
Statistical Copolymers of N–Vinylpyrrolidone and 2–Chloroethyl Vinyl Ether via Radical RAFT Polymerization: Monomer Reactivity Ratios, Thermal Properties, and Kinetics of Thermal Decomposition of the Statistical Copolymers



Abstract
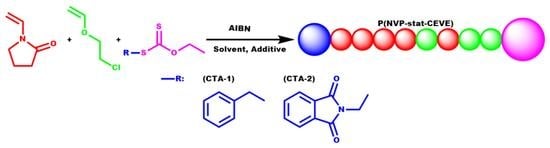
1. Introduction
2. Materials and Methods
2.1. Materials
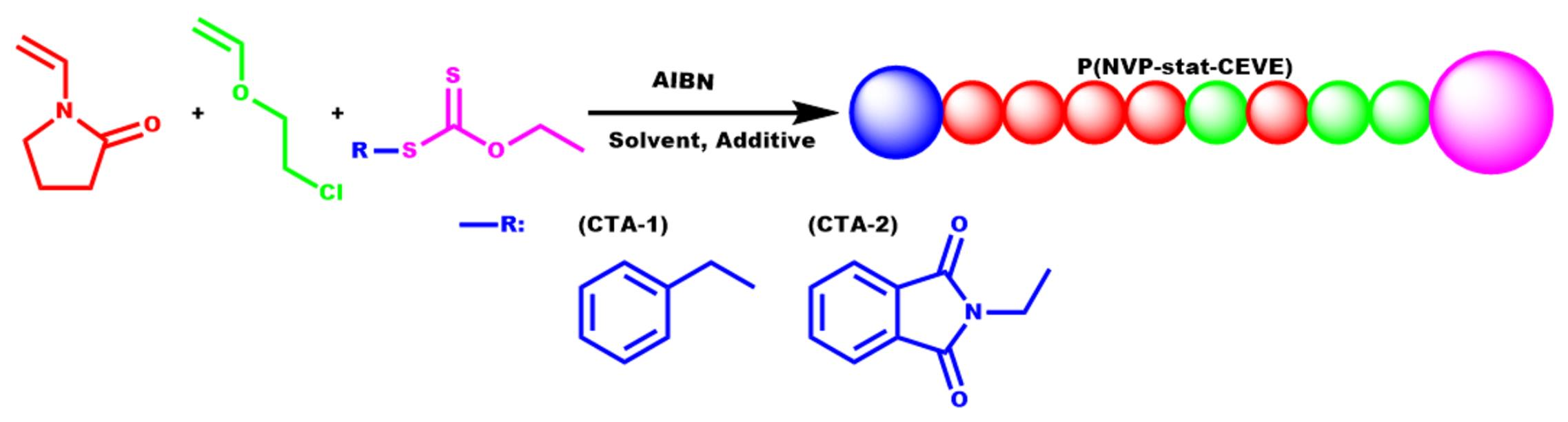
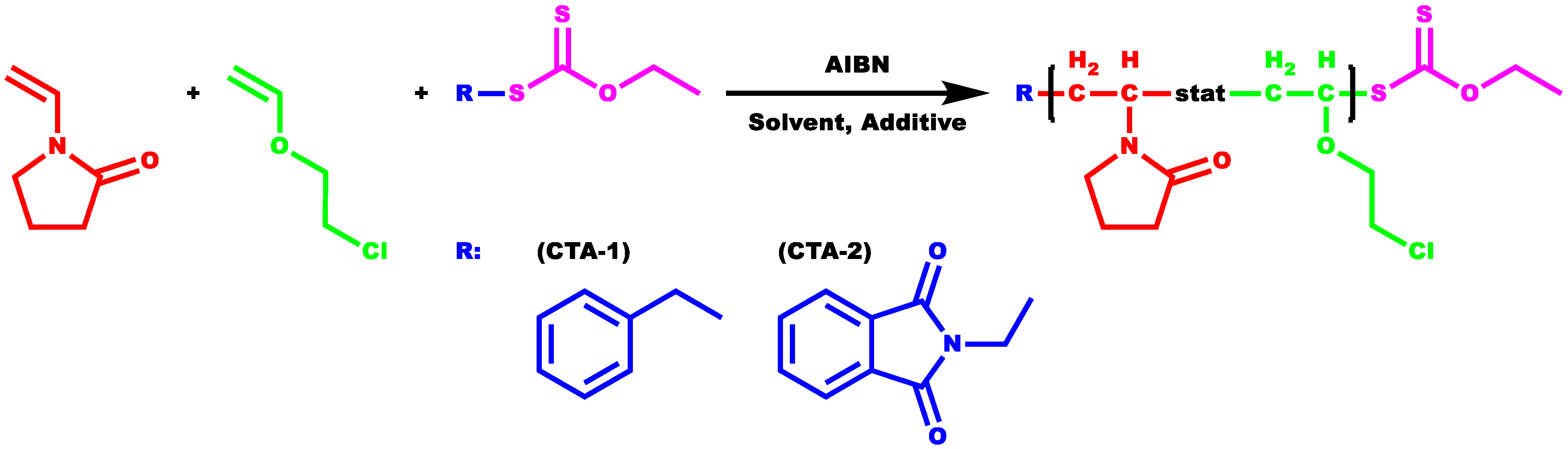
2.2. Copolymerization of NVP and CEVE via Radical RAFT Polymerization
2.3. Reactivity Ratios of NVP and CEVE Using CTA-1 as RAFT Agent
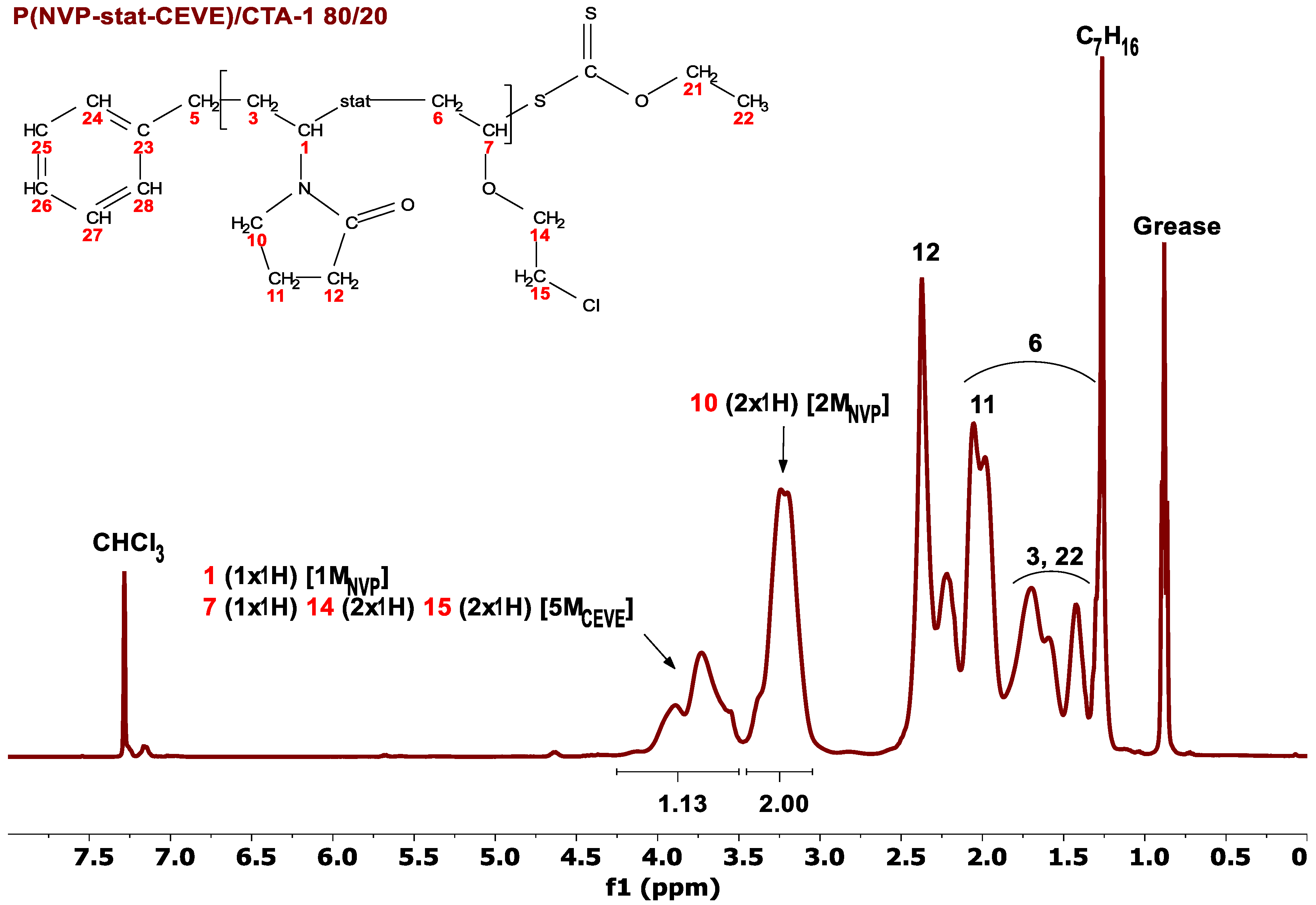
2.4. Characterization Techniques
3. Results and Discussion
3.1. Statistical Copolymers of NVP and CEVE via Radical RAFT Polymerization
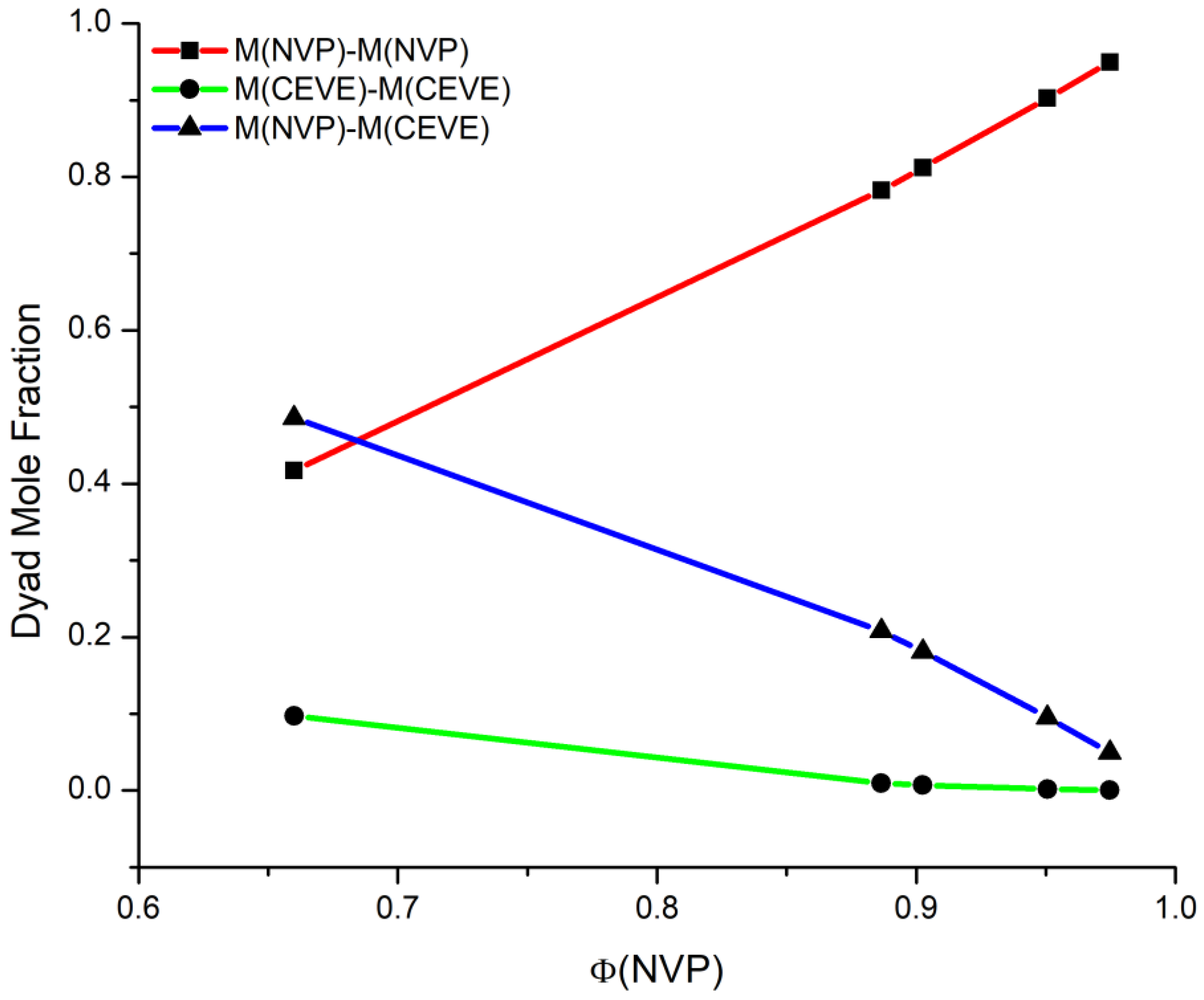
3.2. Monomer Reactivity Ratios and Statistical Analysis of the Copolymers
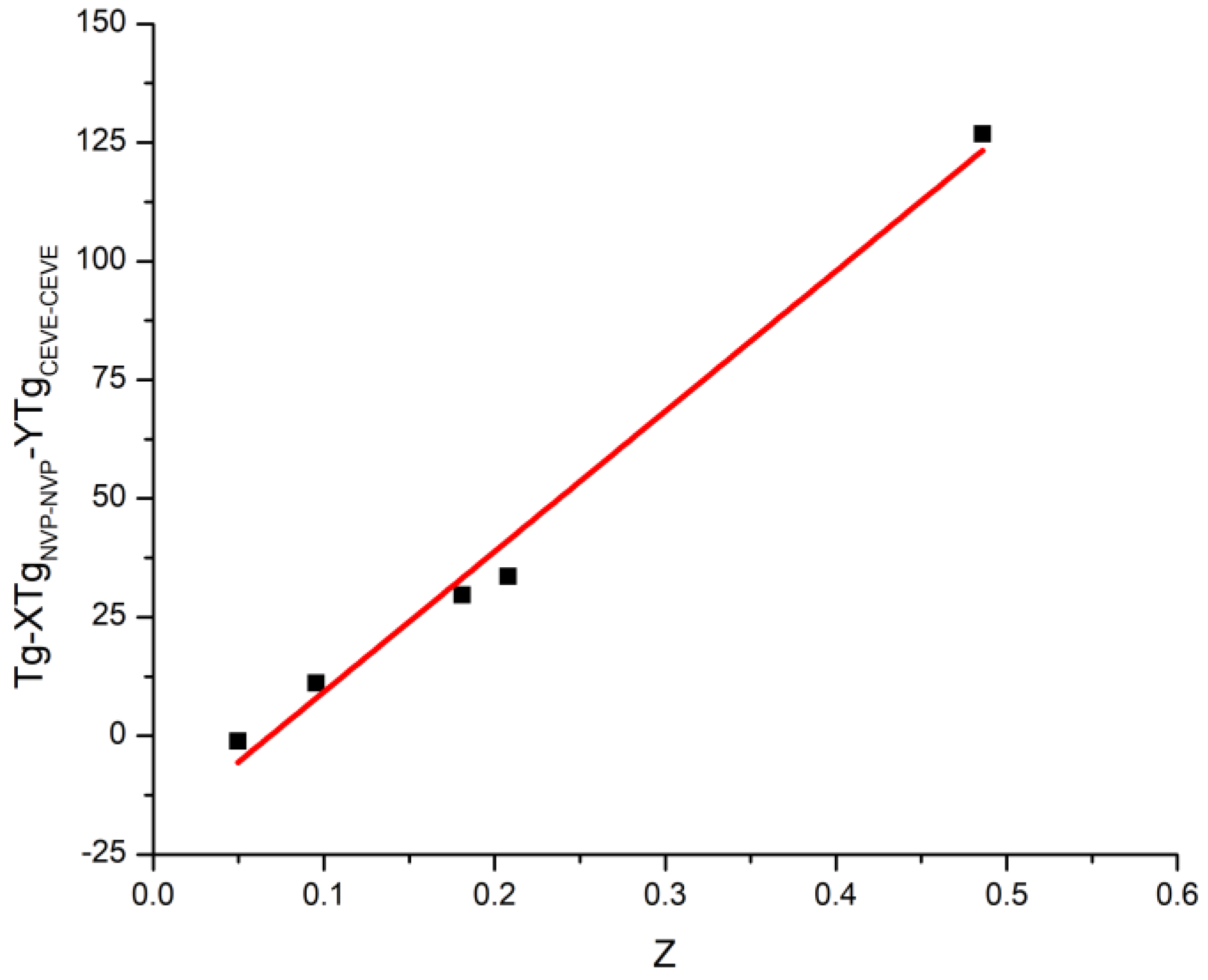
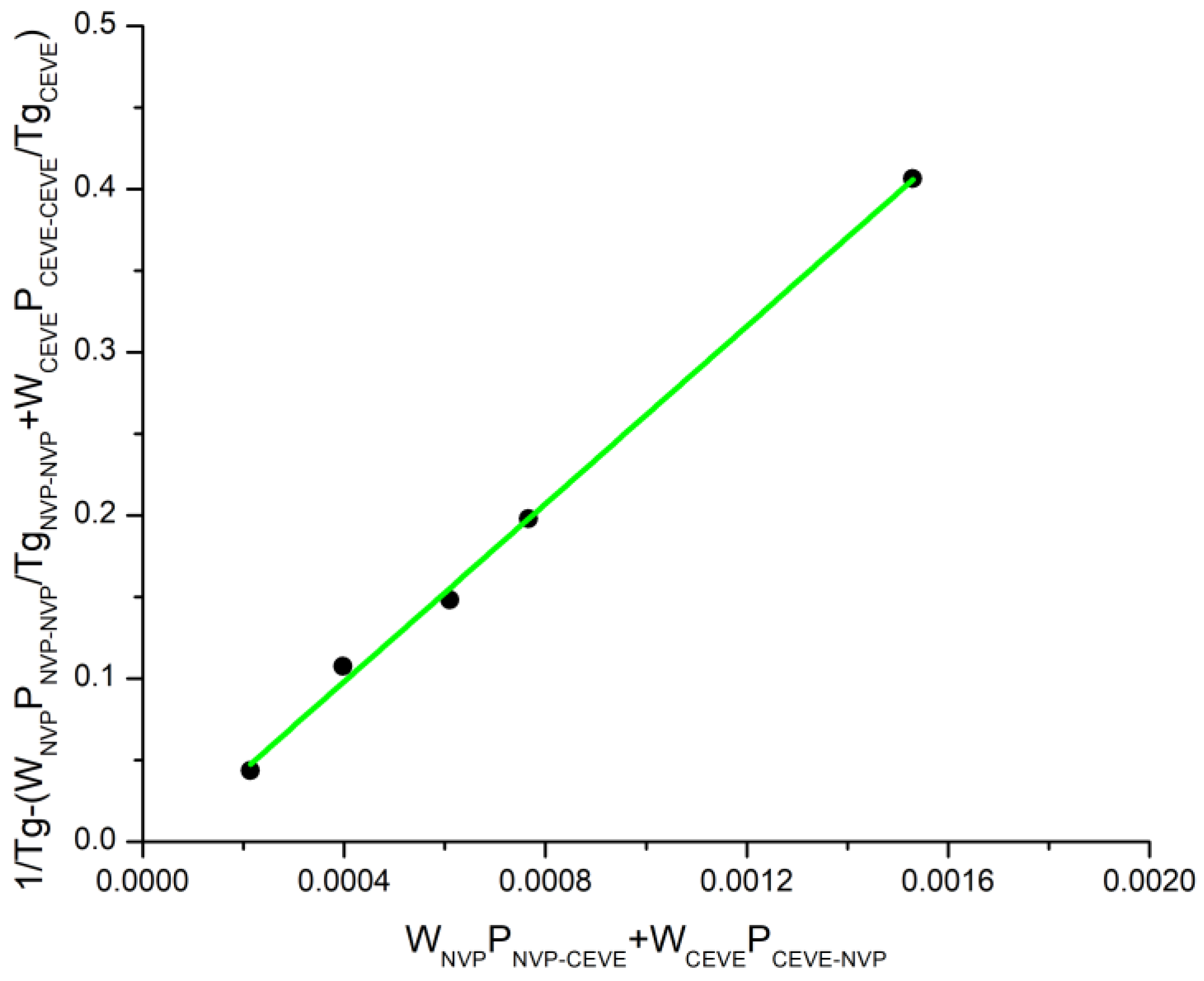
3.3. Glass Transition Temperatures of the Statistical Copolymers
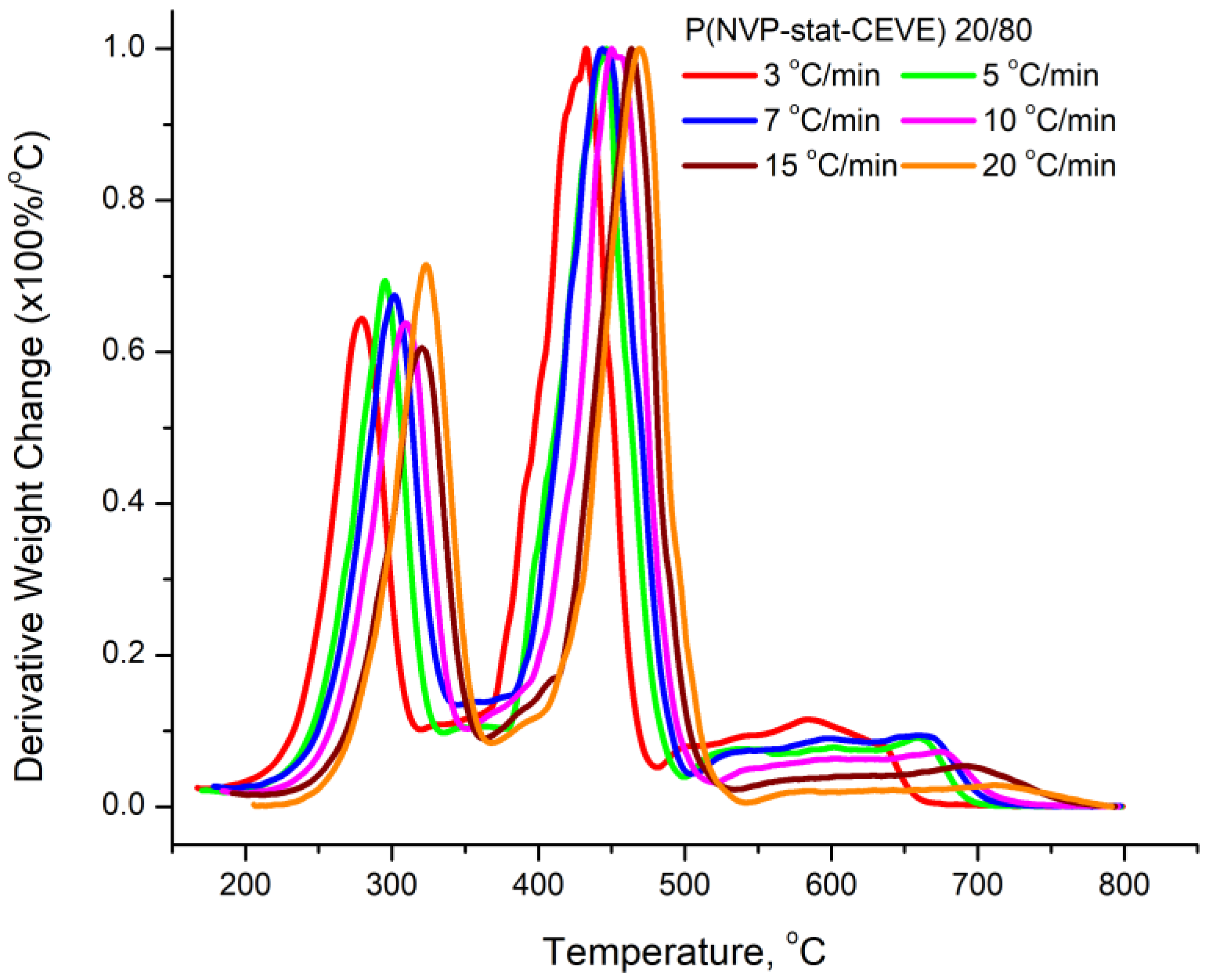
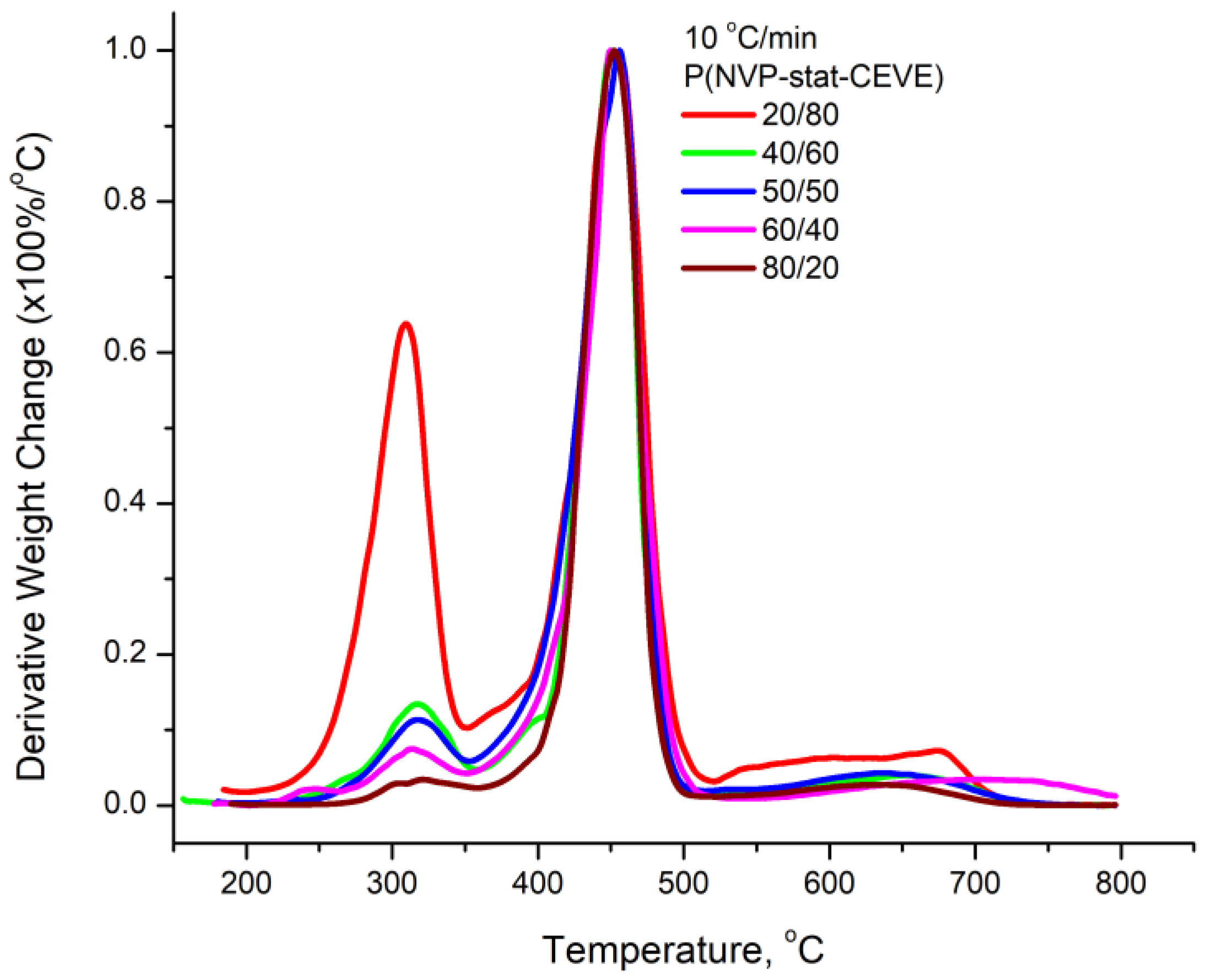
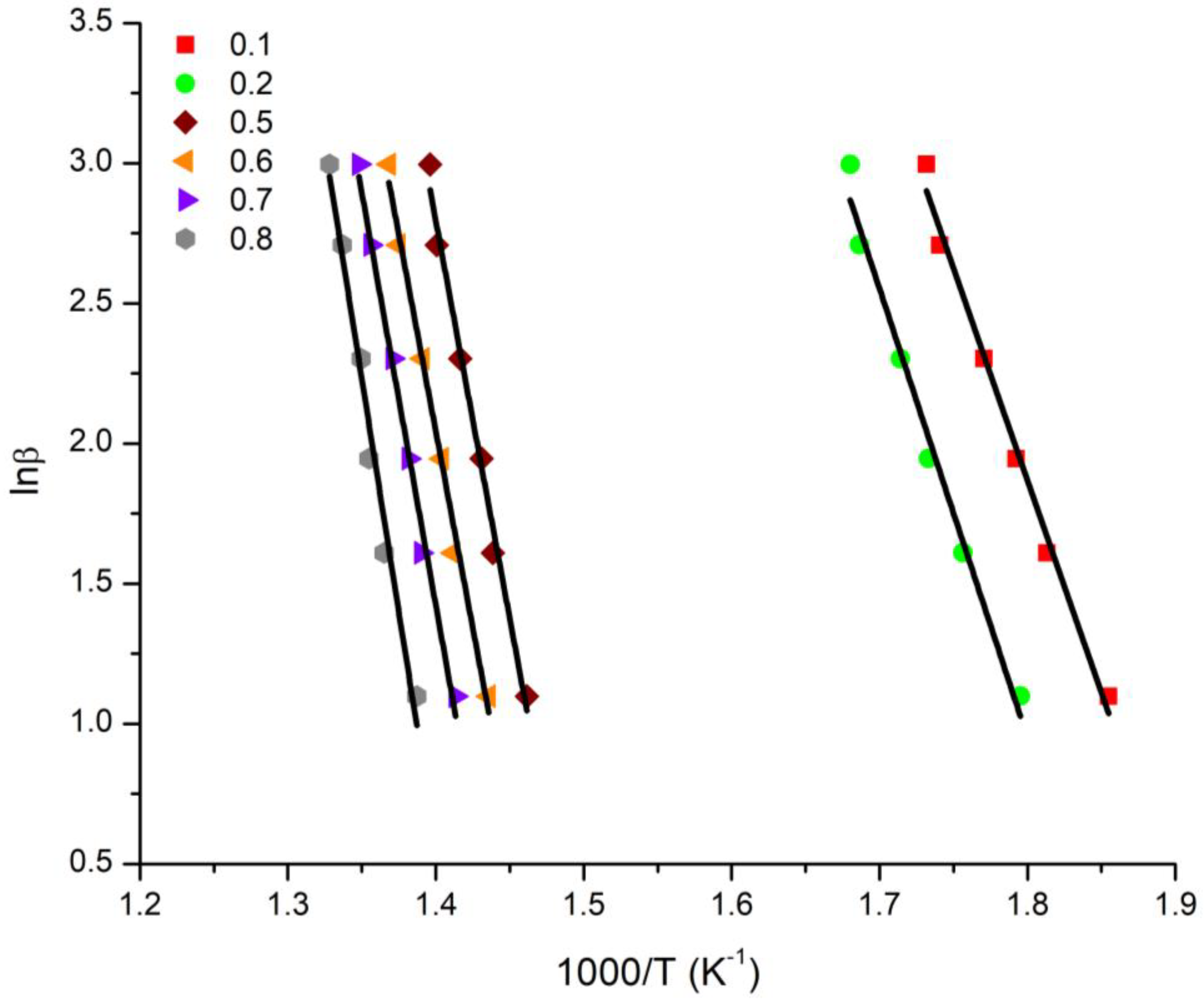
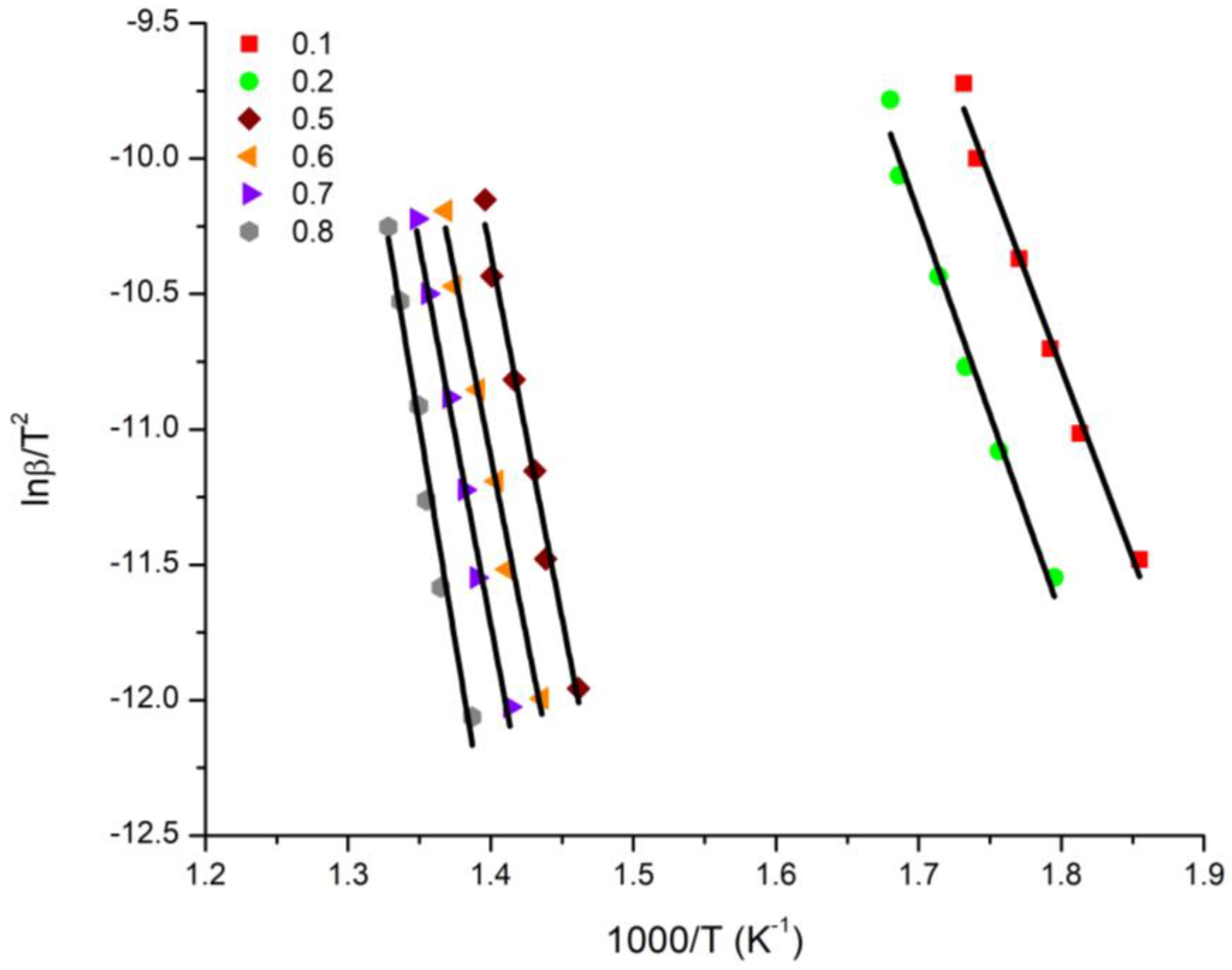
3.4. Kinetics of the Thermal Decomposition of the Statistical Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Teodorescu, M.; Bercea, M. Poly(vinylpyrrolidone)—A Versatile Polymer for Biomedical and beyond Medical Applications. Polym. Plast. Technol. Eng. 2015, 549, 923–943. [Google Scholar] [CrossRef]
- Franco, F.; De Marco, I. The Use of Poly(N-vinyl pyrrolidone) in the Delivery of Drugs: A Review. Polymers 2020, 12, 1114. [Google Scholar] [CrossRef] [PubMed]
- Haaf, F.; Sanner, A.; Straub, F. Polymers of N-vinylpyrrolidone: Synthesis, characterization and uses. Polym. J. 1985, 17, 143–152. [Google Scholar] [CrossRef]
- Boyer, C.; Bulmus, V.; Davis, T.P.; Ladmiral, V.; Liu, J.; Perrier, S. Bioapplications of RAFT Polymerization. Chem. Rev. 2009, 109, 5402–5436. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process—A Third Update. Aust. J. Chem. 2012, 65, 985–1076. [Google Scholar] [CrossRef]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT Agent Design and Synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.J.; Matyjaszewski, K.; Boyer, C. Reversible-deactivation radical polymerization (Controlled/living radical polymerization): From discovery to materials design and applications. Progr. Polym. Sci. 2020, 111, 101311. [Google Scholar] [CrossRef]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Roka, N.; Kokkorogianni, O.; Pitsikalis, M. Statistical copolymers of N-vinylpyrrolidone and 2-(dimethylamino)ethyl methacrylate via RAFT: Monomer reactivity ratios, thermal properties, and kinetics of thermal decomposition. Inc. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3776–3787. [Google Scholar] [CrossRef]
- Roka, N.; Pitsikalis, M. Statistical copolymers of N-vinylpyrrolidone and benzyl methacrylate via RAFT: Monomer reactivity ratios, thermal properties and kinetics of thermal decomposition. J. Macromol. Sci. Part A Pure Appl. Chem. 2018, 55, 222–230. [Google Scholar] [CrossRef]
- Mitsoni, E.; Roka, N.; Pitsikalis, M. Statistical copolymerization of N-vinyl-pyrrolidone and alkyl methacrylates via RAFT: Reactivity ratios and thermal analysis. J. Polym. Res. 2019, 26, 118. [Google Scholar] [CrossRef]
- Kokkorogianni, O.; Kontoes-Georgoudakis, P.; Athanasopoulou, M.; Polizos, N.; Pitsikalis, M. Statistical Copolymers of N-Vinylpyrrolidone and Isobornyl Methacrylate via Free Radical and RAFT Polymerization: Monomer Reactivity Ratios, Thermal Properties, and Kinetics of Thermal Decomposition. Polymers 2021, 13, 778. [Google Scholar] [CrossRef]
- Gikarakis, T.; Pappas, I.; Arvanitaki, P.; Pantazi, E.; Mitsoni, E.; Roka, N.; Pitsikalis, M. Thermal Stability and Kinetics of Thermal Decomposition of Statistical Copolymers of N-Vinylpyrrolidone and Alkyl Methacrylates Synthesized via RAFT Polymerization. J. Chem. 2021, 2021, 6633052. [Google Scholar] [CrossRef]
- Roka, N.; Kokkorogianni, O.; Kontoes-Georgoudakis, P.; Choinopoulos, I.; Pitsikalis, M. Recent Advances in the Synthesis of Complex Macromolecular Architectures Based on Poly(N-vinyl pyrrolidone) and the RAFT Polymerization Technique. Polymers 2022, 14, 701. [Google Scholar] [CrossRef]
- Satas, D. (Ed.) Handbook of Pressure Sensitive Adhesive Technology; Van Nostrand Reinhold: New York, NY, USA, 1989. [Google Scholar]
- Sanchez, M.; Deffieux, A.; Bordenave, L.; Baquey, C.H.; Fontanille, M. Synthesis of Hemocompatible Materials, Part 1: Surface Modification of Polyurethanes Based on Poly(chloroalkylvtnilether)s by RGD Fragments. Clin. Mater. 1994, 15, 253–258. [Google Scholar] [CrossRef]
- Confortini, O.; Du Prez, F.E. Functionalized Thermo-Responsive Poly(vinyl ether) by Living Cationic Random Copolymerization of Methyl Vinyl Ether and 2-Chloroethyl Vinyl Ether. Macromol. Chem. Phys. 2007, 208, 1871–1882. [Google Scholar] [CrossRef]
- Heroguez, V.; Deffieux, A.; Fontanille, M. Living cationic polymerization of 2-chloroethyl vinyl ether: Kinetics, mechanism and application to polymer synthesis. Macromol. Chem.-Macromol. Symp. 1990, 32, 199–216. [Google Scholar] [CrossRef]
- Chabani, M.; Yahiaoui, A.; Hachemaoui, A.; Belbachir, M. New approach for the polymerization of 2-chloroethyl vinyl ether using a maghnite clay as eco-catalyst. J. Appl. Polym. Sci. 2011, 122, 1800–1806. [Google Scholar] [CrossRef]
- Zouganelis, S.; Choinopoulos, I.; Goulas, I.; Pitsikalis, M. Statistical Copolymers of n-Butyl Vinyl Ether and 2-Chloroethyl Vinyl Ether via Metallocene-Mediated Cationic Polymerization. A Scaffold for the Synthesis of Graft Copolymers. Polymers 2019, 11, 1510. [Google Scholar] [CrossRef]
- Uchiyama, M.; Satoh, K.; Kamigaito, M. Cationic RAFT Polymerization Using ppm Concentrations of Organic Acid. Angew. Chem. Int. Ed. 2015, 54, 1924–1928. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, M.; Satoh, K.; Kamigaito, M. A Phosphonium Intermediate for Cationic RAFT Polymerization. Polym. Chem. 2016, 7, 1387–1396. [Google Scholar] [CrossRef]
- McKenzie, T.G.; Fu, Q.; Uchiyama, M.; Satoh, K.; Xu, J.; Boyer, C.; Kamigaito, M.; Qiao, G.G. Beyond Traditional RAFT: Alternative Activation of Thiocarbonylthio Compounds for Controlled Polymerization. Adv. Sci. 2016, 3, 1500394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Z.; Jiang, Y.; Liao, S. Organocatalytic, Stereoselective, Cationic Reversible Addition−Fragmentation Chain-Transfer Polymerization of Vinyl Ethers. J. Am. Chem. Soc. 2022, 144, 679–684. [Google Scholar] [CrossRef]
- Yilmaz, G.; Yagci, Y. Mechanistic transformations involving radical and cationic polymerizations. Chin. J. Polym. Sci. 2020, 38, 205–212. [Google Scholar] [CrossRef]
- Satoh, K.; Fujiki, Y.; Uchiyama, M.; Kamigaito, M. Vinyl Ether/Vinyl Ester Copolymerization by Cationic and Radical Interconvertible Simultaneous Polymerization. In Reversible Deactivation Radical Polymerization: Mechanisms and Synthetic Methodologies; Matyjaszewski, K., Gao, H., Sumerlin, B.S., Tsarevsky, N.V., Eds.; American Chemical Society: Washington, DC, USA, 2018; pp. 324–334. [Google Scholar]
- Guerre, M.; Uchiyama, M.; Folgado, E.; Semsarilar, M.; Améduri, B.; Satoh, K.; Kamigaito, M.; Ladmira, M. Combination of Cationic and Radical RAFT Polymerizations: A Versatile Route to Well-Defined Poly(ethyl vinyl ether)-blockpoly(vinylidene fluoride) Block Copolymers. ACS Macro Lett. 2017, 6, 393–398. [Google Scholar] [CrossRef]
- Guerre, M.; Uchiyama, M.; Lopez, G.; Améduri, B.; Satoh, K.; Kamigaito, M.; Ladmiral, V. Synthesis of PEVE-b-P(CTFE-alt-EVE) block copolymers by sequential cationic and radical RAFT polymerization. Polym. Chem. 2018, 9, 352. [Google Scholar] [CrossRef]
- Kamachi, M.; Tanaka, K.; Kuwae, Y. ESR Studies on Radical Polymerization of Vinyl Ethers. J. Polym. Sci. Part A Polym. Chem. 1986, 24, 925–929. [Google Scholar] [CrossRef]
- Fueno, T.; Kamachi, M.; Ab, M. Initio SCF Study of the Addition of the Methyl Radical to Vinyl Compounds. Macromolecules 1988, 21, 908–912. [Google Scholar] [CrossRef]
- Sugihara, S.; Kawamoto, Y.; Maeda, Y. Direct Radical Polymerization of Vinyl Ethers: Reversible Addition−Fragmentation Chain Transfer Polymerization of Hydroxy-Functional Vinyl Ethers. Macromolecules 2016, 49, 1563–1574. [Google Scholar] [CrossRef]
- Sugihara, S.; Yoshida, A.; Fujita, S.; Maeda, Y. Design of Hydroxy-Functionalized Thermoresponsive Copolymers: Improved Direct Radical Polymerization of Hydroxy-Functional Vinyl Ethers. Macromolecules 2017, 50, 8346–8356. [Google Scholar] [CrossRef]
- Sugihara, S.; Yoshida, A.; Kono, T.; Takayama, T.; Maeda, Y. Controlled Radical Homopolymerization of Representative Cationically Polymerizable Vinyl Ethers. J. Am. Chem. Soc. 2019, 141, 13954–13961. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. Part A Polym. Chem. Ed. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Wan, D.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Xanthate-Mediated Radical Polymerization of N-Vinylpyrrolidone in Fluoroalcohols for Simultaneous Control of Molecular Weight and Tacticity. Macromolecules 2005, 38, 10397–10405. [Google Scholar] [CrossRef]
- Postma, A.; Davis, T.P.; Li, G.; Moad, G.; O’Shea, M.S. RAFT polymerization with phthalimidomethyl trithiocarbonates or xanthates. On the origin of bimodal molecular weight distributions in living radical polymerization. Macromolecules 2006, 39, 5307–5318. [Google Scholar] [CrossRef]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Macromol. Sci. Part A Chem. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Kelen, T.; Tüdos, F. Analysis of the Linear Methods for Determining Copolymerization Reactivity Ratios. I. A New Improved Linear Graphic Method. J. Macromol. Sci. Part A Chem. 1975, 9, 1–27. [Google Scholar] [CrossRef]
- Beginn, U. COPOINT—A simple computer program to determine copolymerization parameters by numerical integration. E-Polymers 2005, 5, 759–773. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Hagiopol, C. Copolymerization: Toward a Systematic Approach; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Igarashi, S. Representation of composition and blockiness of the copolymer by a triangular coordinate system. J. Polym. Sci. Part B Polym. Lett. 1963, 1, 359–363. [Google Scholar] [CrossRef]
- Gibbs, J.H.; Di Marzio, E.A. Molecular interpretation of glass temperature depression by plasticizers. J. Polym. Sci. Part A 1963, 1, 1417–1428. [Google Scholar]
- Fox, T.G.; Flory, P.J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Barton, J.M. Relation of glass transition temperature to molecular structure of addition copolymers. J. Polym. Sci. Part C 1970, 30, 573–597. [Google Scholar] [CrossRef]
- Johnston, N.W. Sequence Distribution-Glass Transition Effects. J. Macromol. Sci. Rev. Macromol. Chem. Phys. Part C. 1976, 14, 215–250. [Google Scholar] [CrossRef]
- Zouganelis, S.; Pitsikalis, M. Cationic Polymerization of 2-Chloroethyl Vinyl Ether Employing Activated Metallocene Complexes as Initiators. Kinetics of Polymerization and Thermal Properties; Industrial Chemistry Laboratory, Department of Chemistry, National and Kapodistrian University of Athens: Athens, Greece, 2023; to be submitted. [Google Scholar]
- Galukhin, A.; Liavitskaya, T.; Vyazovkin, S. Kinetic and mechanistic insights into thermally initiated polymerization of cyanate esters with different bridging groups. Macromol. Chem. Phys. 2019, 220, 1900141. [Google Scholar] [CrossRef]
- Boulkadid, M.K.; Touidjine, S.; Trache, D.; Belkhiri, S. Analytical methods for the assessment of curing kinetics of polyurethane binders for high energy composites. Crit. Rev. Anal. Chem. 2022, 52, 1112–1121. [Google Scholar] [CrossRef]
- Tarchoun, A.F.; Trache, D.; Klapötke, T.M.; Chelouche, S.; Derradji, M.; Bessa, W.; Mezroua, A. A promising energetic polymer from Posidonia oceanica brown alge: Synthesis, characterization, and kinetic modeling. Macromol. Chem. Phys. 2019, 220, 1900358. [Google Scholar] [CrossRef]
- Liqing, L.; Donghua, C. Application of iso-temperature method of multiple rate to kinetic analysis. Dehydration for calcium oxalate monohydrate. J. Therm. Anal. Calorim. 2004, 78, 283–293. [Google Scholar] [CrossRef]
- Lim, A.C.R.; Chin, B.L.F.; Jawad, Z.A.; Hii, K.L. Kinetic analysis of rice husk pyrolysis using Kissinger-Akahira-Sunose (KAS) method. Procedia Engin. 2016, 148, 1247–1251. [Google Scholar] [CrossRef]
- Hayoune, F.; Chelouche, S.; Trache, D.; Zitouni, S.; Grohens, Y. Thermal decomposition kinetics and lifetime prediction of a PP/PLA blend supplemented with iron stearate during artificial aging. Thermochim. Acta 2020, 690, 178700. [Google Scholar] [CrossRef]
- Trache, D.; Abdelaziz, A.; Siouani, B. A simple and linear isoconversional method to determine the pre-exponential factors and the mathematical reaction mechanism functions. J. Therm. Anal. Calorim. 2017, 128, 335–348. [Google Scholar] [CrossRef]
- Ozawa, T. A new method of analyzing thermogravimetric data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. A quick, direct method for the determination of activation energy from thermogravimetric data. J. Polym. Sci. Polym. Lett. Ed. 1966, 4, 323–328. [Google Scholar] [CrossRef]
- Ozawa, T. Kinetic analysis of derivative curves in thermal analysis. J. Therm. Anal. Calorim. 1970, 2, 301–324. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Doyle, C.D. Kinetic analysis of thermogravimetric data. J. Appl. Polym. Sci. 1961, 5, 285–292. [Google Scholar] [CrossRef]
- Coates, A.W.; Redfern, J. Kinetic parameters from thermogravimetric data. Nature 1964, 201, 68–69. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Sample (CTA) NVP/CEVE | Additive (mol) | Mw × 103 (Daltons) f | Đ f | Conversion (%) | % mol NVP g | % mol CEVE g |
|---|---|---|---|---|---|---|---|
| 1 | a (1) 20/80 | - | - | - | 0.00 | - | - |
| 2 | b (1) 20/80 | - | 1.8 | 1.33 | 16.5 | 80 | 20 |
| 3 | b (1) 60/240 | - | 3.8 | 1.51 | 16.4 | 78 | 22 |
| 4 | c (1) 100/400 | - | 5.8 | 1.67 | 21.5 | 70 | 30 |
| 5 | c (1) 150/150 | - | 15.6 | 2.04 | 89.5 | 82 | 18 |
| 6 | c (2) 60/240 | - | 4.8 | 1.54 | 27.0 | 72 | 28 |
| 7 | d (2) 250/250 | - | 24.8 | 2.37 | 90.0 | 88 | 12 |
| 8 | e (1) 60/240 | LiOH (3) | 2.7 | 1.61 | 6.7 | 78 | 22 |
| 9 | e (1) 60/240 | LiOH (6) | 1.9 | 1.44 | 4.0 | 81 | 19 |
| 10 | e (1) 60/240 | LiOH (1.5) | 1.9 | 1.58 | 6.1 | 81 | 19 |
| 11 | e (1) 250/250 | LiOH (3) | 17.1 | 1.84 | 72.5 | 86 | 14 |
| Sample NVP/CEVE | Time (min) | Mw × 103 (Daltons) a | Đ a | Monomers Conversion (%) | % mol NVP b | % mol CEVE b |
|---|---|---|---|---|---|---|
| 20/80 | 255 | 5.2 | 1.51 | 17.90 | 66.00 | 34.00 |
| 40/60 | 90 | 10.2 | 1.40 | 28.10 | 88.60 | 11.40 |
| 50/50 | 30 | 16.0 | 1.65 | 31.60 | 90.20 | 9.80 |
| 60/40 | 20 | 11.2 | 1.76 | 22.20 | 95.10 | 4.90 |
| 80/20 | 10 | 20.8 | 1.80 | 24.50 | 97.50 | 2.50 |
| Method | rNVP | rCEVE |
|---|---|---|
| F–R | 9.50 | 0.004 |
| inv. F–R | 11.66 | 0.21 |
| K–T | 10.26 | 0.09 |
| COPOINT | 10.90 | 0.06 |
| Sample | MNVP–MNVP | MCEVE–MCEVE | MNVP–MCEVE | μNVP | μCEVE |
|---|---|---|---|---|---|
| 20/80 | 0.417 | 0.097 | 0.485 | 3.724 | 1.252 |
| 40/60 | 0.783 | 0.009 | 0.208 | 8.264 | 1.095 |
| 50/50 | 0.812 | 0.007 | 0.181 | 11.896 | 1.063 |
| 60/40 | 0.903 | 0.002 | 0.095 | 17.344 | 1.042 |
| 80/20 | 0.949 | 0.0005 | 0.049 | 44.584 | 1.016 |
| Sample | Experimental Tg (°C) | Gibbs–Di Marzio Tg (°C) | Fox Tg (°C) | Barton Tg (°C) | Johnston Tg (°C) |
|---|---|---|---|---|---|
| 20/80 | 70 | 116.0 | 105.1 | 71.4 | 83.1 |
| 40/60 | 123 | 163.4 | 152.6 | 144.3 | 134.9 |
| 50/50 | 132 | 166.7 | 160.0 | 150.1 | 147.5 |
| 60/40 | 154 | 176.8 | 166.8 | 168.0 | 158.0 |
| 80/20 | 163 | 181.8 | 178.8 | 177.3 | 175.1 |
| Conversion | 20/80 Ea (KJ/mol) | 40/60 Ea (KJ/mol) | 50/50 Ea (KJ/mol) | 60/40 Ea (KJ/mol) | 80/20 Ea (KJ/mol) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | |
| 0.1 | 125.86 | 116.58 | 144.69 | 134.72 | 188.19 | 177.89 | 107.17 | 96.29 | 180.14 | 168.83 |
| 0.2 | 133.07 | 123.49 | 201.18 | 189.95 | 184.12 | 172.84 | 191.57 | 180.07 | 175.17 | 163.57 |
| 0.3 | 252.79 | 242.77 | 194.79 | 183.21 | 190.80 | 179.21 | 194.77 | 183.09 | 178.86 | 167.11 |
| 0.4 | 341.65 | 330.45 | 196.38 | 184.62 | 198.31 | 186.53 | 195.62 | 183.82 | 181.98 | 170.14 |
| 0.5 | 235.80 | 224.16 | 201.38 | 189.50 | 204.28 | 192.38 | 196.54 | 184.63 | 185.59 | 173.65 |
| 0.6 | 233.20 | 221.33 | 207.10 | 195.09 | 210.61 | 198.59 | 197.99 | 185.99 | 189.88 | 177.85 |
| 0.7 | 244.51 | 232.46 | 215.64 | 203.52 | 220.56 | 208.42 | 204.28 | 192.16 | 194.80 | 182.68 |
| 0.8 | 276.42 | 264.17 | 246.38 | 234.12 | 249.91 | 237.62 | 206.33 | 194.10 | 209.30 | 197.06 |
| Average | 208.14 | 197.03 | 200.94 | 189.34 | 205.85 | 194.19 | 198.16 | 186.27 | 186.97 | 175.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plachouras, N.V.; Pitsikalis, M. Statistical Copolymers of N–Vinylpyrrolidone and 2–Chloroethyl Vinyl Ether via Radical RAFT Polymerization: Monomer Reactivity Ratios, Thermal Properties, and Kinetics of Thermal Decomposition of the Statistical Copolymers. Polymers 2023, 15, 1970. https://doi.org/10.3390/polym15081970
Plachouras NV, Pitsikalis M. Statistical Copolymers of N–Vinylpyrrolidone and 2–Chloroethyl Vinyl Ether via Radical RAFT Polymerization: Monomer Reactivity Ratios, Thermal Properties, and Kinetics of Thermal Decomposition of the Statistical Copolymers. Polymers. 2023; 15(8):1970. https://doi.org/10.3390/polym15081970
Chicago/Turabian StylePlachouras, Nikolaos V., and Marinos Pitsikalis. 2023. "Statistical Copolymers of N–Vinylpyrrolidone and 2–Chloroethyl Vinyl Ether via Radical RAFT Polymerization: Monomer Reactivity Ratios, Thermal Properties, and Kinetics of Thermal Decomposition of the Statistical Copolymers" Polymers 15, no. 8: 1970. https://doi.org/10.3390/polym15081970
APA StylePlachouras, N. V., & Pitsikalis, M. (2023). Statistical Copolymers of N–Vinylpyrrolidone and 2–Chloroethyl Vinyl Ether via Radical RAFT Polymerization: Monomer Reactivity Ratios, Thermal Properties, and Kinetics of Thermal Decomposition of the Statistical Copolymers. Polymers, 15(8), 1970. https://doi.org/10.3390/polym15081970