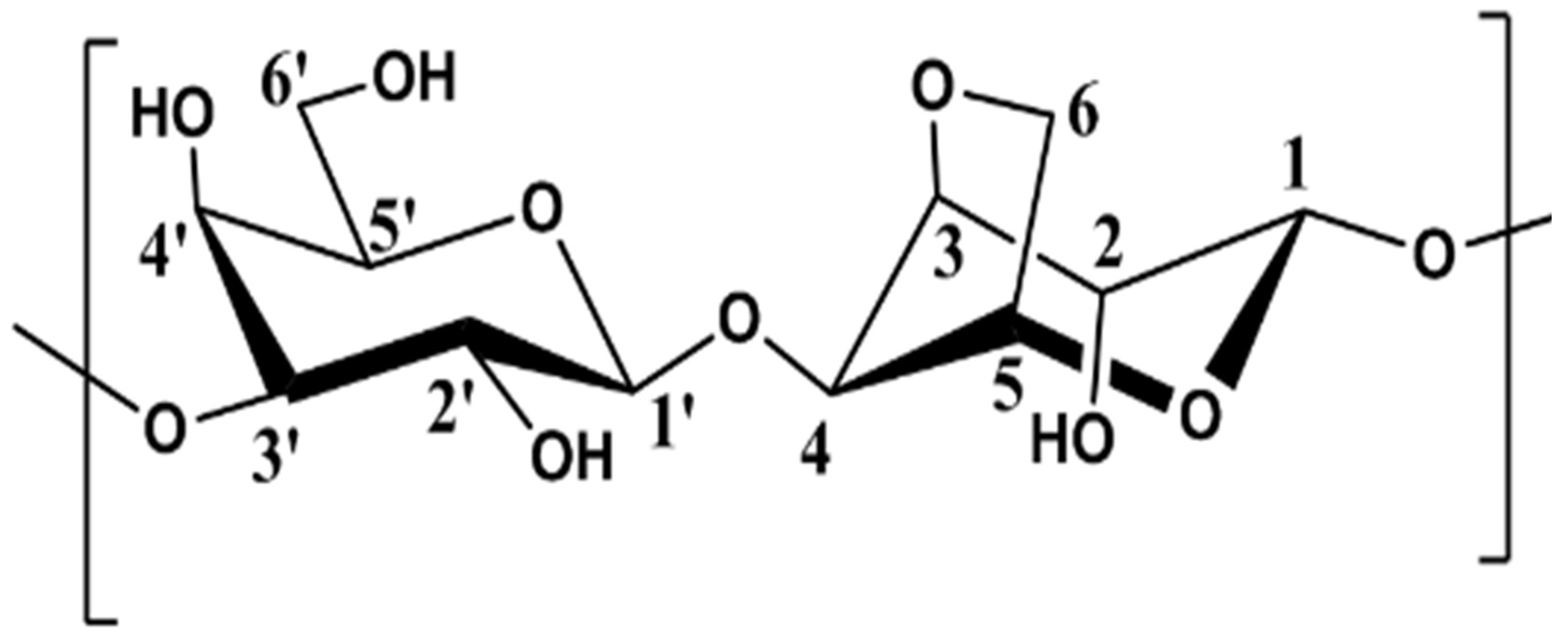
3.1.1. Assignement of 13C and 1H in D2O
3.1.2. Assignement of 13C and 1H in DMSO-d6
3.1.3. Degree of Substitution on Agarose
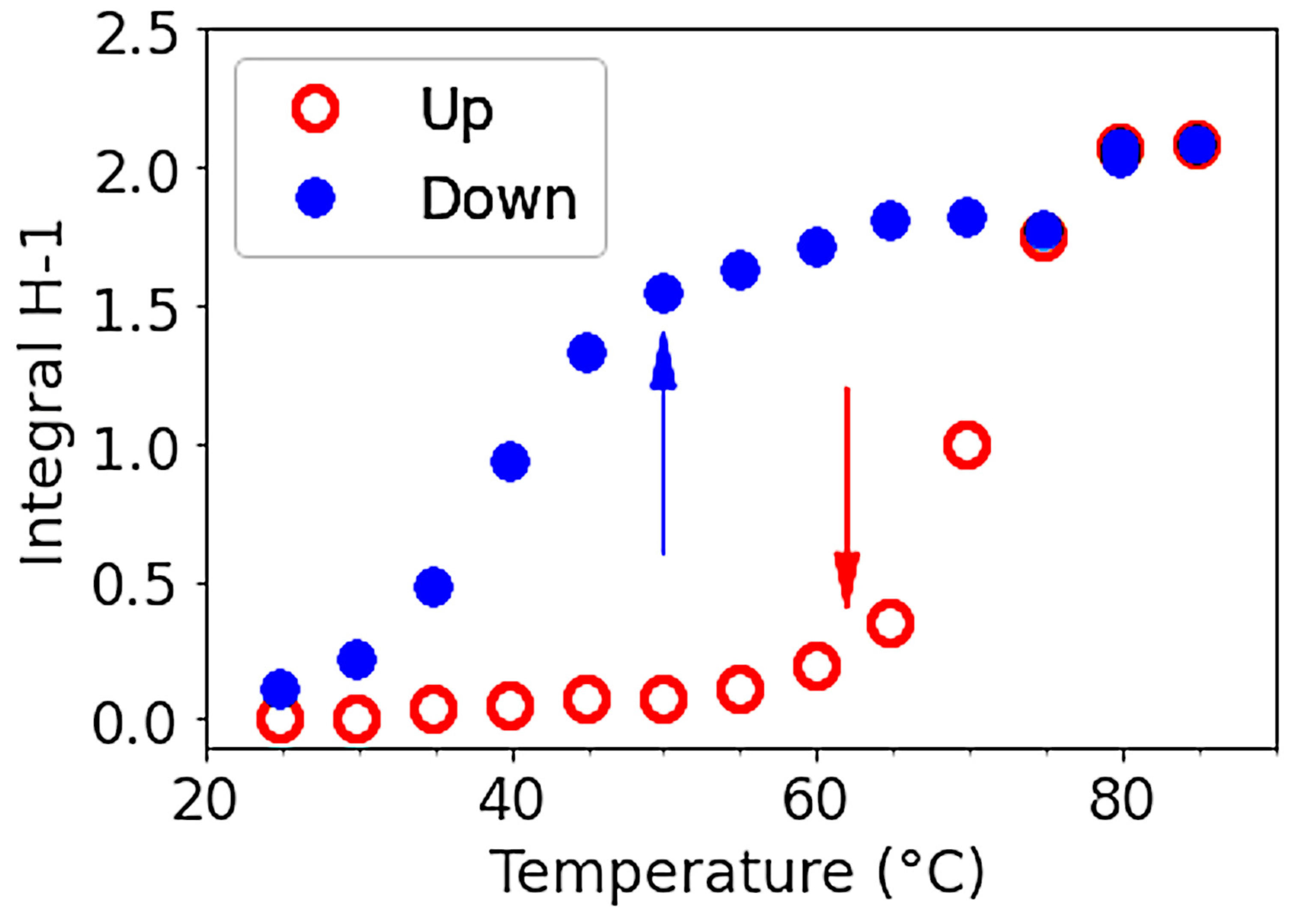
3.1.4. Thermal Hysteresis
3.1.5. Thermal Behaviour of Gel in Ethanol
3.2. Degree of Solvation
3.3. Rheology of Agarose Gels
3.3.1. Comparison of Gels in Water and Ethanol
3.3.2. Hysteresis for Sol-Gel Transition in Water
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vo, H.N.P.; Chaiwong, C.; Zheng, L.; Nguyen, T.M.H.; Koottatep, T.; Nguyen, T.T. Algae-Based Biomaterials for Sustainable Development; Elsevier: Amsterdam, The Netherlands, 2022; Chapter 9; pp. 185–202. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, W.; Lv, G.; Zhu, J.; Wang, W.; Ma, X.; Liu, X. Comprehensive Biotechnology, “the Artificial Organ: Cell Encapsulation”; Elsevier: Amsterdam, The Netherlands, 2011; pp. 673–688. [Google Scholar] [CrossRef]
- Thomas, D.J.; Jessop, Z.M.; Whitaker, I.S. 3D Bioprinting for Reconstructive Surgery; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Djabourov, M.; Nishinari, K.; Ross-Murphy, S.B. Helical Structures from Neutral Biopolymers in Physical Gels from Biological and Synthetic Polymers. Cambridge University Press: Cambridge, UK, 2013; pp. 208–221. [Google Scholar]
- Bulone, D.; Newman, J.; San Biagio, P.L. Mesoscopic gels at low agarose concentration: Perturbation effects of ethanol. Biophys. J. 1997, 72, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Rochas, C.; Rinaudo, M. Mechanism of gel formation in κ-carrageenan. Biopolym. Orig. Res. Biomol. 1984, 23, 735–745. [Google Scholar] [CrossRef]
- Dai, B.; Matsukawa, S. Elucidation of gelation mechanism and molecular interactions of agarose in solution by 1H NMR. Carbohydr. Res. 2013, 365, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Belton, P.S.; Hills, B.P.; Raimbaud, E.R. The effects of morphology and exchange on proton NMR relaxation in agarose gels. Mol. Phys. 1988, 63, 825–842. [Google Scholar] [CrossRef]
- Minghou, J.; Lahaye, M.; Yaphe, W. Structure of agar from Gracilaria spp (Rhodophyta) collected in the People’s Republic of China. Bot. Mar. 1985, 28, 521–528. [Google Scholar] [CrossRef]
- Rodríguez, M.C.; Matulewicz, M.C.; Noseda, M.D.; Ducatti DR, B.; Leonardi, P.I. Agar from Gracilaria gracilis (Gracilariales, Rhodophyta) of the Patagonic coast of Argentina–Content, structure and physical properties. Bioresour. Technol. 2009, 100, 1435–1441. [Google Scholar] [CrossRef]
- Rochas, C.; Lahaye, M. Average molecular weight and molecular weight distribution of agarose and agarose-type polysaccharides. Carbohydr. Polym. 1989, 10, 289–298. [Google Scholar] [CrossRef]
- Bao, X.; Hayashi, K.; Li, Y.; Teramoto, A.; Abe, K. Novel agarose and agar fibers: Fabrication and characterization. Mater. Lett. 2010, 64, 2435–2437. [Google Scholar] [CrossRef]
- Lahaye, M.; Rochas, C. Chemical structure and physico-chemical properties of agar. Hydrobiologia 1991, 221, 137–148. [Google Scholar] [CrossRef]
- Vaca Chávez, F.; Persson, E.; Halle, B. Internal water molecules and magnetic relaxation in agarose gels. J. Am. Chem. Soc. 2006, 128, 4902–4910. [Google Scholar] [CrossRef]
- Andrasko, J. Water in agarose gels studied by nuclear magnetic resonance relaxation in the rotating frame. Biophys. J. 1975, 15, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Aşkin, M.; Yilmaz, A. The Calculation of Correlation Time (τ) for T 1 Spin–Lattice and T 2 Spin–Spin Relaxation Times in Agar Solutions. Spectrosc. Lett. 2004, 37, 217–224. [Google Scholar] [CrossRef]
- Gamini, A.; Toffanin, R.; Murano, E.; Rizzo, R. Hydrogen-bonding and conformation of agarose in methyl sulfoxide and aqueous solutions investigated by 1H and 13C NMR spectroscopy. Carbohydr. Res. 1997, 304, 293–302. [Google Scholar] [CrossRef]
- Nicolaisen, F.M.; Meyland IN, G.E.; Schaumberg, K. 13C-NMR Spectra at 67.9 MHz of aqueous solutions of agarose and partly 6-O-methylated agarose at 95 °C. Acta Chem. Scand. B 1980, 34, 103–107. [Google Scholar] [CrossRef]
- Davies, E.; Huang, Y.; Harper, J.B.; Hook, J.M.; Thomas, D.S.; Burgar, I.M.; Lillford, P.J. Dynamics of water in agar gels studied using low and high resolution 1H NMR spectroscopy. Int. J. Food Sci. Technol. 2010, 45, 2502–2507. [Google Scholar] [CrossRef]
- Rinaudo, M.; Landry, S. On the volume change on non covalent gels in solvent-non solvent mixtures. Polym. Bull. 1987, 17, 563–565. [Google Scholar] [CrossRef]
- Burger, R.; Bigler, P. DEPTQ: Distorsionless enhancement by polarization transfer including the detection of quaternary nuclei. J. Magn. Reson. 1998, 135, 529–534. [Google Scholar] [CrossRef]
- Jouon, N.; Rinaudo, M.; Milas, M.; Desbrieres, J. Hydration of hyaluronic acid as a function of the counterion type and relative humidity. Carbohydr. Polym. 1995, 26, 69–73. [Google Scholar] [CrossRef]
- Yoshida, H.; Hatakeyama, T.; Hatakeyama, H. Characterization of water in polysaccharide hydrogels by DSC. J. Therm. Anal. Calorim. 1993, 40, 483–489. [Google Scholar] [CrossRef]
- Ed-Daoui, A.; Snabre, P. Poroviscoelasticity and compression-softening of agarose hydrogels. Rheol. Acta 2021, 60, 327–351. [Google Scholar] [CrossRef]
- Cardoso MV, C.; Sabadini, E. The gelling of κ-carrageenan in light and heavy water. Carbohydr. Res. 2010, 345, 2368–2373. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Collapse of gels and the critical endpoint. Phys. Rev. Lett. 1978, 40, 820. [Google Scholar] [CrossRef]
- Tanaka, T. Phase transition of gels. In Polyelectrolyte Gels; Properties, Preparation and Applications; Harland, R.A., Prud’homme, R.K., Eds.; ACS Symposium Series No. 480; American Chemical Society: Washington, DC, USA, 1991; pp. 1–21. [Google Scholar]
- Stenram, U.; Acevedo, F.; Larsson-Raznikiewicz, M. Agarose gel isoelectric focusing of gliadins in the presence of ethanol. Food Chem. 1990, 36, 305–310. [Google Scholar] [CrossRef]
- Donley, G.J.; Singh, P.K.; Shetty, A.; Rogers, S.A. Elucidating the G″ overshoot in soft materials with a yield transition via a time-resolved experimental strain decomposition. Proc. Natl. Acad. Sci. USA 2020, 117, 21945–21952. [Google Scholar] [CrossRef] [PubMed]
- Ghebremedhin, M.; Seiffert, S.; Vilgis, T.A. Physics of agarose fluid gels: Rheological properties and microstructure. Curr. Res. Food Sci. 2021, 4, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, E.; Roux, D.; Bleses, D.; Galliard, H.; Caton, F. Optimal fourier rheometry. Rheol. Acta 2012, 51, 413–420. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C1 | C2 | C3 | C4 | C5 | C6 | CH3 | |
| G′ | 102.63 | 70.38 | 82.33 | 68.93 | 75.51 | 61.56 | |
| G″ | 102.63 | 70.38 | 82.33 | 68.93 | 75.51 | 71.89 | 59.2 |
| G | 98.52 | 70.05 | 80.31 | 77.6 | 75.7 | 69.64 | |
| H1 | H2 | H3 | H4 | H5 | H6 | CH3 | |
| G′ | 4.58 | 3.65 | 3.78 | 4.14 | 3.73 | 3.80 | |
| G″ | 4.58 | 3.65 | 3.78 | 4.14 | 3.73 | 3.70 | 3.43 |
| G | 5.16 | 4.13 | 4.54 | 4.68 | 4.57 | 4.22 a–4.04 b |
| C1 | C2 | C3 | C4 | C5 | C6 | CH3 | |
| G′ | 102.5 | 70.13 | 81.35 | 68.11 | 75.54 | 60.8 | |
| G″ | 102.5 | 70.13 | 80.99 | 68.63 | 73.52 | 71.96 | 58.9 |
| G | 97.54 | 70.37 | 80.23 | 76.53 | 75.12 | 68.9 | |
| H1 | H2 | H3 | H4 | H5 | H6 | CH3 | |
| G′ | 4.31 | 3.45 | 3.54 | 3.83 | 3.44 | 3.55 | |
| G″ | 4.31 | 3.45 | 3.47 | 3.76 | 3.63 | 3.54 a–3.46 b | 3.3 |
| G | 5.09 | 3.83 | 4.22 | 4.34 | 4.35 | 3.91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roux, D.C.D.; Jeacomine, I.; Maîtrejean, G.; Caton, F.; Rinaudo, M. Characterization of Agarose Gels in Solvent and Non-Solvent Media. Polymers 2023, 15, 2162. https://doi.org/10.3390/polym15092162
Roux DCD, Jeacomine I, Maîtrejean G, Caton F, Rinaudo M. Characterization of Agarose Gels in Solvent and Non-Solvent Media. Polymers. 2023; 15(9):2162. https://doi.org/10.3390/polym15092162
Chicago/Turabian StyleRoux, Denis C. D., Isabelle Jeacomine, Guillaume Maîtrejean, François Caton, and Marguerite Rinaudo. 2023. "Characterization of Agarose Gels in Solvent and Non-Solvent Media" Polymers 15, no. 9: 2162. https://doi.org/10.3390/polym15092162
APA StyleRoux, D. C. D., Jeacomine, I., Maîtrejean, G., Caton, F., & Rinaudo, M. (2023). Characterization of Agarose Gels in Solvent and Non-Solvent Media. Polymers, 15(9), 2162. https://doi.org/10.3390/polym15092162