
Nanocarrier of α-Tocopheryl Succinate Based on a Copolymer Derivative of (4,7-dichloroquinolin-2-yl)methanol and Its Cytotoxicity against a Breast Cancer Cell Line

 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Experimental
2.1. Materials and Reagents
2.2. Spectroscopic Characterization
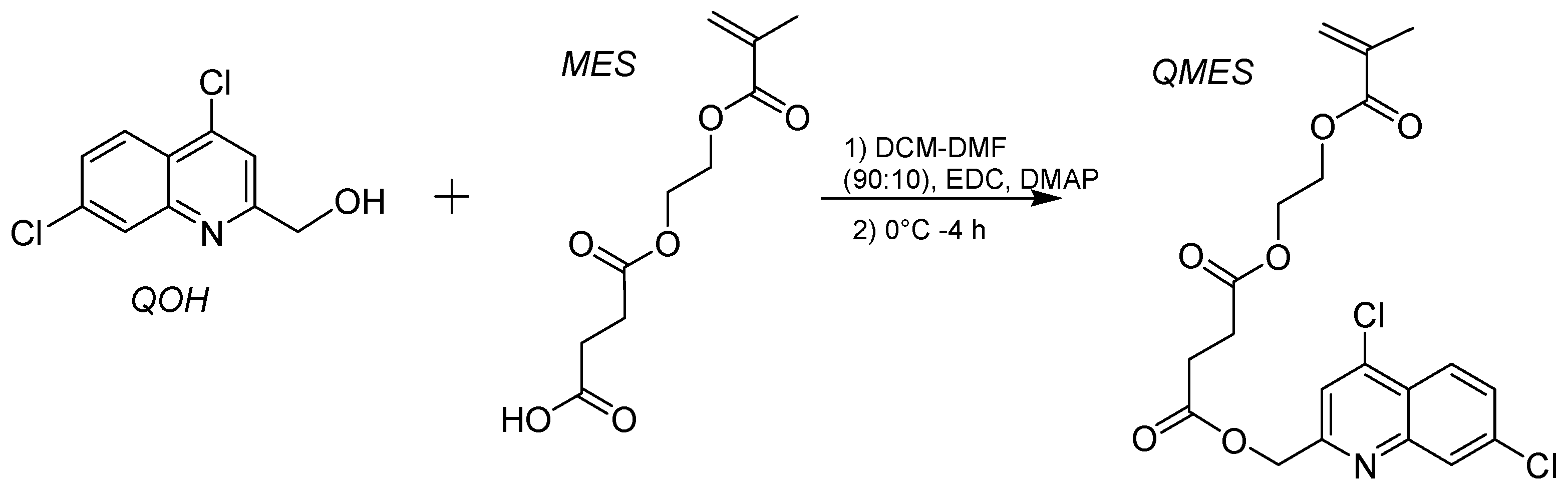
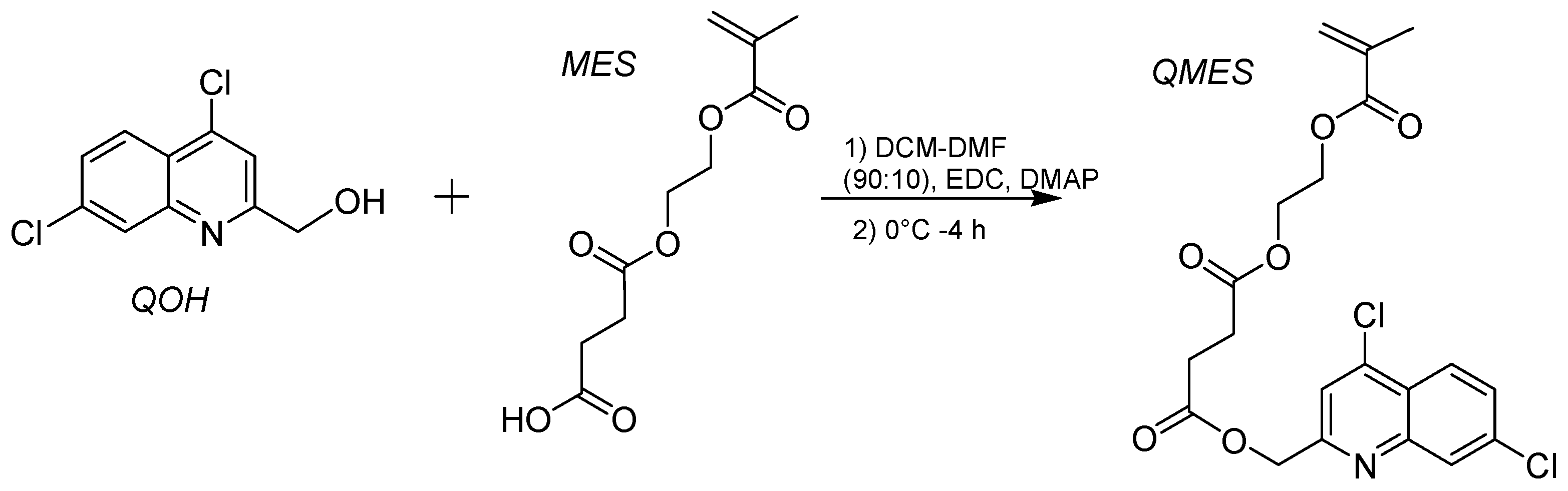
2.3. Synthesis of QMES
2.4. Synthesis of Poly(QMES-co-VP) Copolymers
2.5. Gel Permeation Chromatography (GPC)
2.6. Differential Scanning Calorimetry
2.7. Thermogravimetric Analysis (TGA)
2.8. Preparation and Characterization of Copolymer Nanoparticles by Nanoprecipitation [14,32]
2.9. Determination of Encapsulation Efficiency (EE) of α-TOS
2.10. Cell Culture and Biological Reagents
2.11. Cytotoxicity Assay on MCF-7 Cells and HUVEC Cells
2.12. c6 Loaded NPs Cellular Uptake and Intracellular Location
2.13. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and 1H NMR Analysis of the QMES Monomer
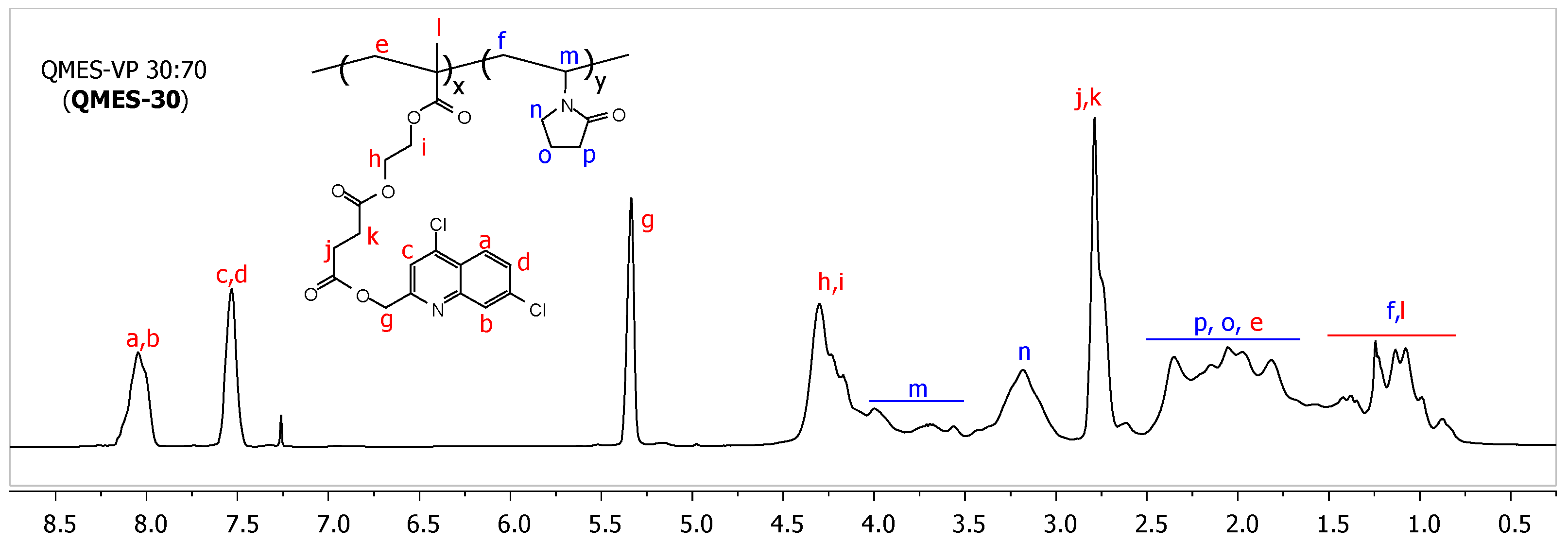
3.2. Synthesis and 1H NMR Analysis of QMES-VP Copolymers
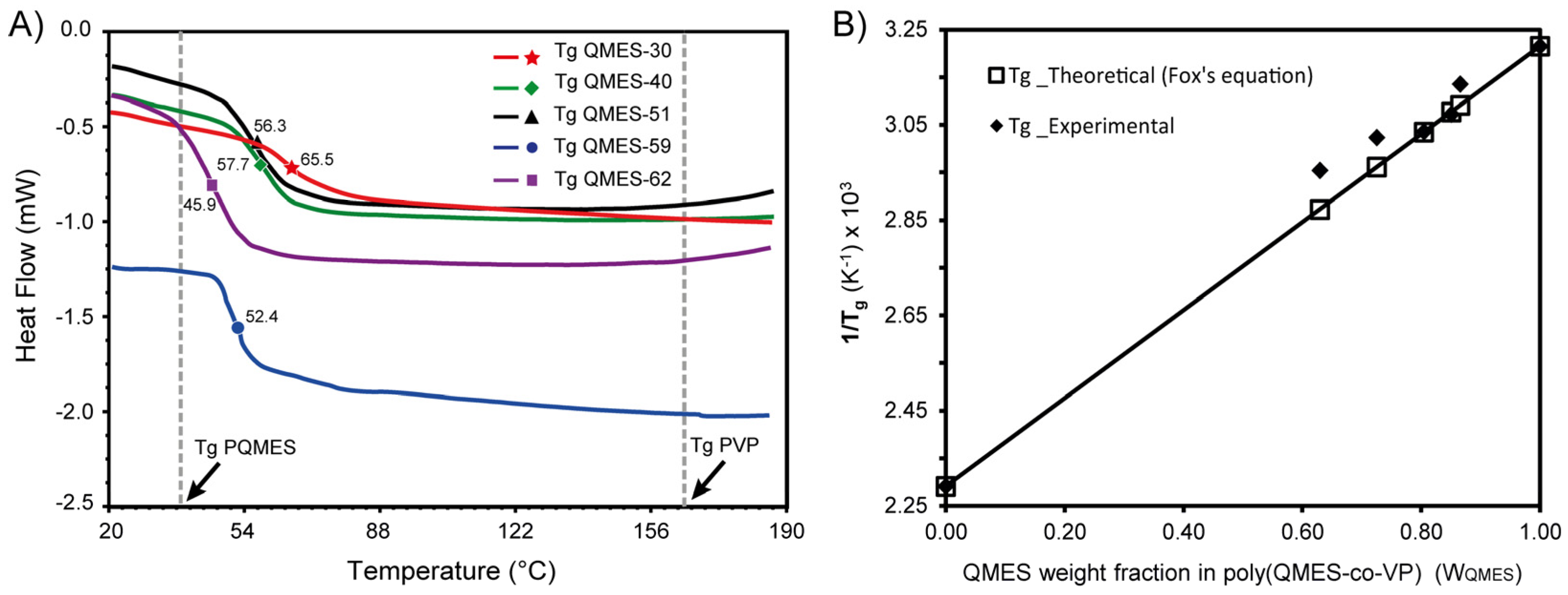
3.3. DSC Analysis of QMES-VP Copolymers and Homopolymers
3.4. GPC Analysis of QMES-VP Copolymers and Homopolymers
3.5. Thermogravimetric Analysis of QMES-VP Copolymers and Homopolymers
3.6. Characterization of α-TOS-Loaded and Empty Poly(QMES-co-VP) Nanoparticles
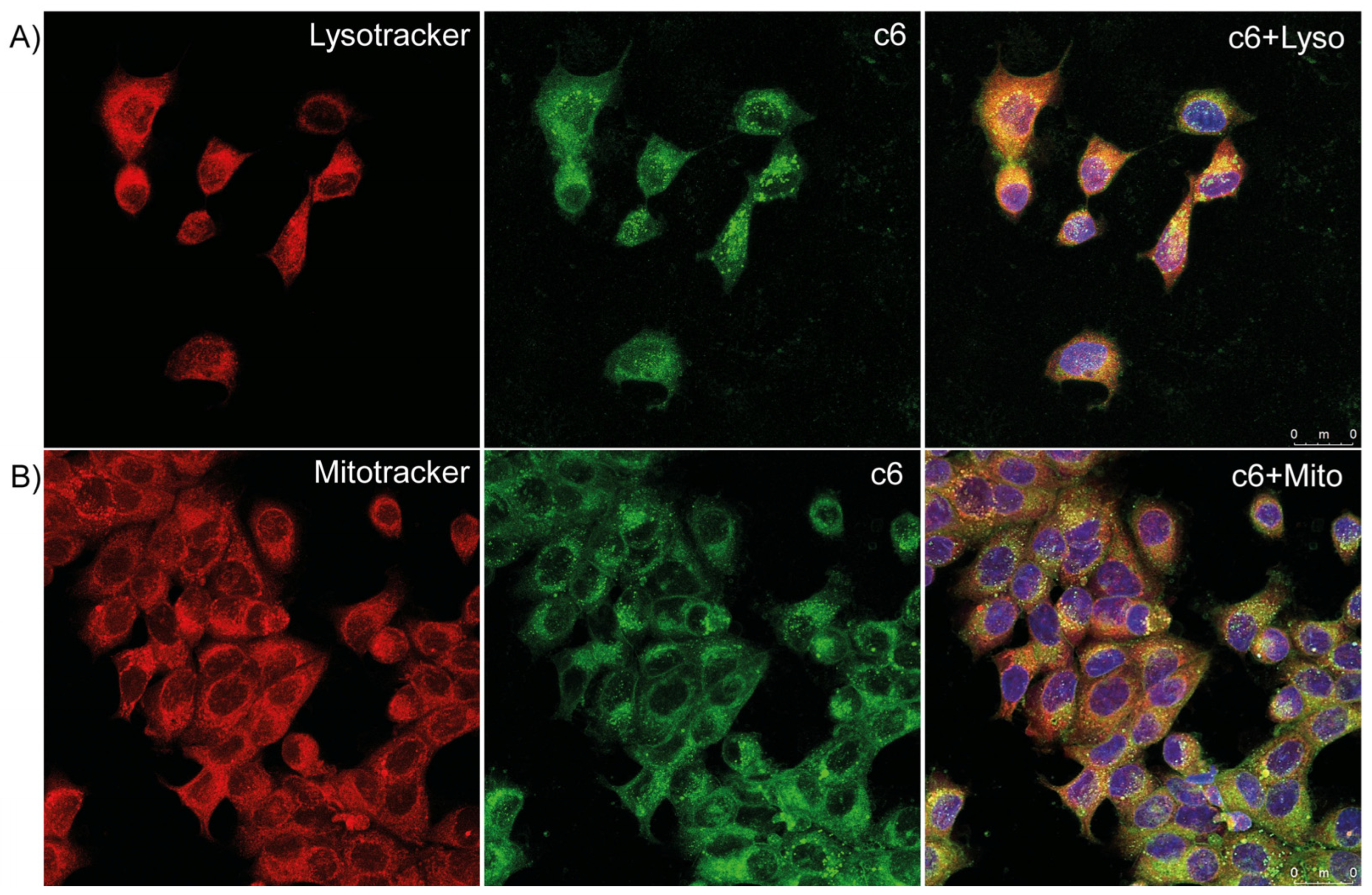
3.7. Uptake and Co-Localization of c6-Loaded NP
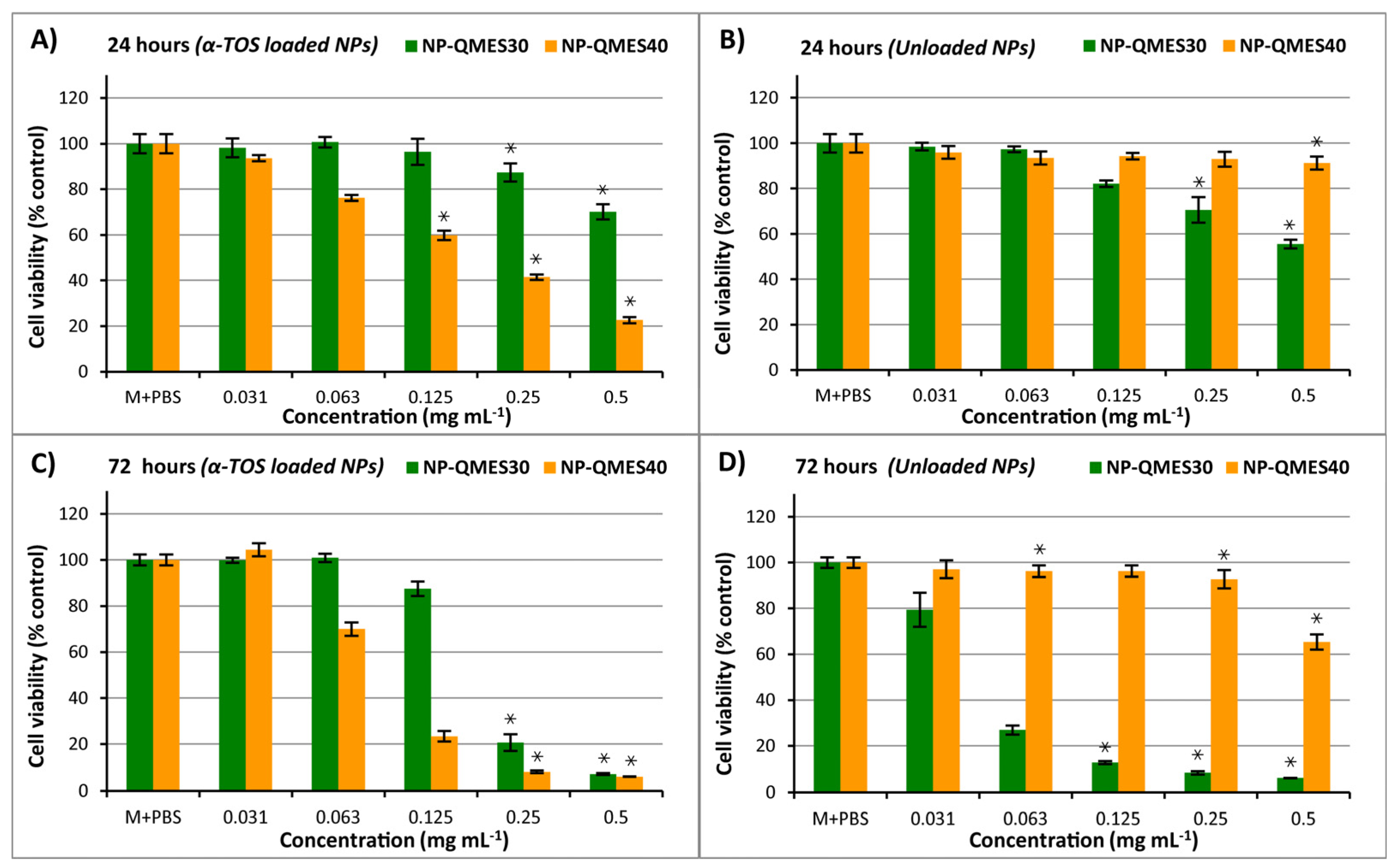
3.8. Cytotoxic Activity of Poly(QMES-co-VP) Nanoparticles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan American Health Organization PAHO/WHO. World Cancer Day 2023: Close the Care Gap. Available online: https://www.paho.org/en/campaigns/world-cancer-day-2023-close-care-gap (accessed on 15 July 2023).
- Schwartz, G.K. (Ed.) Combination Cancer Therapy: Modulators and Potentiators; Humana Press: Totowa, NJ, USA, 2005; ISBN 1-58829-200-2. [Google Scholar]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and Hydroxychloroquine for Cancer Therapy. Mol. Cell. Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine Enhances Temozolomide Cytotoxicity in Malignant Gliomas by Blocking Autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Cho, H.-Y.; Hofman, F.M.; Louie, S.G.; Schonthal, A.H.; Chen, T.C. Quinoline-Based Antimalarial Drugs: A Novel Class of Autophagy Inhibitors. Neurosurg. Focus 2015, 38, E12. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Tao, S.; Rojo de la Vega, M.; Park, S.L.; Vonderfecht, A.A.; Jacobs, S.L.; Zhang, D.D.; Wondrak, G.T. The Antimalarial Amodiaquine Causes Autophagic-Lysosomal and Proliferative Blockade Sensitizing Human Melanoma Cells to Starvation- and Chemotherapy-Induced Cell Death. Autophagy 2013, 9, 2087–2102. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Adasme, M.F.; Heinrich, J.C.; Haupt, V.J.; Daminelli, S.; Zhang, Y.; Schroeder, M. From Malaria to Cancer: Computational Drug Repositioning of Amodiaquine Using PLIP Interaction Patterns. Sci. Rep. 2017, 7, 11401. [Google Scholar] [CrossRef]
- Huang, P.; Li, Q. Combinational Uses of Nitroxoline and Its Analogues with Chemotherapies and Immunotherapies in the Treatment of Cancers. Patent Application No. AU2017240074B2, 2 January 2020. [Google Scholar]
- Prachayasittikul, V.; Chan-On, W.; Nguyen Thi Bich, H.; Songtawee, N.; Suwanjang, W.; Prachayasittikul, S. Quinoline-Based Clioquinol and Nitroxoline Exhibit Anticancer Activity Inducing FoxM1 Inhibition in Cholangiocarcinoma Cells. Drug Des. Devel. Ther. 2015, 9, 2033–2047. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Taggart, J.E.; Zhang, X.; Benbrook, D.M.; Lind, S.E.; Ding, W.-Q. Nitroxoline (8-Hydroxy-5-Nitroquinoline) Is More a Potent Anti-Cancer Agent than Clioquinol (5-Chloro-7-Iodo-8-Quinoline). Cancer Lett. 2011, 312, 11–17. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S.; Ali, R.; Kumar, R.; Jaggi, M.; et al. A Review on Anticancer Potential of Bioactive Heterocycle Quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Sutradhar, K.B.; Lutful Amin, M.; Amin, M.L. Nanotechnology in Cancer Drug Delivery and Selective Targeting. ISRN Nanotechnol. 2014, 2014, 939378. [Google Scholar] [CrossRef]
- Tabero, A.; Xian, W.; Martínez De Pinillos, A.; Acedo, P. Nanoterapias en el campo de la Biomedicina. Revista SEBBM 2017, 192, 24–29. [Google Scholar]
- Palao-Suay, R.; Aguilar, M.R.; Parra-Ruiz, F.J.; Fernández-Gutiérrez, M.; Parra, J.; Sánchez-Rodríguez, C.; Sanz-Fernández, R.; Rodrigáñez, L.; Román, J.S. Anticancer and Antiangiogenic Activity of Surfactant-Free Nanoparticles Based on Self-Assembled Polymeric Derivatives of Vitamin E: Structure–Activity Relationship. Biomacromolecules 2015, 16, 1566–1581. [Google Scholar] [CrossRef] [PubMed]
- Palao-Suay, R.; Rodrigáñez, L.; Aguilar, M.R.; Sánchez-Rodríguez, C.; Parra, F.; Fernández, M.; Parra, J.; Riestra-Ayora, J.; Sanz-Fernández, R.; Román, J.S. Mitochondrially Targeted Nanoparticles Based on α-TOS for the Selective Cancer Treatment. Macromol. Biosci. 2016, 16, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Alven, S.; Nqoro, X.; Buyana, B.; Aderibigbe, B.A. Polymer-Drug Conjugate, a Potential Therapeutic to Combat Breast and Lung Cancer. Pharmaceutics 2020, 12, 406. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. Polymer–Drug Conjugation, Recent Achievements and General Strategies. Prog. Polym. Sci. 2007, 32, 933–961. [Google Scholar] [CrossRef]
- Agrawal, N.; Rohini; Joseph, A.; Mukerjee, A. Polymeric Prodrugs: Recent Achievements and General Strategies. J. Antivir. Antiretrovir. 2013, S15, 1–12. [Google Scholar] [CrossRef]
- Greco, F.; Vicent, M.J. Combination Therapy: Opportunities and Challenges for Polymer–Drug Conjugates as Anticancer Nanomedicines. Adv. Drug Deliv. Rev. 2009, 61, 1203–1213. [Google Scholar] [CrossRef]
- Theodosis-Nobelos, P.; Charalambous, D.; Triantis, C.; Rikkou-Kalourkoti, M. Drug Conjugates Using Different Dynamic Covalent Bonds and Their Application in Cancer Therapy. Curr. Drug Deliv. 2020, 17, 542–557. [Google Scholar] [CrossRef]
- Lee, H.; Chung, H.J.; Park, T.G. Perspectives On: Local and Sustained Delivery of Angiogenic Growth Factors. J. Bioact. Compat. Polym. 2007, 22, 89–114. [Google Scholar] [CrossRef]
- Hickey, J.W.; Santos, J.L.; Williford, J.-M.; Mao, H.-Q. Control of Polymeric Nanoparticle Size to Improve Therapeutic Delivery. J. Control. Release 2015, 219, 536–547. [Google Scholar] [CrossRef]
- Javia, A.; Vanza, J.; Bardoliwala, D.; Ghosh, S.; Misra, L.A.; Patel, M.; Thakkar, H. Polymer-Drug Conjugates: Design Principles, Emerging Synthetic Strategies and Clinical Overview. Int. J. Pharm. 2022, 623, 121863. [Google Scholar] [CrossRef]
- Huang, L.; Song, J.; Chen, B. A Novel Targeting Drug Carrier to Deliver Chemical Bonded and Physical Entrapped Anti-Tumor Drugs. Int. J. Pharm. 2014, 466, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Delplace, V.; Couvreur, P.; Nicolas, J. Recent Trends in the Design of Anticancer Polymer Prodrug Nanocarriers. Polym. Chem. 2014, 5, 1529–1544. [Google Scholar] [CrossRef]
- Esparza-Ruiz, A.; Herrmann, C.; Chen, J.; Patrick, B.O.; Polishchuk, E.; Orvig, C. Synthesis and in Vitro Anticancer Activity of Ferrocenyl-Aminoquinoline-Carboxamide Conjugates. Inorganica Chim. Acta 2012, 393, 276–283. [Google Scholar] [CrossRef]
- Ireland, D.J.; Kissick, H.T.; Beilharz, M.W. Alpha-Tocopheryl Succinate: Toxicity and Lack of Anti-Tumour Activity in Immuno-Competent Mice. Food Chem. Toxicol. 2008, 46, 508–512. [Google Scholar] [CrossRef]
- Koudelka, S.; Turanek Knotigova, P.; Masek, J.; Prochazka, L.; Lukac, R.; Miller, A.D.; Neuzil, J.; Turanek, J. Liposomal Delivery Systems for Anti-Cancer Analogues of Vitamin E. J. Control. Release 2015, 207, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Valle, H.; Viswanathan Mangalaraja, R.; Aguilar, L.; Carrasco, C.; Padilla, N.; Dahrouch, M.; Rivas, B.; Urrutia, H. Antibacterial activity and fluorescence properties of 4,7-dichloro-2-quinolinemethylacrylate. J. Chil. Chem. Soc. 2020, 65, 4784–4789. [Google Scholar] [CrossRef]
- Park, Y.; Fei, X.; Yuan, Y.; Lee, S.; Hur, J.; Park, S.J.; Jung, J.-K.; Seo, S.-Y. Chemoselective Acylation of 2-Amino-8-Quinolinol in the Generation of C2-Amides or C8-Esters. RSC Adv. 2017, 7, 41955–41961. [Google Scholar] [CrossRef]
- Yearick, K.; Ekoue-Kovi, K.; Iwaniuk, D.P.; Natarajan, J.K.; Alumasa, J.; de Dios, A.C.; Roepe, P.D.; Wolf, C. Overcoming Drug Resistance to Heme-Targeted Antimalarials by Systematic Side Chain Variation of 7-Chloro-4-Aminoquinolines. J. Med. Chem. 2008, 51, 1995–1998. [Google Scholar] [CrossRef]
- Valle, H.; Palao-Suay, R.; Aguilar, M.R.; Román, J.S.; Becerra, J.; Rivas, B.; Mangalaraja, R.V. Nanoparticles of 4,7-Dichloro-2-Quinolinemethylacrylate-Based Copolymers and Their Potential Cytotoxic Activity on Human Breast Carcinoma Cells. J. Appl. Polym. Sci. 2019, 136, 47545. [Google Scholar] [CrossRef]
- Rudin, A. The Elements of Polymer Science and Engineering, 2nd ed.; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Brostow, W.; Chiu, R.; Kalogeras, I.M.; Vassilikou-Dova, A. Prediction of Glass Transition Temperatures: Binary Blends and Copolymers. Mater. Lett. 2008, 62, 3152–3155. [Google Scholar] [CrossRef]
- Wong, C.L.; Kim, J.; Torkelson, J.M. Breadth of Glass Transition Temperature in Styrene/Acrylic Acid Block, Random, and Gradient Copolymers: Unusual Sequence Distribution Effects. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 2842–2849. [Google Scholar] [CrossRef]
- Kuo, S.-W.; Xu, H.; Huang, C.-F.; Chang, F.-C. Significant Glass-Transition-Temperature Increase through Hydrogen-Bonded Copolymers. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 2313–2323. [Google Scholar] [CrossRef]
- Molyneux, P.; Cornarakis-Lentzos, M. The Interaction of Polyvinylpyrrolidone with Aromatic Compounds in Aqueous Solution Part IV. Evaluation of the Co-Operativity Parameter, and the Methylene-Group Contribution to the Binding Strength, for the Alkyl Parahydroxybenzoates. Colloid Polym. Sci. 1979, 257, 855–873. [Google Scholar] [CrossRef]
- Pound, G. Reversible Addition Fragmentation Chain Transfer (RAFT) Mediated Polymerization of N-Vinylpyrrolidone. Ph.D. Thesis, Stellenbosch University, Stellenbosch, South Africa, 2008. [Google Scholar]
- Brandup, J.; Immergut, E.H.; Grulke, E.A. Polymer Handbook, 4th ed.; Abe, A., Bloch, D.R., Eds.; John Wiley and Sons: New York, NY, USA, 1999. [Google Scholar]
- Dikusar, M.A.; Kubrakova, I.V.; Chinarev, A.A.; Bovin, N.V. Polymerization of 4-Nitrophenyl Acrylate under Microwave Heating Conditions. Russ. J. Bioorg. Chem. 2001, 27, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I. Synthesis and Characterisation of Polymeric Materials via RAFT Polymerisation. Ph.D. Thesis, University of Durham, Durham, UK, 2013. [Google Scholar]
- Deng, W. Poly(Acrylonitrile-Co-1-Vinylimidazole): A New Melt Processable Carbon Fiber Precursor. Ph.D. Thesis, Clemson University, Clemson, SC, USA, 2010. [Google Scholar]
- Moad, G.; Li, G.; Pfaendner, R.; Postma, A.; Rizzardo, E.; Thang, S.; Wermter, H. RAFT Copolymerization and Its Application to the Synthesis of Novel Dispersants—Intercalants—Exfoliants for Polymer—Clay Nanocomposites. In Controlled/Living Radical Polymerization; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2006; Volume 944, pp. 35–514. ISBN 9780841239913. [Google Scholar]
- Odian, G. Principles of Polymerization, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; ISBN 0-471-27400-3. [Google Scholar]
- Tkachenko, V.; Vidal, L.; Josien, L.; Schmutz, M.; Poly, J.; Chemtob, A. Characterizing the Core-Shell Architecture of Block Copolymer Nanoparticles with Electron Microscopy: A Multi-Technique Approach. Polymers 2020, 12, 1656. [Google Scholar] [CrossRef]
- Li, L.; Raghupathi, K.; Song, C.; Prasad, P.; Thayumanavan, S. Self-Assembly of Random Copolymers. Chem. Commun. 2014, 50, 13417–13432. [Google Scholar] [CrossRef] [PubMed]
- Iversen, T.G.; Skotland, T.; Sandvig, K. Endocytosis and Intracellular Transport of Nanoparticles: Present Knowledge and Need for Future Studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Ren, S.; Sun, X.; Wang, H.; Nguyen, T.H.; Sadeghipour, N.; Xu, X.; Kang, C.S.; Liu, Y.; Xu, H.; Wu, N.; et al. Design, Synthesis, and Biological Evaluation of Polyaminocarboxylate Ligand-Based Theranostic Conjugates for Antibody-Targeted Cancer Therapy and Near-Infrared Optical Imaging. ChemMedChem 2018, 13, 2606–2617. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and Cancer: More Ore to Be Mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef]
- Yuan, J.; Lovejoy, D.B.; Richardson, D.R. Novel Di-2-Pyridyl–Derived Iron Chelators with Marked and Selective Antitumor Activity: In Vitro and in Vivo Assessment. Blood 2004, 104, 1450–1458. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | fQMES | FQMES | WQMES | Yield (%) | Tg * (°C) | Tg_Fox ≠ (°C) | Mw (KDa) | Mn (KDa) | Ð |
|---|---|---|---|---|---|---|---|---|---|
| PVP | 0 | 0 | 0 | 55.2 | 163.4 | 163.4 | 18.6 | 13.1 | 1.42 |
| QMES-30 | 0.20 | 0.30 | 0.63 | 56.2 | 65.5 | 75.1 | 18.7 | 9.50 | 1.96 |
| QMES-40 | 0.30 | 0.40 | 0.73 | 61.6 | 57.7 | 64.7 | 23.7 | 11.4 | 2.09 |
| QMES-51 | 0.50 | 0.51 | 0.80 | 68.9 | 56.3 | 56.5 | 29.1 | 13.4 | 2.18 |
| QMES-59 | 0.70 | 0.59 | 0.85 | 76.4 | 52.4 | 52.0 | 31.2 | 13.9 | 2.25 |
| QMES-62 | 0.80 | 0.62 | 0.87 | 84.4 | 45.9 | 50.5 | 32.9 | 14.1 | 2.34 |
| PQMES | 1 | 1 | 1 | 82.2 | 38.0 | 38.0 | 26.7 | 13.3 | 2.01 |
| Copolymer | TgA1 (°C) | ΔW1 | TgA2 (°C) | ΔW2 |
|---|---|---|---|---|
| PVP | --- | --- | 438.7 | 89.8 |
| QMES-30 | 302.8 | 32.8 | 400.8 | 46.0 |
| QMES-40 | 303.4 | 33.9 | 392.9 | 37.8 |
| QMES-51 | 305.2 | 34.8 | 388.5 | 38.3 |
| QMES-59 | 294.2 | 34.3 | 385.1 | 36.0 |
| QMES-62 | 291 | 32.9 | 370.6 | 33.4 |
| PQMES | 287.5 | 42.9 | 368.9 | 28.2 |
| Sample | FQMES | [NPs] (mg mL−1) | α-TOS (%) | c6 (%) | Dh (nm) | PDI | ZP (mV) | EE (%) | * LE (mg mg−1) |
|---|---|---|---|---|---|---|---|---|---|
| NP-QMES-30 | 0.30 | 1 | 0 | 0 | 118.9 ± 2.5 | 0.056 ± 0.017 | −3.2 ± 0.5 | --- | --- |
| NP-QMES-30.TOS | 0.30 | 1 | 10 | 0 | 128.6 ± 5.8 | 0.063 ± 0.020 | −15.3 ± 1.5 | 52 ± 3 | 0.052 |
| NP-QMES-30.c6 | 0.30 | 1 | 0 | 1 | 124.4 ± 2.7 | 0.120 ± 0.020 | 2.5 ± 0.2 | --- | --- |
| NP-QMES-40 | 0.40 | 1 | 0 | 0 | 177.5 ± 3.4 | 0.031 ± 0.010 | −2.8 ± 0.4 | --- | --- |
| NP-QMES-40.TOS | 0.40 | 1 | 10 | 0 | 148.8 ± 8.6 | 0.060 ± 0.015 | −17.6 ± 3.2 | 65 ± 5 | 0.065 |
| NP-QMES-40.c6 | 0.40 | 1 | 0 | 1 | 247.3 ± 2.7 | 0.191 ± 0.012 | 2.2 ± 0.5 | --- | --- |
| NP-QMES-51 | 0.51 | 1 | 0 | 0 | 312.2 ± 5.2 | 0.331 ± 0.050 | --- | --- | --- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valle, H.; Palao-Suay, R.; Aguilar, M.R.; Lerma, T.A.; Palencia, M.; Mangalaraja, R.V.; Guzmán, L.; Pérez Sotelo, D.; Becerra, J. Nanocarrier of α-Tocopheryl Succinate Based on a Copolymer Derivative of (4,7-dichloroquinolin-2-yl)methanol and Its Cytotoxicity against a Breast Cancer Cell Line. Polymers 2023, 15, 4342. https://doi.org/10.3390/polym15224342
Valle H, Palao-Suay R, Aguilar MR, Lerma TA, Palencia M, Mangalaraja RV, Guzmán L, Pérez Sotelo D, Becerra J. Nanocarrier of α-Tocopheryl Succinate Based on a Copolymer Derivative of (4,7-dichloroquinolin-2-yl)methanol and Its Cytotoxicity against a Breast Cancer Cell Line. Polymers. 2023; 15(22):4342. https://doi.org/10.3390/polym15224342
Chicago/Turabian StyleValle, Hernán, Raquel Palao-Suay, María Rosa Aguilar, Tulio A. Lerma, Manuel Palencia, Ramalinga Viswanathan Mangalaraja, Leonardo Guzmán, Dairo Pérez Sotelo, and José Becerra. 2023. "Nanocarrier of α-Tocopheryl Succinate Based on a Copolymer Derivative of (4,7-dichloroquinolin-2-yl)methanol and Its Cytotoxicity against a Breast Cancer Cell Line" Polymers 15, no. 22: 4342. https://doi.org/10.3390/polym15224342
APA StyleValle, H., Palao-Suay, R., Aguilar, M. R., Lerma, T. A., Palencia, M., Mangalaraja, R. V., Guzmán, L., Pérez Sotelo, D., & Becerra, J. (2023). Nanocarrier of α-Tocopheryl Succinate Based on a Copolymer Derivative of (4,7-dichloroquinolin-2-yl)methanol and Its Cytotoxicity against a Breast Cancer Cell Line. Polymers, 15(22), 4342. https://doi.org/10.3390/polym15224342