1. Introduction
The persistence length (
lp) is a core parameter in polymer science. In lay terms,
lp reflects how easily a linear chain can bend, with
lp decreasing with increasing chain flexibility. For instance,
lp increases from 0.48 nm for flexible poly(ethylene oxide) [
1] to 1.8 nm for more rigid bisphenol A polycarbonate [
2]. Because stiffer polymers have a larger modulus (
G), theoretical work has aimed to relate
lp to
G to use
lp for a given polymer as a predictor of the viscoelastic properties expected for its solution [
3,
4,
5,
6,
7]. Computational methods yield
lp by measuring the distance over which a vector tangent to the main chain loses its orientation as it is moved along the chain with respect to the tangent vector obtained at a reference point [
8]. Experimentally,
lp is measured by building conformational plots from measurements of the radius of gyration (
RG) [
9,
10] or the intrinsic viscosity ([
η]) [
11], obtained by scattering or viscosity experiments, respectively, as a function of the molecular weight of polymer samples prepared with a narrow molecular weight distribution (MWD). For many polymers, that cannot be produced with a narrow MWD, gel permeation chromatography (GPC) instruments equipped with a combination of differential refractive index, static light scattering, and viscosity detectors can be employed to generate conformation plots by taking advantage of GPC’s ability to yield
RG and [
η] as a function of polymer molecular weight [
12,
13].
Despite its importance in polymer science, lp remains unknown for most polymers because many polymers cannot be obtained with a narrow MWD and require GPC analysis for lp determination. Unfortunately, GPC instruments are typically operated in a given solvent, which is not always suitable for all polymer types. Poorly soluble polymers induce interactions between the polymers and the packing material of the GPC column that result in distorted GPC traces, preventing the determination of lp by GPC analysis. Consequently, alternative experimental methods are required to determine lp for polydisperse polymer samples in solvents where they can be fully dissolved.
The interest in scattering or viscosity techniques for determining lp resides in their ability to use RG or [η] to probe the local density generated inside the macromolecular volume defined by the polymer under study. Since a more flexible polymer can pack more structural units (SU) inside the same macromolecular volume occupied by a stiffer polymer, the polymer coil generated by the stiffer polymer is less dense than the polymer coil generated by the more flexible polymer, thus enabling the determination of lp from conformation plots established with RG or [η]. This discussion suggests that in theory, any technique capable of probing the local density of a polymer coil in solution should be able to yield lp for that polymer.
One such technique was recently presented using a methodology based on pyrene excimer formation (PEF) between an excited and a ground-state pyrenyl label covalently attached to a macromolecule. Since PEF is a chemical reaction, its efficiency depends on the local concentration ([
Py]
loc) of ground-state pyrenes found within the macromolecular volume [
14]. This dependency was recently established by demonstrating that the average rate constant (<
k>) for PEF between pyrenyl labels attached to a macromolecule is directly proportional to [
Py]
loc [
15]. Since the experimentalist knows where the pyrenyl labels are attached on the pyrene-labeled macromolecule (PyLM), [
Py]
loc reflects the local density of a PyLM, thus enabling the application of PEF to determine
lp. The PEF-based methodology that was developed to determine
lp uses polymers that were randomly labeled with pyrene and whose fluorescence decays were fitted according to the fluorescence
blob model (FBM) [
16]. Within the framework of the FBM, an excited pyrenyl label only probes a subvolume, also referred to as a
blob, of the much larger polymer coil. The
blob is then used as a unit volume to compartmentalize the polymer coil into a cluster of
blobs among which the pyrenyl labels distribute themselves randomly with a Poisson distribution. As for any
blob-based method, the use of
blobs shifts the study from the entire polymer to that of a
blob, eliminating problems associated with polydisperse samples, that often plague scattering studies [
17,
18] since a large or small polymer can be described by many or few identical
blobs [
19]. The FBM analysis of the fluorescence decays yields the average number <
n> of pyrenyl groups per
blob, which is used to determine the number
Nblob of SU per
blob. Since a flexible polymer can pack more SU inside a
blob than a stiffer polymer,
Nblob is larger for a flexible polymer than for a stiffer polymer and thus responds to
lp.
With this insight, the dependency of
Nblob on the flexibility, and thus
lp, of a polymer was recently taken advantage of to determine
lp for a series of poly(oligo(ethylene glycol) methyl ether methacrylate)s, that were labeled with a 1-pyrenebutyl derivative and are referred to as PyBut-PEG
nMA [
20]. However, only
lp values that are smaller than the dimension of a
blob probed by an excited pyrenyl label, can be measured with sufficient accuracy. Since the polymer backbone is much less mobile than the pyrenyl label, the linker connecting the pyrene moiety to the polymer backbone defines the size of the
blob, which can be viewed as a sphere with an equivalent diameter of 3.0 nm in the case of the 1-pyrenebutyl derivative [
20]. This meant that
lp no greater than 2.0 nm could be measured with the PyBut-PEG
nMA samples [
20]. While a range of
lp values up to 2.0 nm covers a sizeable group of polymers that can be as flexible as PEO with an
lp of 0.48 nm [
1], to bisphenol A polycarbonate with an
lp of 1.8 nm [
2], the ability to determine an
lp larger than 2.0 nm would enable the study of stiffer backbones. With this in mind, this report describes how labeling PEG
nMA with a 1-pyrenemethoxy-penta(ethylene glycol) derivative to yield PyEG
5-PEG
nMA samples enlarged the diameter of a
blob from 3.0 nm for the PyBut-PEG
nMA samples to 5.4 nm in
N,
N-dimethylformamide, thus enabling the measurement of
lp values of up to 4.0 nm. The increase in
lp values recovered with the PyEG
5-PEG
nMA samples from 2.0 to 4.0 nm represents a significant improvement in the range of
lp values that can be determined from this PEF-based method and it will enable the study of stiffer polymer backbones.
2. Materials and Methods
2.1. Materials
Copper(II) bromide (Sigma, St Louis, MI, USA, 99%), Celite (Sigma), dichloromethane (DCM, Sigma, ≥99.8%), diethylether (Sigma, ≥99%), N,N-dimethylformamide (DMF, Sigma, ≥99.8%), dimethyl sulfoxide (DMSO, Sigma, ≥99.9%), 4-(dimethylamino)pyridine (DMAP, Sigma, ≥99%), ethyl acetate (Sigma, ≥99.7%), ethyl-α-bromoisobutyrate (Sigma, 98%), tetra(ethylene glycol) methyl ether (EG4, PurePEG, San Diego, CA, USA, ≥97%), penta(ethylene glycol) (Fisher, Hampton, NJ, USA, ≥95%), penta(ethylene glycol) methyl ether (EG5, PurePEG, ≥95%), hepta(ethylene glycol) methyl ether (EG7, TCI, Portland, OR, USA, ≥97%), 1,1,4,7,10,10-hexamethyl-triethylenetetramine (HMTETA, Sigma, ≥97%), methacrylic anhydride (Sigma, 94%), 1-pyrenemethanol (Sigma, 98%), sodium chloride (Sigma), sodium hydride (NaH, Sigma, 60% dispersion in mineral oil), sodium hydroxide (NaOH, Sigma, pellets, ≥97%), sodium sulfate (Sigma, anhydrous, ≥99%), tetrahydrofuran (Sigma, ≥99%), tetrahydrofuran optima (Fisher, ≥99.9%), and triethylamine (Sigma, ≥99.5%) were used as received.
Tri(ethylene glycol) methyl ether methacrylate (EG3MA, Sigma, 93%) and two oligo(ethylene glycol) methyl ether methacrylates (EG9MA with number average molecular weight (Mn) = 500 g/mol and EG19MA with Mn = 950 g/mol, Sigma) were dissolved in DCM, washed with 2 M NaOH, and dried with sodium sulfate before use. The radical initiator 2,2′-azo-bis-isobutyronitrile (AIBN, Sigma, 98%) was recrystallized in ethanol three times. p-Toluenesulfonyl chloride (Sigma, ≥98%) was dissolved in diethyl ether and washed with 2 M NaOH. The organic phase was extracted and dried with sodium sulfate. Unless otherwise specified, all other chemicals were purchased from commercially available sources and used as received.
2.2. Preparation of Penta(Ethylene Glycol) Mono p-Toluene Sulfonate (Tos1-EG5OH)
Penta(ethylene glycol) (EG
5) (2.00 g, 8.39 mmol) was added to a round bottom flask (RBF) equipped with a magnetic stirrer with freshly distilled DCM. Re-crystallized
p-toluenesulfonyl chloride (1.76 g, 9.23 mmol) and triethyl amine (1.74 mL, 12.6 mmol) were added to the RBF and left to stir overnight. The next day, the reaction mixture was washed three times with a saturated solution of aqueous sodium chloride. The organic layer was extracted and dried with sodium sulfate. Silica gel chromatography was used to purify the singly tosylated EG
5 (Tos
1-EG
5OH) from the doubly tosylated EG
5 and unmodified EG
5 using ethyl acetate as the eluent. The Tos
1-EG
5OH fraction was dried in vacuo (vacuum oven from VWR, Radnor, PA, USA) and its chemical composition was verified using
1H NMR (
Figure S1 in SI).
2.3. Preparation of 1-Pyrenemethyl Ether Penta(Ethylene Glycol) (PyEG5OH)
1-Pyrenemethanol (1.18 g, 5.10 mmol) was added to a RBF with 50 mL of dried and distilled DMF. The solution was stirred and kept under a nitrogen atmosphere. Sodium hydride (NaH) (0.20 g, 5.10 mmol) was added to the RBF and the solution was allowed to stir for 1 h during which time the color of the solution changed from yellow to dark red/purple. Tos
1-EG
5OH (1.00 g, 2.55 mmol) was then added. The RBF was placed in an oil bath at 55 °C and left to stir overnight. After the RBF was removed from the oil bath and allowed to cool, 5 mL of Milli-Q water was added to the reaction solution to quench any unreacted NaH. Milli-Q water (50 mL) was then added to the reaction mixture, which was washed with 50 mL of ethyl acetate. The organic phase was collected and dried with sodium sulfate. The crude product was purified by silica gel chromatography using ethyl acetate as the eluent. The chemical composition of the purified PyEG
5OH product was confirmed using
1H NMR (
Figure S2 in SI).
2.4. Methacrylation of Oligo(Ethylene Glycol) Methyl Ethers (EGnOHs) and PyEG5OH
The same protocol was used to prepare the methacrylated oligo(ethylene glycol) methyl ethers (EGnMAs, where n = 4, 5, 7) and PyEG5MA. The synthesis of EG5MA is described in more detail hereafter.
EG
5OH (2.00 g, 7.93 mmol) and DMAP (0.0968 g, 0.793 mmol) were added to a RBF with 25 mL of freshly distilled DCM. The RBF was then placed in an ice water bath and the solution was stirred as methacrylic anhydride was added dropwise (1.18 mL, 7.93 mmol). The reaction was left to stir overnight. The reaction mixture was then washed three times with 2 M NaOH. The organic phase was extracted and dried with sodium sulfate. The crude product was purified by silica gel chromatography using ethyl acetate as the eluent. The chemical composition of the purified EG
5MA macromonomer was characterized by
1H NMR (
Figure S3 in SI).
2.5. Random Copolymerization Using Conventional Radical Polymerization
The pyrene-labeled poly(oligo(ethylene glycol) methyl ether methacrylate)s (PyEG
5-PEG
nMA) were prepared by conventional radical polymerization of methyl methacrylate (EG
0MA), tri(ethylene glycol) methyl ether methacrylate (EG
3MA), tetra(ethylene glycol) methyl ether methacrylate (EG
4MA), penta(ethylene glycol) methyl ether methacrylate (EG
5MA), hepta(ethylene glycol) methyl ether methacrylate (EG
7MA), and two oligo(ethylene glycol) methyl ether methacrylates (EG
9MA and EG
19MA) with PyEG
5MA. The chemical structure of the PyEG
5-PEG
nMA samples is shown in
Table 1. The moles of PyEG
5MA used in the polymerization were varied to obtain different molar percentages of pyrene-labeling, ranging from 1 to 10 mol % of PyEG
5MA, incorporated into the PyEG
5-PEG
nMA samples. The polymerization of PyEG
5-PEG
0MA labeled with 2 mol % of PyEG
5MA is described in more detail hereafter.
PyEG5MA (0.02 g, 0.04 mmol) and methyl methacrylate (EG0MA, 0.20 g, 2.00 mmol) were dissolved in 6.8 mL of THF such that the overall methacrylate concentration was approximately 0.3 M. The AIBN initiator (2.00 μg, 0.01 μmol) was added to the monomer solution from a stock solution and the mixture was placed in the polymerization tube. The tube was kept on ice before being degassed with nitrogen (Praxair, Danburry, CT, USA, N4.0) for 30 min. After sealing the tube, it was left in an oil bath at 65 °C. The polymerization was terminated after a conversion of 20% or less was reached, as determined by 1H NMR analysis, to minimize an eventual composition drift. The polymer was recovered by precipitating 5–6 times the polymer solution in THF into diethyl ether to remove any unreacted monomer. The precipitated product was then dried in a vacuum oven overnight at room temperature.
2.6. Random Copolymerization Using Initiators for Continuous Activator Regeneration Atom Transfer Radical Polymerization (ICAR-ATRP)
Three of the PyEG
5-PEG
19MA samples were prepared using ICAR-ATRP [
21]. The protocol described for free radical copolymerization was applied to prepare PyEG
5-PEG
19MA using ethyl-α-bromoisobutyrate, Cu(II)Br/HMTETA, and AIBN as initiator, catalyst/ligand system, and radical source, respectively. An example of the ICAR-ATRP of PyEG
5-PEG
19MA is provided in more detail hereafter.
A solution of PyEG5MA (0.02 g, 0.04 mmol) and EG19MA (1.00 g, 1.05 mmol) in 3.6 mL of THF, where the overall methacrylate concentration equaled 0.3 M, was transferred to the polymerization tube. A stock solution of Cu(II)Br (1.18 mg, 5.28 μmol) and HMTETA (4.3 μL, 15.8 μmol) was prepared in 10 mL of THF from which 10 μL was added to the polymerization tube. Ethyl-α-bromoisobutyrate (10.0 μL, 68.1 μmol) was added to 1 mL of THF from which 7.8 μL was added to the polymerization tube. AIBN (5.00 mg, 0.03 mmol) was added to 10 mL of THF to make a 3.05 mM stock solution. The solution was further diluted to 0.3 mM from which 0.2 mL was added to the polymerization tube, which was placed on ice and degassed for 30 min with nitrogen (Praxair, N4.0). The tube was then sealed and heated to 60 °C in an oil bath for 20 h. Before the polymer was precipitated, the polymer solution in THF was filtered through a silica gel and Celite plug three times to remove copper. The polymer was further purified by 5–6 precipitations into diethyl ether.
2.7. Chemical Composition and Molecular Weight Distribution
The chemical composition of the PyEG
5-PEG
nMA polymers was confirmed by the analysis of the
1H NMR spectra acquired on a Bruker 300 MHz high resolution spectrometer (Bruker, Billerica, MA, USA). A sample
1H NMR spectrum of PyEG
5-PEG
5MA is provided in
Figure S4 in the SI. The molecular weight distribution (MWD) of each PyEG
5-PEG
nMA sample was determined by GPC analysis using either THF or DMSO. The pyrene contents (in mol%),
Mn, and dispersity (
Ð) of each sample are listed in
Table 2.
2.8. Pyrene Content of PyEG5-PEGnMA Samples
The pyrene content expressed as the molar fraction (
x) of the PyEG
5MA monomer incorporated in the copolymers, equivalent to the molar fraction of structural units bearing a pyrenyl labels, was calculated with Equation (1).
In Equation (1),
λPy,
M, and
MPy are the pyrene content of the polymer expressed in mol of pyrene per gram of polymer and the molar mass of the EG
nMA and PyEG
5MA monomers, respectively. λ
Py was determined as follows. A polymer solution was prepared in THF with a known mass concentration (
m) of a PyEG
5-PEG
nMA sample. The pyrene content of the polymer (λ
Py) was calculated from the ratio
Abs/(
m ×
ε), where
Abs is the absorption at 344 nm of the PyEG
5-PEG
nMA solution in THF and
ε is the molar absorption coefficient of 1-pyrenemethanol in THF (ε(344 nm) = 42,700 M
−1·cm
−1) [
22].
2.9. Gel Permeation Chromatography (GPC)
Absolute molecular weights were obtained for PyEG5-PEG0MA, PyEG5-PEG3MA, and PyEG5-PEG5MA by injecting 1 mg/mL solutions of the samples dissolved in THF into a Viscotek GPC (Viscotek, Houston, TX, USA) equipped with a differential refractive index, static light scattering (low and right angle), and UV–Vis absorption detector and three 300 × 8 mm2 PolyAnalytik Superes linear mixed-bed columns (PolyAnalytik, London, ON, Canada). A flow rate of 1 mL/min of THF at 35 °C was used. The system was calibrated with a 1 mg/mL THF solution of a polystyrene (PS) standard with M = 90 × 103 g·mol−1 and Ð = 1.04.
However, PyEG5-PEG7MA, PyEG5-PEG9MA, and PyEG5-PEG19MA were found to interact with the column set of the GPC instrument in THF resulting in distorted GPC traces. As a result, the absolute molecular weights of these samples were obtained by injecting 2 mg/mL polymer solutions in DMSO into a TOSOH EcoSEC High Temperature GPC instrument equipped with a triple detection system and two 300 × 7.8 mm2 TOSOH TSKgel Alpha-M 13 μm columns (Tosoh, Tokyo, Japan). This detection system included an in-line differential refractometer, a Wyatt Dawn Heleos8 MALLS detector (wavelength, λ = 660 nm) (Wyatt, Santa Barbara, CA, USA), and a viscometer. A flow rate of 0.6 mL/min of DMSO at 60 °C was used. The system was calibrated with a 1 mg/mL solution of pullulan standard in DMSO with Mw = 47.1 × 103 g·mol−1 and Ð = 1.07.
The specific refractive index increment (
dn
/dc) of each polymer in THF and DMSO was calculated using the differential refractometers of the GPC instruments. Sample GPC traces can be found in
Figure S5 in SI.
2.10. Atomic Force Microscopy (AFM)
AFM images were obtained with a Digital Instruments Dimension 3100 AFM (Digital Instruments, Santa Barbara, CA, USA) at room temperature using a silicon cantilever in the tapping mode. The samples were prepared by spin coating a few drops of a dilute solution of polymer dissolved in tetrahydrofuran (THF) (10 mg/L) onto a freshly cleaved mica surface at 2000 rpm.
2.11. UV–Vis Spectroscopy
A Varian Cary 100 Bio spectrophotometer (Varian, Palo Alto, CA, USA) was used to acquire the absorption spectra of the polymer solutions.
2.12. Steady-State Fluorescence (SSF) Measurements
All fluorescence spectra were acquired on a Horiba QM-400 spectrofluorometer equipped with a Xenon arc lamp (HORIBA Canada, Burlington, ON, Canada). The SSF spectra were acquired for polymer solutions in aerated DMSO with a 2.5 × 10−6 M pyrene concentration equivalent to an absorbance of ~0.1 at 344 nm. The solutions were excited at 344 nm and scanned from 350 to 600 nm using 1 nm slit widths for both the excitation and emission monochromator. Dividing the florescence intensity of the excimer (IE) by the fluorescence intensity of the monomer (IM), calculated by integrating the area underneath the spectrum from 500 to 510 nm and from 375 to 381 nm, respectively, yielded the IE/IM ratio, which was used to quantify the efficiency of pyrene excimer formation (PEF).
2.13. Time Resolved Fluorescence (TRF) Measurements
All fluorescence decays were obtained with an IBH time-resolved fluorometer (IBH, Glasgow, SCT, UK). The solutions were excited at 344 nm and the monomer and excimer fluorescence decays were acquired with 20,000 counts at the decay maximum over 1024 channels at 375 and 510 nm using cut-off filters at 370 and 495 nm, respectively. A time-per-channel of either 1.02 ns/ch or 2.04 ns/ch was employed for the decay acquisition. A Ludox solution was used for the instrument response function (IRF), which was obtained by setting the emission monochromator at 344 nm. The IRF was convoluted with the FBM equations shown as
Equations (S1) and (S2) in SI and the convolution result was compared to the experimental decay.
2.14. The Fluorescence Blob Model (FBM) Analysis
The FBM compartmentalizes a polymer into equally sized
blobs, where the volume of a
blob is the volume probed by a pyrenyl label, while it remains excited [
14,
16]. The four pyrene species
Pydiff*,
Pyk2*,
Pyagg*, and
Pyfree* are considered to represent PEF, which occurs via a dynamic and static pathway. Dynamic PEF takes place sequentially.
Pydiff* represents an excited pyrenyl group, whose diffusion in solution is controlled by the polymer backbone and side chain dynamics.
Pydiff* diffuses inside a
blob populated by other ground-state pyrenes until
Pydiff* becomes close enough to a ground-state pyrene molecule for
Pydiff* to turn into
Pyk2*. The diffusive motions of two pyrenyl groups inside a
blob are described by the rate constant
kblob. Rapid rearrangement of
Pyk2* and the nearby ground-state pyrene with the large rate constant
k2 (
k2~10 ×
kblob) results in the formation of an excimer made of two pyrenyl labels, that are well (
E0*), or poorly (
D*) stacked and emit with their natural lifetimes
τE0 and
τD, respectively. Static PEF occurs through direct excitation of a pyrene aggregate resulting in the instantaneous formation of the
E0* or
D* species. The species
Pyagg* combines the two pyrenyl species
E0* and
D* formed instantaneously from the direct excitation of a pyrene aggregate. Finally, those excited pyrenes, that are isolated along the polymer backbone, do not form excimers, and emit with their natural lifetime
τM, and are referred to as
Pyfree*. During the decay analysis, the decays are fit twice, initially with a floating
k2 using the program
globmis90lbg for all samples of a same PyEG
5-PEG
nMA series prepared with different pyrene contents. All
k2 values obtained for a same polymer series are then averaged and the averaged
k2 value is then fixed in a second analysis with the program
globmis90obg. The parameters retrieved from the FBM analysis with a fixed
k2 have much lower error bars. The molar fractions
fMdiff,
fMk2,
fMfree, where the index
M indicates that they were derived from the monomer decays, and
fEk2,
fEdiffE0,
fEE0,
fEdiffD, and
fED, where the index
E indicates that they were derived from the excimer decays, were combined to yield the molar fractions
fdiff (=
fdiffE0 +
fdiffD),
fk2,
fagg (=
fE0 +
fD), and
ffree for the pyrene species
Pydiff*,
Pyk2*,
Pyagg*, and
Pyfree*, respectively. The average number (<
n>) of ground state pyrene molecules inside a
blob and the rate constant (
kblob) describing the diffusive encounters of two structural units bearing a pyrenyl label inside a
blob were also obtained from the FBM analysis. The number (
Nblob) of structural units encompassed within a
blob was calculated using
fMfree, <
n>, and
x according to Equation (2).
Each pair of monomer and excimer fluorescence decays acquired for a given PyEG
5-PEG
nMA sample was fit globally with
Equations (S1) and (S2) in SI according to the FBM. The functions described by
Equations (S1) and (S2) were convoluted with the IRF and the convolution product was compared to the experimental decays for optimization of the parameters with the Marquardt–Levenberg algorithm [
23]. A fit was deemed acceptable when the χ
2 was lower than 1.3 and when both the residuals and autocorrelation of the residuals were randomly distributed around zero.
2.15. Flow Chart Depicting the Methodology Applied for Determining the Persistence Length
The strategy applied to determine the persistence length by PEF is depicted in
Figure 1 and
Figure 2.
Figure 1 represents the experimental process to determine <
Nblob> for each PyEG
5-PEG
nMA series and
Nblob∞, which is the <
Nblob> value obtained for a hypothetical PyEG
5-PEG
nMA sample having an infinitely long side chain (
n → ∞).
Figure 2 is a geometrical construction describing the mathematical process applied to determine
lp from <
Nblob> according to the Kratky–Porod equation [
24]. In
Figure 1, the fluorescence decays of the pyrene monomer and excimer shown in the left panel are acquired and fitted globally according to the FBM illustrated in the middle panel to determine
Nblob for different PyEG
5-PEG
nMA samples of a same series with
n = 0, 3, 4, 5, 7, 9, and 19. The
Nblob values obtained for several pyrene contents of a same PyEG
5-PEG
nMA series are averaged to obtain <
Nblob>. These <
Nblob> values are plotted as a function of the molecular weight of a structural unit (
MW(SU)) in the right panel of
Figure 1. For small
MW(SU),
Nblob is large indicating a coiled conformation reflecting a small
lp. As
MW(SU) increases,
Nblob decreases as the chain conformation changes from a coiled to a worm-like conformation. For very large
MW(SU), the polymer chain achieves an extended conformation on the length-scale of the
blob,
Nblob does not change any more with increasing
MW(SU), and its value corresponds to that expected for an extended polymer with infinitely long side chains (
Nblob∞).
Once <
Nblob> and
Nblob∞ are determined, their values are introduced into Equation (3), which is a modified version of the Kratky–Porod equation (KPE) for worm-like chains to account for the fact that it is applied to the chain segment inside a
blob instead of the entire chain. The left part of the KPE in Equation (3) represents the square of the end-to-end distance (<
rEE2>
blob) of the chain fragment encompassed inside a
blob. Since the pyrene moiety is connected to the polymethacrylate backbone with the same linker for all PyEG
5-PEG
nMA constructs, all PyEG
5-PEG
nMA samples share a same
blob regardless of side chain length. According to this reasoning, <
rEE2>
blob takes the same value for all PyEG
5-PEG
nMA samples including those samples that have an infinitely long side chain for which the polymethacrylate backbone is fully extended. The fully extended polymethacrylate backbone corresponds to a hypothetical PyEG
5-PEG
nMA sample, for which
n and
MW(SU) take infinitely large values and
Nblob tends to
Nblob∞ as shown in the right panel of
Figure 1. For a fully extended chain segment inside a
blob, <
rEE2>
blob equals the product (
Nblob∞ ×
b)
2, where
b is the length of a methacrylate structural unit equal to 0.25 nm [
25,
26]. Since all PyEG
5-PEG
nMA share the same
blob with <
rEE2>
blob = (
Nblob∞ ×
b)
2, the left-hand side of the KPE is known and the function on the right-hand side of the equation can be solved for
lp as represented in
Figure 2. The <
Nblob> value obtained for each PyEG
5-PEG
nMA series is entered into the right-hand side of the KPE, which is plotted as a function of
lp in
Figure 2. The abscissa of the intercept between the horizontal dashed line representing <
rEE2> = (
Nblob∞ ×
b)
2 and the line corresponding to the right-hand side of the KPE yields
lp for the PyEG
5-PEG
nMA series under consideration.
3. Results and Discussion
A series of pyrene-labeled poly(oligo(ethylene glycol) methyl ether methacrylate)s (PyEG
5-PEG
nMA with
n = 0, 3, 4, 5, 7, 9, and 19) were synthesized using a grafting through technique by mainly free radical copolymerization of the same penta(ethylene glycol) 1-pyrenemethyl ether methacrylate (PyEG
5MA) and different oligo(ethylene glycol) methyl ether methacrylate (EG
nMA) macromonomers. Their chemical structure, the number (
NS) of atoms in each side chain, and the molecular weight of the structural unit (
MW(SU)) were presented in
Table 1. The number average molecular weight (
Mn) and dispersity (
Ð) of all PyEG
5-PEG
nMA samples were determined by gel permeation chromatography and are listed in
Table 2. Variations in
Mn and
Ð were observed from sample-to-sample in
Table 2 due to the relative purity and reactivity of the different monomers. Nevertheless, the
Mn values were sufficiently large to ensure that all polymer samples were constituted of many
blobs, which enabled the analysis of the fluorescence decays with the FBM, that could handle these samples, whose
Ð values greater than 1.0 indicate that they are polydisperse. The design of the PyEG
5-PEG
nMA constructs was carefully considered. It was established in an earlier study with pyrene-labeled poly(
n-butyl methacrylate)s, that the motion of the pyrenyl group became uncorrelated from the motion of the main chain, when a 1-pyrenemethoxy derivative was connected to the main polymethacrylate backbone by a linker made of two or more ethylene glycol units [
27]. The use of a penta(ethylene glycol) linker for the PyEG
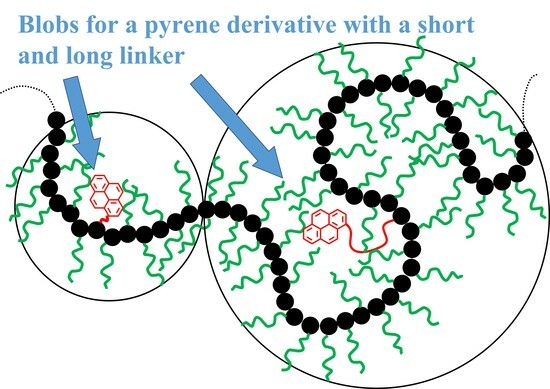
5-PEG
nMA samples thus ensured that an excited pyrenyl label would probe a well-defined sub-volume (
Vblob) of the PBBs, referred to as a
blob within the FBM framework, that would be unaffected by any main chain motion. In turn, this condition implied that each PBB was being probed over the same length scale defined by the same
Vblob for all PyEG
5-PEG
nMA constructs considered in this study.
The SSF spectra for all pyrene contents of each PyEG
5-PEG
nMA series were acquired in acetonitrile, tetrahydrofuran (THF),
N,
N-dimethylformamide (DMF), and dimethyl sulfoxide (DMSO) and are presented in
Figures S6–S9 in the SI. The spectra for all pyrene contents of the PyEG
5-PEG
4MA series in each solvent are shown in
Figure 3.
The spectra were normalized to the first peak of the monomer emission,
I1, which is the 0-0 transition of pyrene. They showed the characteristic fluorescence peaks between 375 and 410 nm for the pyrene monomer with the broad and structureless excimer emission centered at 480 nm. It is apparent from
Figure 3, that more excimer is produced in acetonitrile than in THF, DMF, and DMSO, with DMSO producing the least amount of excimer. The
IE/
IM ratio was calculated to quantify the efficiency of pyrene excimer formation (PEF) for the different constructs in different solvents. The
IE/
IM ratio is proportional to the local concentration of pyrene, [
Py]
loc, and the rate constant for PEF through diffusive encounters,
kdiff, as indicated by Equation (4).
The
IE/
IM ratios were plotted as a function of pyrene content for each PyEG
5-PEG
nMA sample in
Figure S10. They yielded straight lines over a wide range of pyrene contents and the slope of these lines (
m(
IE/
IM)) was plotted as a function of
MW(SU) in
Figure 4. In each solvent, the slope
m(
IE/
IM) decreased as
MW(SU) increased for the PyEG
5-PEG
nMA samples with
n equal to 0, 3, 4, and 5, respectively. This decrease was attributed to an extension of the polymer backbone, that resulted from increased crowding of the volume surrounding the main chain with increasing
MW(SU). Main chain extension reduced the number of encounters between the pyrenyl terminals of the PyEG
5 side chains, which was associated with a decrease in [
Py]
loc in Equation (3). The decrease in
m(
IE/
IM) continued until an
MW(SU) of 408 g/mol for PyEG
5-PEG
7MA was reached, after which
m(
IE/
IM) seemed to plateau for
MW(SU) values of 500 and 950 g/mol for PyEG
5-PEG
9MA and PyEG
5-PEG
19MA, respectively. The plateau region observed for
NS values larger than 400 g/mol indicated that a further increase in side chain length would not result in an increase in main chain extension, probably because the main chain was, or was close to being, fully extended on the length scale probed by an excited pyrene. The
m(
IE/
IM)-
vs.-
MW(SU) trends shown in
Figure 4 suggested that the steric hindrance generated by the side chains influence a region inside the polymeric bottlebrush (PBB) volume, that is close to the main chain and where the shorter EG
n side chains have the strongest effect. As the side chains become long enough to expand past the local region close to the main chain and into the mostly empty space away from the main chain, their effect on the main chain becomes less important, resulting in the plateau observed for large side chain lengths in the
m(
IE/
IM)-
vs-
MW(SU) plot in
Figure 4. Similar saturation effects with increasing side chain length have already been reported for PBBs [
20,
28].
The
m(
IE/
IM) slopes in
Figure 4 were also found to be larger in acetonitrile, followed by THF, DMF, and DMSO. This trend reflects the influence of the solvent viscosity,
η. The viscosity of acetonitrile, THF, DMF, and DMSO at 25 °C equals 0.37, 0.46, 0.79, and 1.99 mPa·s, respectively [
29]. Since
kdiff is inversely proportional to solvent viscosity [
30], acetonitrile with the lowest
η yielded the largest
kdiff values in Equation (4) and the largest
m(
IE/
IM) slopes in
Figure 4A. Similarly, DMSO being the most viscous solvent yielded the lowest
m(
IE/
IM) slopes in
Figure 4D. THF and DMF with their intermediate
η values resulted in intermediate
m(
IE/
IM) slopes. As was pointed out in earlier reports [
31,
32], solvent viscosity, while important, is not the only parameter affecting
kdiff. The probability
p, of forming an excimer upon an encounter between an excited and a ground-state pyrenyl label, depends also on the solvent, and its value can offset the relationship expected between
kdiff and
η−1 [
30]. Consequently, the interpretation of the parameter
m(
IE/
IM) obtained from the analysis of the steady-state fluorescence spectra offers only a qualitative description of the fluorescence results.
A more quantitative measure of polymer stiffness, such as the persistence length (
lp), can only be retrieved from PEF measurements through the global analysis of the monomer and excimer decays acquired with the PyEG
5-PEG
nMA samples as was performed earlier with the PyBut-PEG
nMA samples [
20]. The determination of
Nblob for the PyEG
5-PEG
nMA samples represents the second step in the procedure applied to obtain
lp as described in
Figure 1. The FBM analysis of the decays yields the number
Nblob of methacrylate units that can pack inside a
blob, which is the volume probed by an excited pyrenyl label. Lower
Nblob values are obtained for stiffer chains, that bend less efficiently. The fluorescence decays were acquired in acetonitrile, THF, DMF, and DMSO and the FBM yielded
Nblob, which was plotted as a function of pyrene content in
Figure 5A–D.
Within experimental error,
Nblob remained constant with pyrene content in
Figure 5A–D.
Nblob was averaged over all pyrene contents for all the samples of the same PyEG
5-PEG
nMA series in the same solvent to yield <
Nblob>, which was plotted as a function of the molecular weight of a structural unit (
MW(SU)) in
Figure 5E–H. The plots shown in
Figure 5E–H display some interesting features. For each solvent, <
Nblob> was found to decrease with increasing side chain length reflecting the increased extension of the PEG
nMA backbone with increasing side chain length. <
Nblob> reached a plateau value (
Nblob∞) for the largest side chains indicating that the polymethacrylate backbone appeared fully extended over the length scale probed by an excited pyrenyl label. Finally, <
Nblob> for the PyEG
5-PEG
nMA samples with an 18-atom-long linker connecting pyrene to the polymethacrylate backbone was significantly larger than <
Nblob> obtained earlier for the PyBut-PEG
nMA samples with a 6-atom-long spacer between pyrene and the polymethacrylate backbone as indicated by the difference between the dashed and solid lines in
Figure 5E–H. These differences in <
Nblob> between the PyBut-PEG
nMA and PyEG
5-PEG
nMA samples reflect the longer reach of the pyrene derivative used for the latter series.
Another interesting feature in the plots shown in
Figure 5E–H was that for the same pyrene content, <
Nblob> decreased with increasing solvent viscosity. While this effect had also been observed for the PyBut-PEG
nMA samples [
20], it was much more pronounced for the PyEG
5-PEG
nMA samples. This effect could be better visualized in
Figure 6A, where <
Nblob> was plotted as a function of
NS−2, with
NS being the number of non-hydrogen atoms in the PEG
nMA side chains equal to 3 + 3 ×
n for a given PEG
nMA sample. The <
Nblob>-
vs-
NS−2 plots in
Figure 6A yielded straight lines, except for the <
Nblob> value of PyEG
5-PEG
0MA, which departed from the linear behavior in all solvents. The largest <
Nblob> values were found in acetonitrile, followed by THF, DMF, and DMSO, where the lowest <
Nblob> values were obtained.
The value of <
Nblob> for poly(methyl methacrylate) in all solvents was lower than that expected from the straight lines shown in
Figure 6A. This is probably because for infinite
NS−2, the polymethacrylate backbone still retains some residual stiffness preventing it from collapsing and packing an infinite number of methacrylate monomers when
NS is infinitely small. Consequently, a limit must be reached experimentally, that prevents <
Nblob> from taking an infinite value for infinitely small side chains, as would be otherwise predicted from the straight lines shown in
Figure 6A. It is thus reasonable that the <
Nblob> values obtained for the PyEG
5-PEG
nMA samples having shorter side chains, such as for poly(methyl methacrylate), did not obey the linear <
Nblob>-vs-
NS−2 found for the PyEG
5-PEG
nMA samples with longer side chains in
Figure 6A. Extrapolating the straight lines in
Figure 6A to the Y-intercept yielded
Nblob∞ representing the number of methacrylate units encompassed inside a
blob for a fully extended polymethacrylate backbone.
Nblob∞ was plotted as a function of solvent viscosity in
Figure 6B for the PyEG
5-PEG
nMA samples along with the
Nblob∞ values found earlier for the PyBut-PEG
nMA samples. The determination of
Nblob∞ represents the third step in the methodology developed to determine
lp as shown in
Figure 1.
Nblob∞ for the PyEG
5-PEG
nMA samples was much larger than for the PyBut-PEG
nMA samples [
20] reflecting the longer reach of the linker for the PyEG
5 derivative [
27]. The difference between the
Nblob∞ values obtained for the PyBut-PEG
nMA and PyEG
5-PEG
nMA samples decreased with increasing viscosity since a larger solvent viscosity hinders the deployment of the pyrenyl labels at the end of the long EG
5 linker in the PyEG
5-PEG
nMA samples during the finite time that the pyrenyl label remains excited. This effect is much less pronounced for the PyBut-PEG
nMA samples for which the much shorter butyl linker enables the full deployment of the pyrenyl label while it remains excited. Indeed, the
Nblob∞ dependency on solvent viscosity for the PyBut-PEG
nMA samples in
Figure 6B is much weaker than that for the PyEG
5-PEG
nMA samples.
The slopes of the straight lines obtained in
Figure 6A were plotted as a function of solvent viscosity in
Figure 6C. The slopes showed little dependency on solvent viscosity. The
Nblob∞ values and the slopes for the PyEG
5-PEG
nMA samples could be fitted with power laws, whose empirical expressions are given as Equations (5) and (6), respectively. In turn, the equations for
Nblob∞ and the slopes could be rearranged to yield the bending function (
fb(
η,
MW(
SU))) in Equation (7). Multiplying
Nblob∞ by the bending function yielded <
Nblob> in Equation (8), which was found to properly describe the experimental <
Nblob> values in
Figure 5E–H for
MW(SU) greater than 200 g/mol.
The parametrization of the <
Nblob> values with Equation (8) could now be applied to predict the persistence length (
lp) for any hypothetical molar mass
MW(SU) greater than 200 g/mol of a PyEG
5-PEG
nMA sample in any solvent as depicted in
Figure 2. This was achieved by solving for
lp in Equation (3) representing the Kratky–Porod equation [
24] modified to represent the polymer segment made of <
Nblob> structural units of contour length <
Nblob> ×
b, where
b is the length of a methacrylate monomer typically taken to equal 0.25 nm [
25,
26], inside a
blob with an end-to-end distance <
rEE2>
blob [
20]. In turn, since all PyEG
5-PEG
nMA constructs use the same pyrene derivative, the excited pyrenyl label probes the same volume for all the samples, including those fully extended PyEG
5-PEG
nMA samples with infinitely long side chains for which <
rEE2>
blob is simply equal to
Nblob∞ ×
b, where the expression of
Nblob∞ was given in Equation (5).
Since the left-hand side of Equation (3) is known, Equation (3) could be solved to retrieve
lp for any <
Nblob> value obtained with Equation (8) for any solvent viscosity and
MW(SU) greater than 200 g/mol. The resulting
lp-
vs-
NS2 plots are shown in
Figure 6.
lp increased linearly with increasing
NS2 in all solvents considered. The linear increase in
lp with
NS2 agrees with theoretical predictions [
33]. The predicted trends obtained with
lp for the PyEG
5-PEG
nMA samples showed a much closer agreement with the experimental data points compared to the trends obtained with
lp for the PyBut-PEG
nMA samples, probably because the longer linker of the PyEG
5 derivative resulted in larger <
Nblob> values which were retrieved with better accuracy.
As for the
lp values obtained with the PyBut-PEG
nMA samples [
20], solvent viscosity affected the
lp values retrieved for the PyEG
5-PEG
nMA samples. However, the effect of solvent viscosity on
lp was opposite between the two polymer series. Whereas an increase in solvent viscosity led to an increase in
lp for the PyBut-PEG
nMA samples, it was accompanied by a decrease in
lp for the PyEG
5-PEG
nMA samples. The reason for the opposite trends resided in the different spacers connecting pyrene to the polymethacrylate backbone. In the case of the PyBut-PEG
nMA samples, the volume of a
blob (
Vblob) was little affected by solvent viscosity [
20], as indicated by the small changes in
Nblob∞ with solvent viscosity observed in
Figure 6B. The small dependency of
Nblob∞ on solvent viscosity enabled the short 6-atom-long spacer to fully deploy, allowing the pyrenyl label to probe a constant
Vblob regardless of solvent viscosity. An increase in solvent viscosity for the PyBut-PEG
nMA series resulted in weaker PEF, which was erroneously attributed to a stiffening of the chain, resulting in an increase in
lp based on the Kratky–Porod equation. In contrast,
Vblob was much more strongly affected by solvent viscosity for the longer 18-atom-long penta(ethylene glycol) spacer of the PyEG
5-PEG
nMA sample, as illustrated by the significant decrease in
Nblob∞ in
Figure 6B. The inability of the PyEG
5 derivative to fully deploy while a pyrenyl label remained excited meant that the excited pyrene probed a smaller
Vblob with increasing solvent viscosity. Since the density of a
blob with a polymer segment of size
Nblob increases with decreasing
Nblob as
Nblob/
Nblob3/2 =
Nblob−0.5 [
15], a smaller
blob appeared denser, yielding a smaller
lp for the PyEG
5-PEG
nMA samples as observed in
Figure 7A–D.
Since effects induced by solvent viscosity on <
Nblob> were eliminated when working in a solvent with a viscosity of 0.74 mPa.s approaching that of 0.79 mPa.s for DMF according to molecular mechanics optimizations (MMO) [
20], the
lp values obtained in DMF were expected to best represent the persistence length of PEG
nMA. As it turned out, the
lp values retrieved for the PyBut-PEG
nMA and PyEG
5-PEG
nMA samples showed excellent agreement in DMF in
Figure 7C. Furthermore, the
lp values retrieved in DMF matched very closely those reported for a series of poly(alkyl methacrylate)s of similar
MW(SU) [
11], where the poly(alkyl methacrylate)s have been shown to behave similarly to PEG
nMA over the short length scales probed by an excited pyrenyl label [
20]. The concurring trends presented in
Figure 7C for the
lp values retrieved for several polymethacrylates by different procedures provide solid validation of the PEF-based method for measuring the persistence length of these polydisperse polymethacrylate samples.
Another interesting observation in
Figure 7C was that the straight lines representing the
lp-
vs-
NS2 trends in
Figure 7C did not pass through the origin for an infinitely short side chain. The non-zero Y-intercept is a result of the intrinsic stiffness of the polymethacrylate backbone, which prevents
lp from reaching zero for an infinitely short side chain. In turn, this conclusion implies that <
Nblob> cannot take infinite values for infinitely short side chains as expected from the <
Nblob>-
vs-
NS−2 straight lines shown in
Figure 6A. Instead, <
Nblob> should reach a constant value for shorter side chains approaching the <
Nblob> value expected for the unsubstituted polymethacrylate backbone. This most certainly explains why the <
Nblob> value for the PyPEG
5-PEG
0MA series did not fall on the straight lines in
Figure 6A.
In order to properly predict the <
Nblob> values that should be obtained for polymethacrylates having short side chains, Equations (9) and (10) were used to parametrize the intercepts and slopes of the
lp-
vs-
NS2 straight lines in
Figure 6A–D. Combining Equations (9) and (10) yielded the persistence length for any
MW(SU) and solvent viscosity, which could then be employed with Equation (3) to extract <
Nblob>. The resulting plots of <
Nblob> as a function of
MW(SU) are shown in
Figure 8A–D for acetonitrile, THF, DMF, and DMSO.
The new trends pass through most of the data points including the <
Nblob> value for PyEG
5-PEG
0MA. Instead of <
Nblob> tending to infinity for infinitely small side chains, <
Nblob> goes through an inflection point as it approaches
MW(SU) = 100 g/mol for poly(methyl methacrylate) before passing through a maximum and intercepting the
Y-axis at a finite, non-zero value. The small dip observed for short side chains before the maximum in the plots of
Figure 8 is certainly an artefact resulting from the mathematical handling of the Kratky–Porod equation with the linear relationship between
lp and
NS2 in
Figure 7 to predict <
Nblob>. Despite this mathematical artefact, the plots of <
Nblob>-
vs-
MW(SU) in
Figure 8 provide a physically more realistic depiction of the behavior of <
Nblob> expected as the side chains become infinitely short, because <
Nblob> no longer diverges to infinity as would be otherwise predicted with Equation (8).
The backbone conformation of PEG
19MA was further investigated by atomic force microscopy (AFM) to visualize individual PEG
19MA macromolecules, which were prepared without pyrene. This sample had a number (
Mn) and weight (
Mw) average molecular weight of 134,000 and 193,000 g·mol
−1, respectively. Individual polymer molecules were observed in
Figure 9 ranging in length from 20 to 90 nm, in diameter from 10 to 20 nm, and in height from 0.5 to 1 nm. These results are consistent with the expected dimensions of these macromolecules considering that a fully extended PEG
19MA macromolecule would have a number average contour length of ~35 nm and an average width of ~16 nm.
Furthermore, the AFM image shown in
Figure 9 clearly demonstrates the presence of isolated macromolecules with no indication of aggregation. This observation eliminates the possibility that PEG
19MA could aggregate, as has been found for PBBs prepared with longer poly(ethylene oxide) side chains, which have been shown to crystalize resulting in the formation of crystalsomes [
34].
Figure 9 demonstrates that this is not the case for PEG
19MA. Although the chains observed in
Figure 9 show some curvature, the steric hindrance generated by their side chains prevents them from adopting a fully coiled conformation.
The image shown in
Figure 9 complements the conclusions drawn from the plateau reached for
m(
IE/
IM) in
Figure 4 and for
Nblob in
Figure 5E–H obtained by steady-state and time-resolved fluorescence, respectively. These plateaus were defined mostly by the
m(
IE/
IM) and
Nblob values obtained with samples from the PyEG
5-PEG
19MA series, and they were rationalized by evoking the stiffening and extension of the polymer backbone. Such a stiffening is clearly visible in the AFM picture where the PEG
19MA macromolecules appear as WLCs.








 ) PyEG5-PEG0MA, (
) PyEG5-PEG0MA, ( ) PyEG5-PEG3MA, (
) PyEG5-PEG3MA, ( ) PyEG5-PEG4MA, (
) PyEG5-PEG4MA, ( ) PyEG5-PEG5MA, (
) PyEG5-PEG5MA, ( ) PyEG5-PEG7MA, (
) PyEG5-PEG7MA, ( ) PyEG5-PEG9MA, and (
) PyEG5-PEG9MA, and ( ) PyEG5-PEG19MA and (E–H) <Nblob> as a function of MW(SU) in (A,E) acetonitrile, (B,F) THF, (C,G) DMF, and (D,H) DMSO. Lines: prediction for the <Nblob> values for (solid) the PyEG5-PEGnMA samples with Equation (8) and (dashed) the PyBut-PEGnMA samples.
) PyEG5-PEG19MA and (E–H) <Nblob> as a function of MW(SU) in (A,E) acetonitrile, (B,F) THF, (C,G) DMF, and (D,H) DMSO. Lines: prediction for the <Nblob> values for (solid) the PyEG5-PEGnMA samples with Equation (8) and (dashed) the PyBut-PEGnMA samples.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






