While they do not correspond to any chemically specific polymer, generic polymer models are widely used in theoretical and simulation studies in the field of polymer physics as they capture two essential features of all polymers: chain connectivity and non-bonded excluded-volume interactions. Compared to atomistic models that can represent specific polymers used in experiments, molecular simulations of generic models can reach much larger length scales and much longer time scales, and theoretical studies of generic models can also be performed. Depending on whether or not the excluded-volume interactions in generic models prevent complete overlapping of polymer segments, they can be classified into hard-core models (such as those based on the hard-sphere chain model, the Kremer–Grest model [
1], and the various self- and mutual-avoiding walk models on a lattice) and soft-core models (such as those used in the dissipative particle dynamics (DPD) simulation [
2], fast Monte Carlo simulations [
3,
4,
5,
6,
7], field-theoretic simulation (FTS) [
8], variants of FTS under the partial saddle-point approximation [
9], single-chain-in-mean-field simulation [
10] and hybrid particle field molecular dynamics simulation [
11] both under the quasi-instantaneous field approximation [
10]). Taking the study of polymer melts as an example, while hard-core models have been used in conventional molecular simulations for a long time, they have the disadvantage that their chain lengths
N used in such simulations (as limited by the computational cost) are too short compared to those in typical experiments; in other words, such conventional simulations significantly exaggerate the fluctuations in polymer melts compared to experiments [
6,
7,
12]. In contrast, simulations of the more recently proposed soft-core models can readily reach the extent of fluctuations in typical experiments by increasing the chain number density (or equivalently the segment number density
ρ at finite
N) instead of
N [
6,
7,
12].
In this Letter we focus on a simple but important class of generic models for compressible homopolymer melts (or equivalently homopolymer solutions in an implicit solvent) in the continuum, with the excluded-volume interaction between polymer segments described by a short-range, isotropic and purely repulsive pair potential
βunb(
r), where
β ≡ 1/
kBT with
kB being the Boltzmann constant and
T the thermodynamic temperature of the system; this is the basis of more complicated polymer models having attractions and/or more species. The hard- and soft-core models can then be classified according to whether
diverges or not. This classification becomes clear after we write the total dimensionless non-bonded interaction energy for a system of
n chains each having
N segments in volume
V under the commonly used pairwise additivity as
where
ri denotes the spatial position of the
ith segment in the system,
is the segment volume fraction at
r, and the last term deducting the self-interaction of segments gives an unimportant constant; while molecular simulations of this system can be performed at finite
ρ ≡
nN/
V for any
βunb(
r) (along with a chain-connectivity model), for a
homogeneous system (i.e.,
) the widely used polymer self-consistent field (SCF) theory [
13] gives the dimensionless internal energy per chain due to the non-bonded interaction
(due to its mean-field approximation that neglects the system fluctuations and correlations), which diverges if
does (i.e., for the hard-core models). It is then clear that the SCF theory can only be applied to soft-core models, where one can define the dimensionless excluded-volume interaction parameter
ε > 0 via
unb(
r) =
εu0(
r) with the normalized pair potential
u0(
r) satisfying
. Another necessary condition for applying the SCF theory (i.e., having finite
βUnb/
n) is
ε ∝
ρ−1.
Here we compare the correlation effects on the structural and thermodynamic properties of hard- and soft-core generic polymer models, which has rarely been reported [
14], to further reveal their differences. For this purpose we choose the polymer reference interaction site model (PRISM) theory proposed by Schweizer and Curro [
15], which has been applied to many polymeric systems, including homopolymer melts, solutions, blends, block copolymers, nanocomposites, polyelectrolytes, etc. [
16,
17,
18,
19] It can be considered as the most successful molecular-level theory to date for studying the correlations in homogeneous polymeric systems.
For the above homopolymer melts, the PRISM equation is given by
where
is the interchain total segment pair correlation function (PCF) with
and
σ the segment diameter (i.e., the range of
βunb(
r)),
is the normalized (i.e.,
) intrachain segment PCF,
is the interchain direct segment PCF,
denotes the 3D Fourier transform of a radial function
with
q being the wavenumber (in units of 1/
σ), and
is the dimensionless segment number density. For given
N,
and
ω, to solve for both
h and
c, a closure providing an approximate relation between them is needed; here we take the commonly used Percus–Yevick (PY) closure [
20]
which works well for our class of generic models where
is short-range and purely repulsive.
To be more specific, we consider two commonly used generic polymer models: the tangent hard-sphere chain (THSC) and the DPD models; the former is a hard-core model that uses
with
specifying the dimensionless bonded potential between two adjacent segments on the same chain and the hard-sphere (HS) potential
for
and 0 otherwise as
, and the latter is a soft-core model that uses
and the DPD potential
for
and 0 otherwise as
with the dimensionless interaction parameter
chosen to mimic the compressibility of water [
2]. In the thermodynamic limit, the structural and thermodynamic properties of these two models are controlled only by
N and
; typically, molecular simulations of the DPD model uses
or 5.
Finally, we note that is needed as an input for PRISM calculations. While in general the chain conformations characterized by depend on , for simplicity here we use the ideal-chain conformations by setting to with for the THSC model and for the DPD model, where , q is the wave vector for the 3D Fourier transform, and q=|q|.
For the two generic models that we consider here, the PY closure gives
. Since all previously reported numerical methods for PRISM calculations [
21,
22,
23,
24] are not optimal in this case, we first propose an efficient numerical approach as follows. We uniformly discretize the real-space interval [0, 1] into
m subintervals (thus
into
subintervals) each of length
, where
denotes the real-space cut-off, and take
(
i = 0, …,
m−1 for the DPD model and
i = 0, …,
m with
for the THSC model) as the independent variables to be solved. Our approach has three steps:
Given the initial guess of the independent variables and
, for the DPD model we calculate
(
j = 1, …,
M−1) via the discrete sine transform of type I (DST) [
25], which has the computational complexity of
O(
Mln
M) and gives
, the reciprocal-space cut-off
qc =
qM =
mπ and
; for the THSC model, due to the discontinuities in both
and its 1st-order derivative at
, we use an auxiliary function
for
and
otherwise with
and
(calculated via the fourth-order backward finite-difference formula [
26]), which is continuous in both its value and 1st-order derivative, to calculate
(
j = 1, …,
M) via the DST, which gives
. We also calculate
for the DPD model and
for the THSC model via the Romberg integration (RI) [
27].
We calculate (j = 0, …, M) obtained from Equation (1) with being the interchain indirect segment PCF (note that for the DPD model while for the THSC model), then for the DPD model (j = 1, …, M−1) via the DST (which gives ); for the THSC model, we use another auxiliary function to calculate (j = 1, …, M) via the DST, which gives . We also calculate for both models via the RI.
We calculate
(
i = 0, …,
m–1 for the DPD model and
i = 0, …,
m for the THSC model), then use the residual errors of Equation (2) (which becomes
for the THSC model) to converge the independent variables via the Anderson mixing [
28], which has the computational complexity of
O(
m) and can quickly converge a large set of nonlinear equations to a high accuracy.
We use the convergence criterion of
εc < 10
−10 with
εc denoting the maximum absolute value of the residual errors of the PY closure over all
(
i = 0, …,
m–1 for the DPD model and
i = 0, …,
m for the THSC model), and choose the values of
m (=4096 for the THSC model and 512 for the DPD model) and
(
if
N < 100 and
otherwise, rounded to the nearest integer, to capture the correlation-hole effect [
29]) such that the discretization errors are comparable to
εc. Our numerical approach has the least number of independent variables to be iteratively solved, greatly reduces
m (thus
M) both by analytically treating the discontinuities in the THSC model and by taking the inverse Fourier transform only for
(which decays toward 0 with increasing
q much faster than both
and
), and is essential for us to accurately solve the PRISM-PY theory for
N as large as 10
6 (where for the THSC model
M is about 8.2 × 10
6!). To the best of our knowledge, analytically treating the discontinuities caused by the HS potential has not been reported in numerical calculations of even the widely studied Ornstein–Zernike (OZ) equation [
30] (to which Equation (1) reduces for
N = 1); in
Supplemental Material we show that our numerical approach gives several orders of magnitude more accurate results than pyPRISM [
19], a recently developed Python-based open-source framework for PRISM calculations.
In the limit of
N→∞ and
σ→0 at finite root-mean-square end-to-end distance of the ideal chain
, the THSC model becomes the hard-core Gaussian thread model [
31] (HC CGC-
δ, where
Re,0 is taken as the unit of length); to compare the PRISM-PY results of these two models, we define two dimensionless parameters:
and the invariant degree of polymerization [
32]
;
controls the fluctuations in polymer melts, and for the THSC model it is easy to show that
∝
N at large
N.
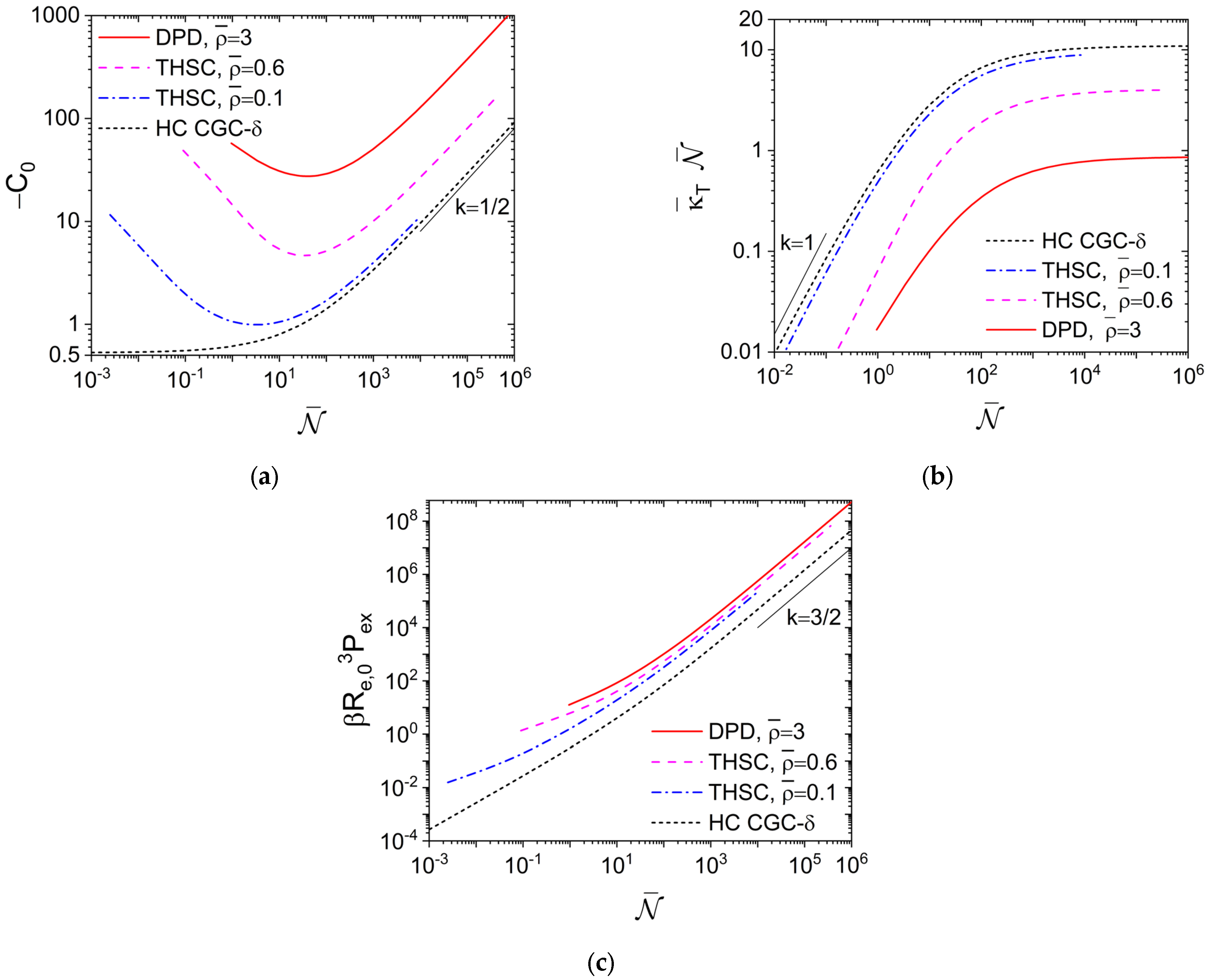
Figure 1a shows how
C0 varies with
for the THSC and HC CGC-
δ models; for the latter model,
is the only parameter, the PRISM-PY equation is given by Equation (18) in our previous work [
14] and the corresponding numerical results for
≥ 100 are shown in figure 8b there. We see that, while −
C0 increases monotonically with increasing
for the HC CGC-
δ model, it exhibits a minimum for the THSC model. At given
due to its
N→∞ the HC CGC-
δ model corresponds to the limit of
of the THSC model as implied in
Figure 1a. At large
, we see that
in all cases. This is in accordance with an asymptotic value of
at given
for the THSC model, while
for the HC CGC-
δ model.
Figure 1a also shows that the DPD model at
gives qualitatively the same behavior of
C0 vs.
as that for the THSC model.
With the normalized isothermal compressibility
given by the compressibility equation, where
ρc ≡
n/
V is the chain number density and
is the isothermal compressibility with
P denoting the system pressure,
Figure 1b presents essentially the same data as in
Figure 1a, but in a way that can be compared with real polymers used in experiments. As shown in figure 2 of our previous work [
14],
for polyethylene (at 180 °C) and 0.119 for polystyrene (at 280 °C), independent of their
≥10
3; this is consistent with
at large
shown in
Figure 1a. On the other hand, while
is expected for very small
, the smallest
(given by
N = 2) is about 0.0025, 0.090 and 0.95, respectively, for the THSC model at
and 0.6 and the DPD model at
. Clearly, both hard- and soft-core models can be used to describe the excluded-volume interactions in real polymers, and experimental values of
can be achieved by adjusting
, for example, in the THSC and DPD models. We attribute the largest
at the same
given by the HC CGC-
δ model to its
σ→0, and note that the DPD model at
is actually “harder” (i.e., more difficult to compress) than the hard-core models studied here.
Figure 1c shows that at large
, the dimensionless excess (virial) pressure due to the interchain repulsion
scales with
for the THSC model; this is due to the same scaling of
Re,03 with
N and also found for the HC CGC-
δ model (where
). We also see that the HC CGC-
δ model gives a much smaller
βRe,03Pex than the THSC model at the same
, again due to its
σ→0.
Figure 1c further shows that at large
, the DPD model at
gives the same scaling of
with
as the hard-core models; at the same
, it has even the largest
, in accordance with its smallest
shown in
Figure 1b.
Note that for both the THSC and DPD models,
is varied by changing
N at fixed
in
Figure 1, which makes
and
N to be approximately on the same order; it is therefore very difficult, if possible at all, to reach via this way in molecular simulations even a relatively small
-value of 10
4 used in experiments. As aforementioned, molecular simulations of soft-core models can readily reach
-values used in experiments by increasing
at fixed
N [
6,
7,
12]. For the DPD model at large
,
and the PY closure approaches the random-phase approximation (RPA) closure [
33,
34]
, which gives
and
independent of
N. We then obtain
from the compressibility equation.
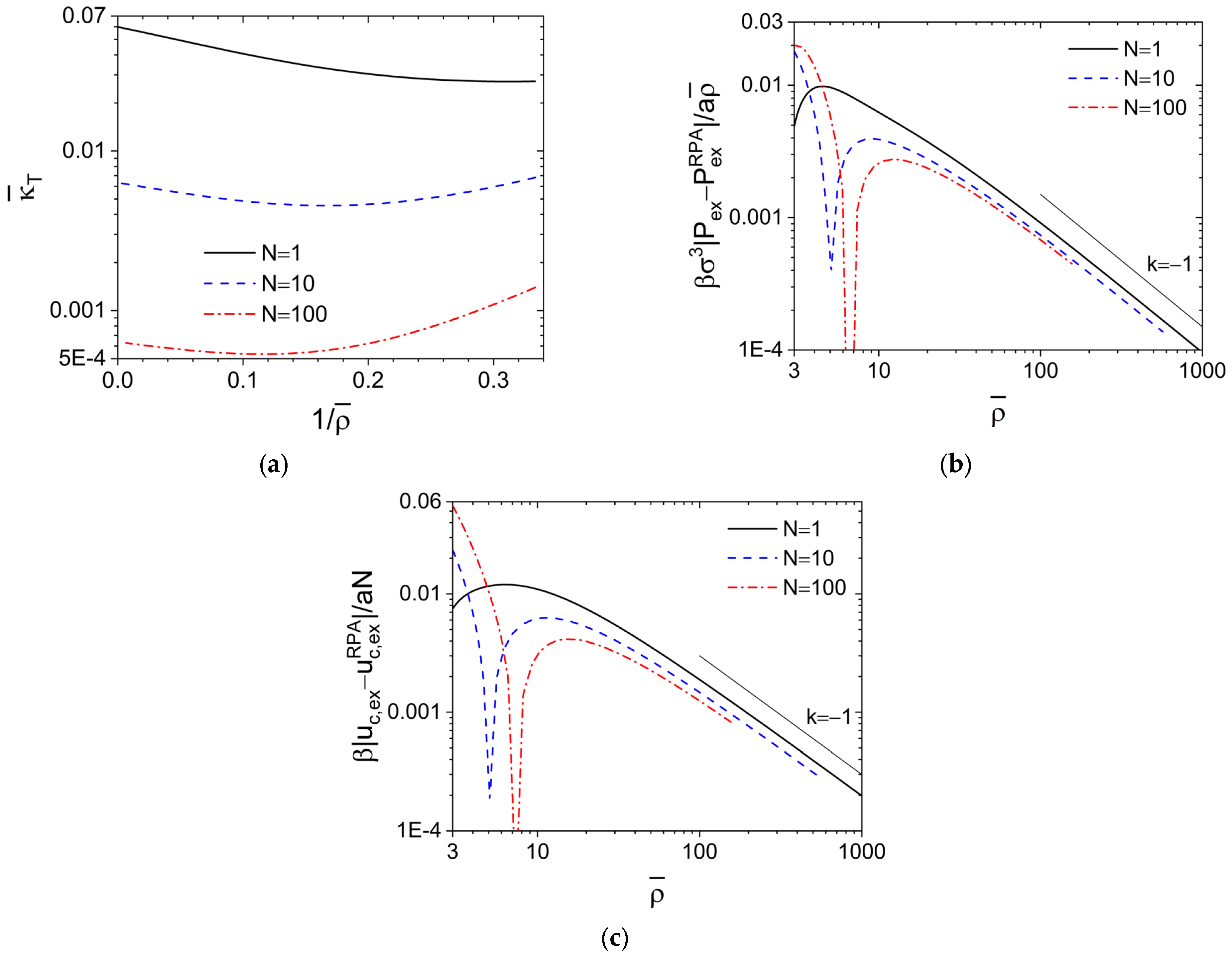
Figure 2a shows
vs.
obtained via the compressibility equation from our PRISM-PY calculations of the DPD model at various
N, where each curve exhibits a minimum with its location shifting to smaller
with increasing
N and the intercept of each (extrapolated) curve with the left axis (i.e., in the limit of
) gives the corresponding
. Clearly, the difference between
and
is entirely due to that between the PY and RPA closures.
A
C0 vs.
plot (not shown) can be obtained from
Figure 2a for the DPD model. In particular, the RPA closure gives
, indicating that
at large
; this is in clear contrast to
for the hard-core models and the DPD model at
shown in
Figure 1a, but consistent with the soft-core Gaussian thread (SC CGC-
δ) model (which is equivalent to the well-known Edwards model [
35]) shown in figure 8a of our previous work [
14], where
N→∞ and
σ→0 at finite
Re,0 and
is used with a
finite dimensionless parameter
controlling the repulsion strength between polymer segments. The behavior of soft-core models at large
, therefore, depends on how
is varied, i.e., whether by changing
N at fixed
(thus the excluded-volume interaction parameter
ε is fixed) or by changing
at fixed
N (thus
ε is also varied as
); in the former case correlations exist even in the limit of
N→∞, while in the latter case the SCF theory becomes exact in the limit of
(at finite
N) where neither fluctuations nor correlations exist.
As aforementioned, with increasing at fixed N, the PY closure approaches the RPA closure, which gives thus according to Equation (1). In the limit of , we have and , thus the SCF results of and independent of N, where denotes the dimensionless excess internal energy per chain due to the interchain repulsion. On the other hand, the differences between the SCF and RPA results as given by and are independent of .
Finally,
Figure 2b shows that
at large
; note that
at large
while
at small
, which leads to the cusp of each curve shown in the figure with its location (i.e., the
-value at which
) increasing with increasing
N (the cusp at
N = 1 is located around
). We also note that
, 0.119 and 0.182 for
N = 1, 10 and 100. Therefore, with increasing
, both
and
approach
. Similar results are found for
as shown in
Figure 2c, where
at large
while
at small
(with the cusp at
N = 1 located around
); also note that
, 0.253 and 0.322 for
N=1, 10 and 100. In particular, the PRISM-RPA theory with
is equivalent to the Gaussian-fluctuation theory neglecting non-Gaussian fluctuations in the system and gives a correction
to the SCF result, while the PRISM-PY theory captures non-Gaussian fluctuations in an approximate way and gives a leading-order correction
to the Gaussian-fluctuation result. These are consistent with our previous study of compressible [
36] and incompressible [
37] homopolymer melts using fast lattice Monte Carlo simulations [
6,
7]. Given this and the agreement of our
Figure 1b with experimental results at large
, we do not expect that the use of more accurate
can qualitatively change our PRISM-PY results here.
To summarize, we have compared the correlation effects on the structural and thermodynamic properties of hard-core models (i.e., the THSC model and its limit of
N→∞ at finite
Re,0 (or equivalently
at given
), the HC CGC-
δ model [
31]) and soft-core models (i.e., the DPD model and its limit of
N→∞ at finite
Re,0, the Edwards model [
35]) for compressible homopolymer melts (or equivalently homopolymer solutions in an implicit solvent) given by the PRISM-PY theory. The behavior of soft-core models at large
depends on how
is varied, i.e., whether by changing
N at fixed
(thus
ε is fixed) or by changing
at fixed
N (thus
ε is also varied as being inversely proportional to
). In the former case, correlations exist even in the limit of
N→∞, and both the hard-core and the DPD models give
at large
, consistent with real polymers used in experiments; it is, however, very difficult to reach via this way in molecular simulations even a relatively small
-value of 10
4 used in experiments. This problem is solved in the latter case, where the widely used polymer SCF theory becomes exact in the limit of
(at finite
N), the Gaussian-fluctuation theory gives a correction
to the SCF result, and the PRISM-PY theory captures non-Gaussian fluctuations in the system in an approximate way and gives a leading-order correction
to the Gaussian-fluctuation result, consistent with our previous simulations [
36,
37]. The soft-core models, however, give
at large
, suggesting that it would be difficult, if possible at all, for the various recently proposed simulation methods [
3,
4,
5,
6,
7,
8,
9,
10,
11] to capture both the fluctuations and correlations in experimental systems. We also proposed an efficient numerical approach, which enables us to accurately solve the PRISM-PY theory for
N as large as 10
6; numerical calculations of such theories can, therefore, capture both the fluctuations and correlations in experimental systems.



{kind=link}
{kind=link}

