
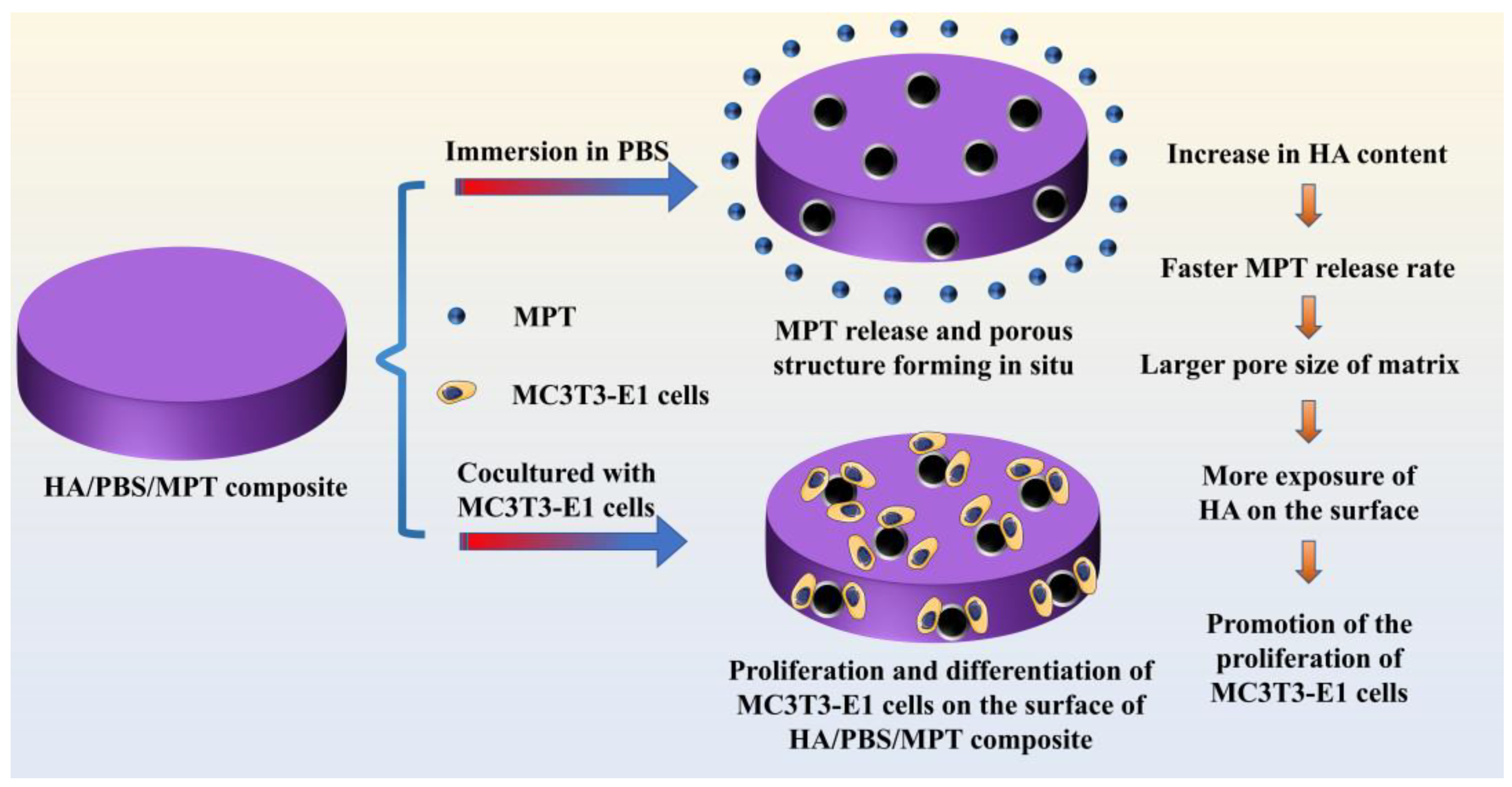
Hydroxyapatite/Poly (Butylene Succinate)/Metoprolol Tartrate Composites with Controllable Drug Release and a Porous Structure for Bone Scaffold Application


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. The Preparation of Composite Samples
2.3. The Characterization of Composite Samples
2.4. In Vitro Drug Release Test
2.5. In Vitro Cytocompatibility Evaluation
2.6. Statistics Analysis
3. Results
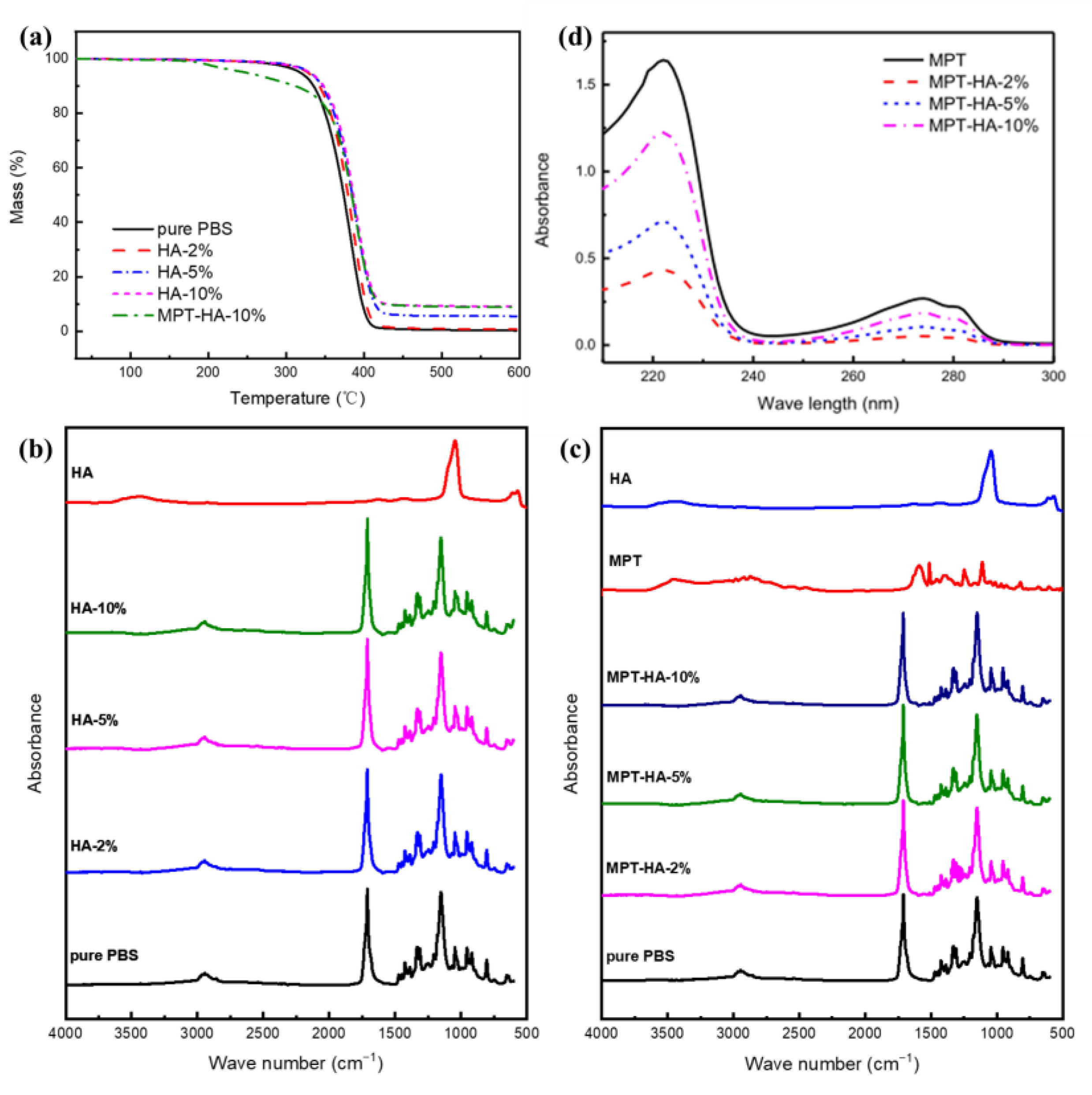
3.1. Thermal and Component Stability during Heat Treatment
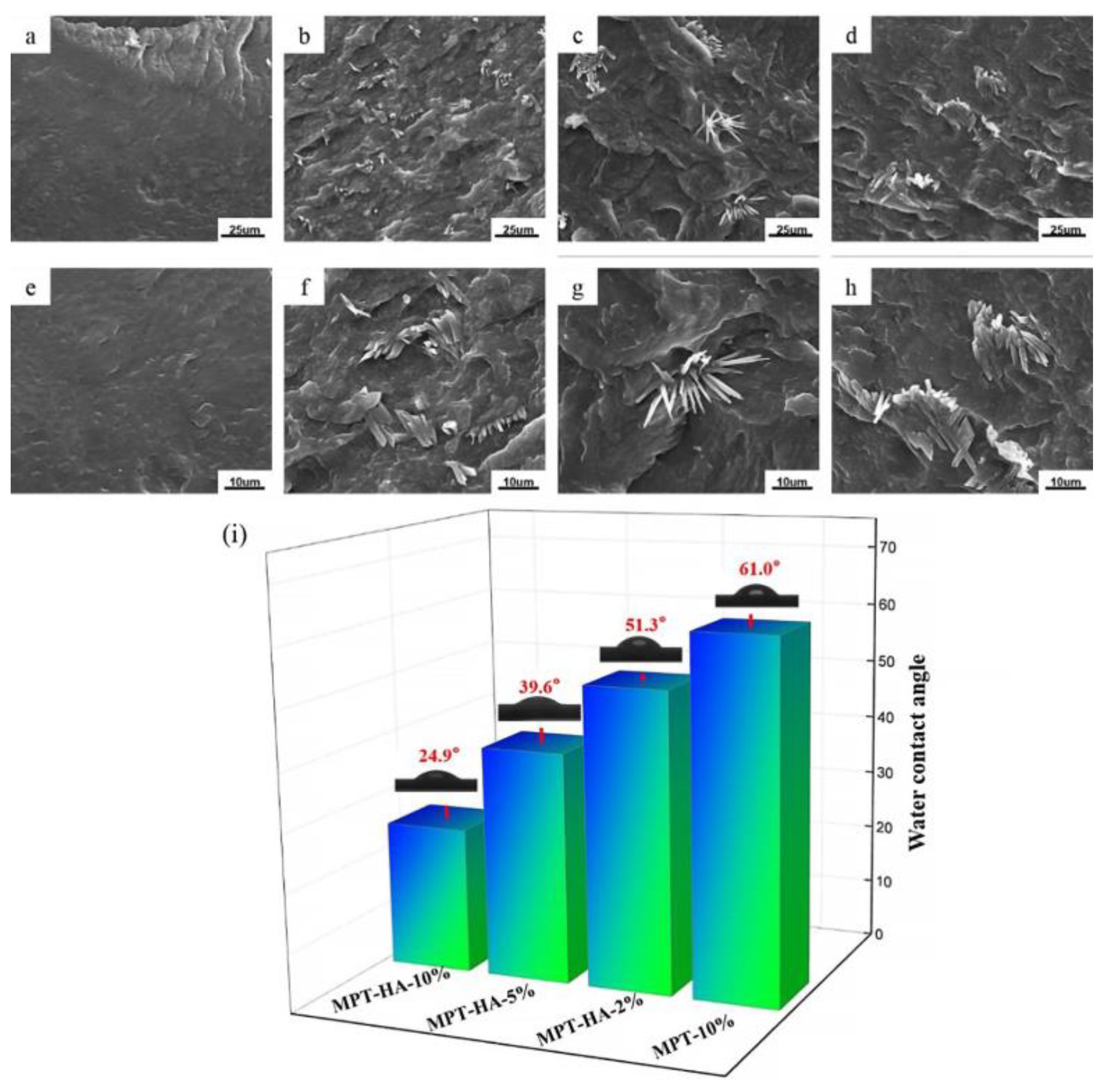
3.2. The Morphology and Hydrophilic Contact Angle Test
3.3. Crystal Morphology of Samples Evaluation
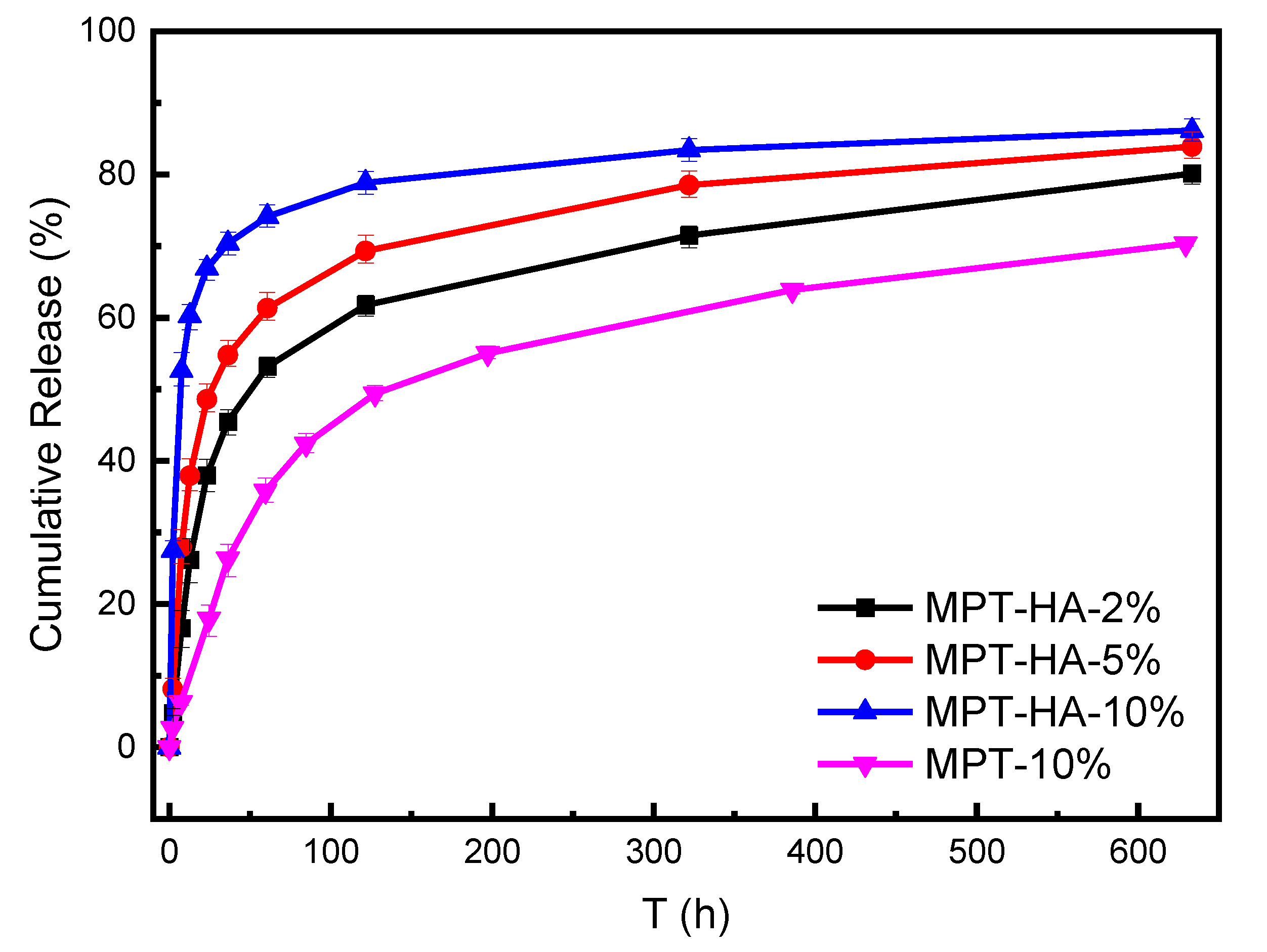
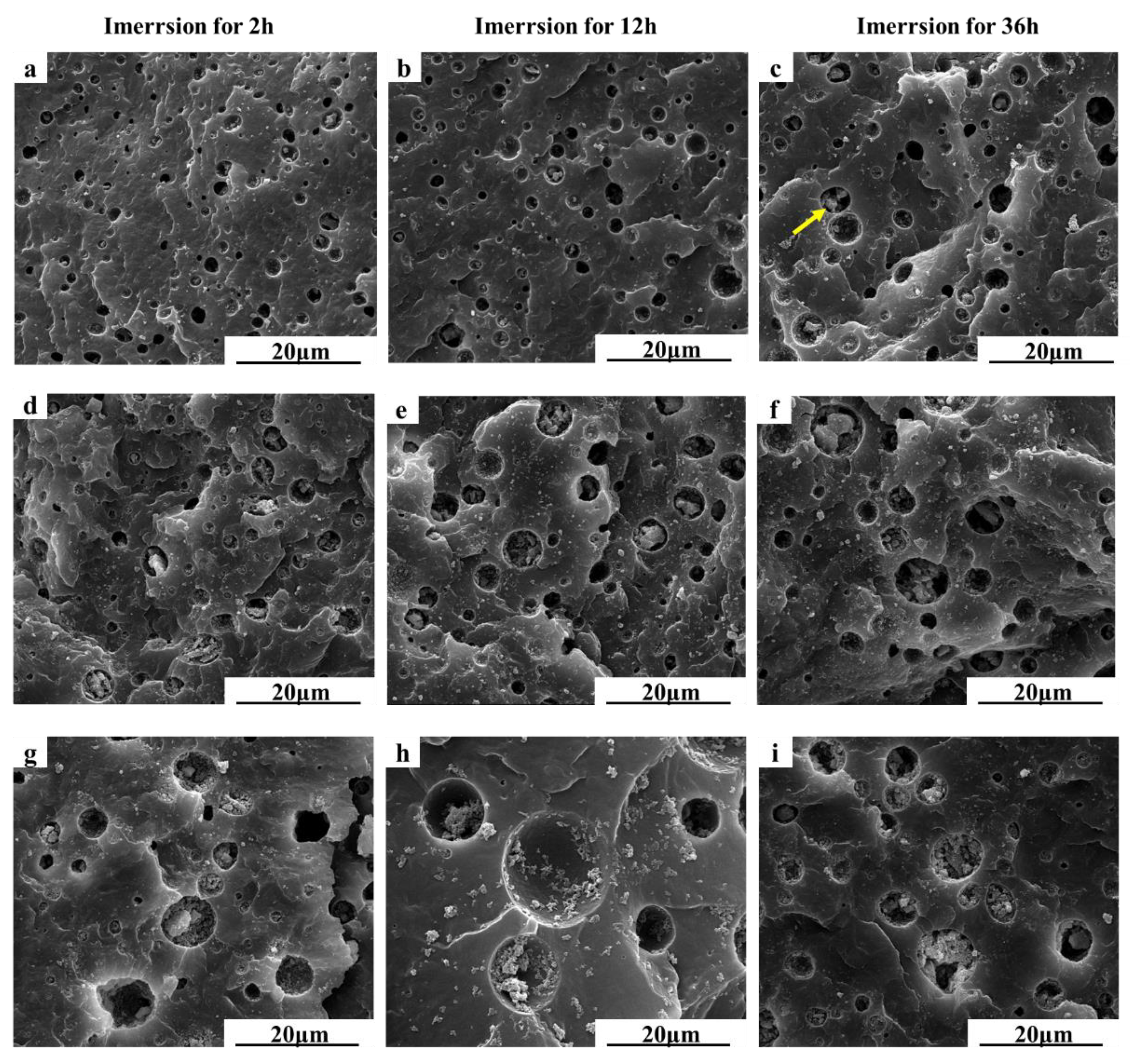
3.4. In Vitro Drug Release Test and Morphology Evaluation
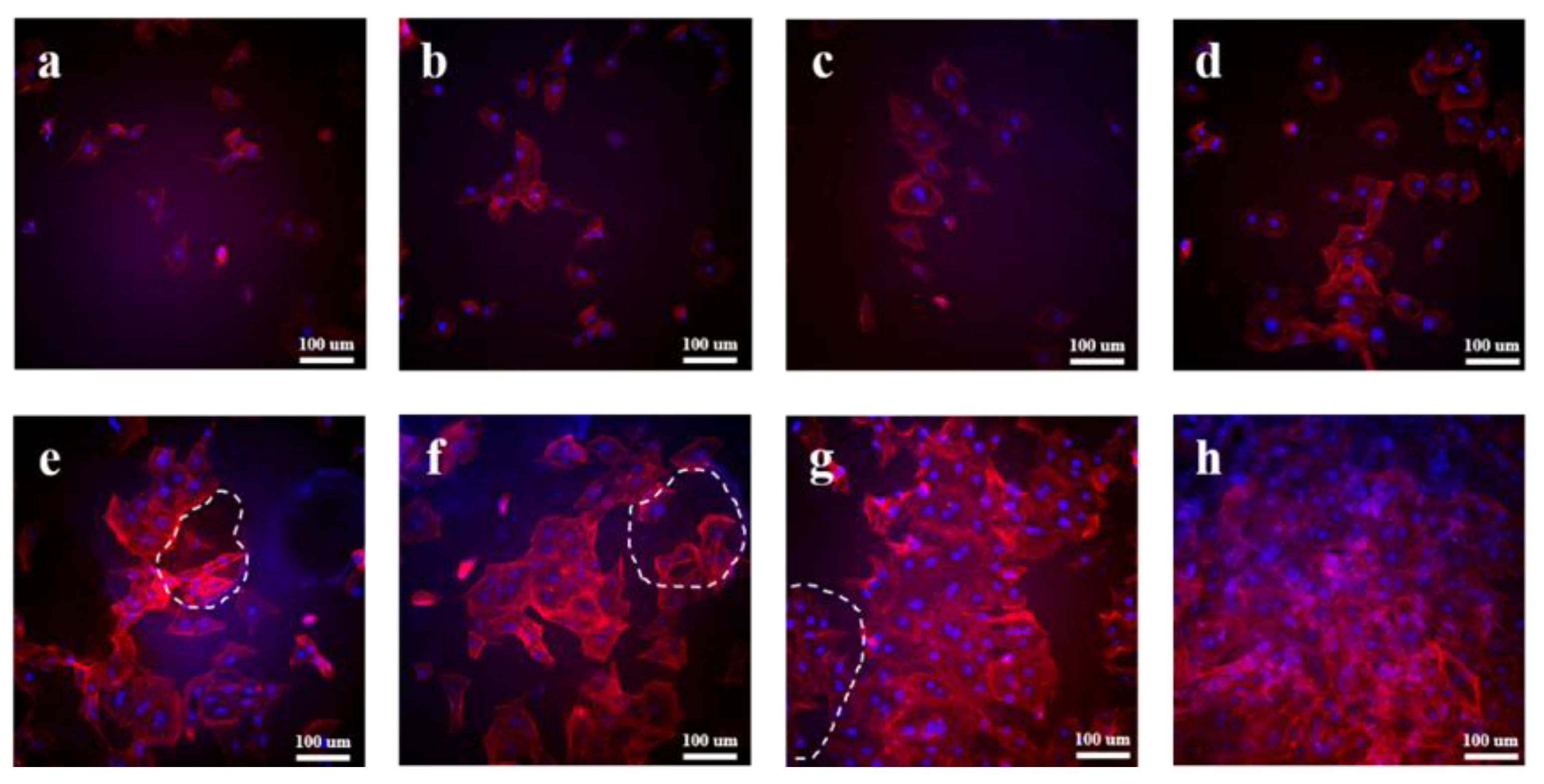
3.5. In Vitro Cytocompatibility Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.F.; Fang, J.; Zhu, W.W.; Zhong, C.X.; Ye, D.D.; Zhu, M.Y.; Lu, X.; Zhao, Y.S.; Ren, F.Z. Bioinspired Highly Anisotropic, Ultrastrong and Stiff, and Osteoconductive Mineralized Wood Hydrogel Composites for Bone Repair. Adv. Funct. Mater. 2021, 31, 10. [Google Scholar] [CrossRef]
- Bao, Z.T.; Gu, Z.P.; Xu, J.B.; Zhao, M.; Liu, G.T.; Wu, J. Acid-responsive composite hydrogel platform with space-controllable stiffness and calcium supply for enhanced bone regeneration. Chem. Eng. J. 2020, 396, 9. [Google Scholar] [CrossRef]
- Shuai, C.J.; Yang, W.J.; Feng, P.; Peng, S.P.; Pan, H. Accelerated degradation of HAP/PLLA bone scaffold by PGA blending facilitates bioactivity and osteoconductivity. Bioact. Mater. 2021, 6, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.N.; Ren, G.; Young, K.; Pina, S.; Reis, R.L.; Oliveira, J.M. Scaffold Fabrication Technologies and Structure/Function Properties in Bone Tissue Engineering. Adv. Funct. Mater. 2021, 31, 2010609. [Google Scholar] [CrossRef]
- Feng, P.; Jia, J.Y.; Liu, M.Y.; Peng, S.P.; Zhao, Z.Y.; Shuai, C.J. Degradation mechanisms and acceleration strategies of poly (lactic acid) scaffold for bone regeneration. Mater. Des. 2021, 210, 18. [Google Scholar] [CrossRef]
- Miszuk, J.; Liang, Z.P.; Hu, J.; Sanyour, H.; Hong, Z.K.; Fong, H.; Sun, H.L. Elastic Mineralized 3D Electrospun PCL Nanofibrous Scaffold for Drug Release and Bone Tissue Engineering. ACS Appl. Bio Mater. 2021, 4, 3639–3648. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M.; Salmasi, A.A.; Asghari, S.; Yekta, H.J.; Moghadas, B.K.; Shahriari, S.; Saber-Samandari, S.; Khandan, A. Optimization and fabrication of alginate scaffold for alveolar bone regeneration with sufficient drug release. Int. Nano Lett. 2021, 11, 295–305. [Google Scholar] [CrossRef]
- Shi, H.; Zhou, Z.Q.; Li, W.D.; Fan, Y.; Li, Z.H.; Wei, J.C. Hydroxyapatite Based Materials for Bone Tissue Engineering: A Brief and Comprehensive Introduction. Crystals 2021, 11, 18. [Google Scholar] [CrossRef]
- Du, M.Z.; Chen, J.D.; Liu, K.H.; Xing, H.R.; Song, C. Recent advances in biomedical engineering of nano-hydroxyapatite including dentistry, cancer treatment and bone repair. Compos. Pt. B-Eng. 2021, 215, 23. [Google Scholar] [CrossRef]
- Dubinenko, G.; Zinoviev, A.; Bolbasov, E.; Kozelskaya, A.; Shesterikov, E.; Novikov, V.; Tverdokhlebov, S. Highly filled poly(l-lactic acid)/hydroxyapatite composite for3Dprinting of personalized bone tissue engineering scaffolds. J. Appl. Polym. Sci. 2021, 138, 10. [Google Scholar] [CrossRef]
- Rogina, A.; Antunovic, M.; Milovac, D. Biomimetic design of bone substitutes based on cuttlefish bone-derived hydroxyapatite and biodegradable polymers. J. Biomed. Mater. Res. Part B 2019, 107, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Pal, U. 3D hydroxyapatite scaffold for bone regeneration and local drug delivery applications. J. Drug Deliv. Sci. Technol. 2019, 53, 11. [Google Scholar] [CrossRef]
- Ramu, M.; Ananthasubramanian, M.; Kumaresan, T.; Gandhinathan, R.; Jothi, S. Optimization of the configuration of porous bone scaffolds made of Polyamide/Hydroxyapatite composites using Selective Laser Sintering for tissue engineering applications. Bio-Med. Mater. Eng. 2018, 29, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, F.; Gigli, M.; Bloise, N.; Chen, H.L.; Bruni, G.; Munari, A.; Moroni, L.; Lotti, N.; Visai, L. Influence of the nanofiber chemistry and orientation of biodegradable poly(butylene succinate)-based scaffolds on osteoblast differentiation for bone tissue regeneration. Nanoscale 2018, 10, 8689–8703. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.Q.; He, Y.X.; Zhou, H.; Gao, P.X.; Xu, L.; Han, Z.L.; Yang, L.; Wang, M. Chondroitin Sulfate/Polycaprolactone/Gelatin Electrospun Nanofibers with Antithrombogenicity and Enhanced Endothelial Cell Affinity as a Potential Scaffold for Blood Vessel Tissue Engineering. Nanoscale Res. Lett. 2021, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Dikici, B.A.; Malayeri, A.; Sherborne, C.; Dikici, S.; Paterson, T.; Dew, L.; Hatton, P.; Asencio, I.O.; MacNeil, S.; Langford, C.; et al. Thiolene- and Polycaprolactone Methacrylate-Based Polymerized High Internal Phase Emulsion (PolyHIPE) Scaffolds for Tissue Engineering. Biomacromolecules 2022, 23, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Nerantzaki, M.; Filippousi, M.; Van Tendeloo, G.; Terzopoulou, Z.; Bikiaris, D.; Goudouri, O.M.; Detsch, R.; Gruenewald, A.; Boccaccini, A.R. Novel poly(butylene succinate) nanocomposites containing strontium hydroxyapatite nanorods with enhanced osteoconductivity for tissue engineering applications. Express Polym. Lett. 2015, 9, 773–789. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specimen | PBS, wt% | HA, wt% | MPT, wt% |
|---|---|---|---|
| PBS | 100 | / | / |
| HA-2% | 98 | 2 | / |
| HA-5% | 95 | 5 | / |
| HA-10% | 90 | 10 | / |
| MPT-HA-2% | 88 | 2 | 10 |
| MPT-HA-5% | 85 | 5 | 10 |
| MPT-HA-10% | 80 | 10 | 10 |
| MPT-10% | 90 | / | 10 |
| Specimen | Td, °C | R, % |
|---|---|---|
| PBS | 348.4 | 0.3 |
| HA-2% | 355.1 | 1.1 |
| HA-5% | 358.1 | 5.5 |
| HA-10% | 359.9 | 9.1 |
| MPT-HA-10% | 181.2 | 8.8 |
| Specimen | Tc, °C | Tm, °C | ΔHm, J/g | Xc, % |
|---|---|---|---|---|
| PBS | 56.9 | 102.1 | 55.0 | 27.5 |
| HA-2% | 56.4 | 101.1 | 52.3 | 26.7 |
| HA-5% | 55.2 | 100.2 | 49.4 | 26.0 |
| HA-10% | 55.0 | 99.6 | 45.2 | 25.1 |
| MPT-HA-2% | 56.5 | 100.8 | 46.3 | 26.3 |
| MPT-HA-5% | 55.1 | 100.4 | 43.7 | 25.7 |
| MPT-HA-10% | 54.8 | 99.3 | 39.4 | 24.6 |
| Specimen | 5 h | 20 h | 50 h | 150 h | 300 h |
|---|---|---|---|---|---|
| MPT-10% | 4.7% | 15.1% | 32.0% | 51.4% | 59.9% |
| MPT-HA-2% | 10.9% | 34.5% | 49.8% | 63.1% | 70.4% |
| MPT-HA-5% | 18.3% | 45.6% | 58.4% | 70.6% | 77.5% |
| MPT-HA-10% | 40.6% | 65.3% | 72.5% | 79.5% | 82.9% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Pan, R.; Zhou, Y.; Liu, G.; Chen, R.; Guo, S. Hydroxyapatite/Poly (Butylene Succinate)/Metoprolol Tartrate Composites with Controllable Drug Release and a Porous Structure for Bone Scaffold Application. Polymers 2023, 15, 4205. https://doi.org/10.3390/polym15214205
Yang H, Pan R, Zhou Y, Liu G, Chen R, Guo S. Hydroxyapatite/Poly (Butylene Succinate)/Metoprolol Tartrate Composites with Controllable Drug Release and a Porous Structure for Bone Scaffold Application. Polymers. 2023; 15(21):4205. https://doi.org/10.3390/polym15214205
Chicago/Turabian StyleYang, Hongming, Rui Pan, Yuan Zhou, Guiting Liu, Rong Chen, and Shaoyun Guo. 2023. "Hydroxyapatite/Poly (Butylene Succinate)/Metoprolol Tartrate Composites with Controllable Drug Release and a Porous Structure for Bone Scaffold Application" Polymers 15, no. 21: 4205. https://doi.org/10.3390/polym15214205
APA StyleYang, H., Pan, R., Zhou, Y., Liu, G., Chen, R., & Guo, S. (2023). Hydroxyapatite/Poly (Butylene Succinate)/Metoprolol Tartrate Composites with Controllable Drug Release and a Porous Structure for Bone Scaffold Application. Polymers, 15(21), 4205. https://doi.org/10.3390/polym15214205