
Advancing Quantification of Water-Extractable Arabinoxylan in Beer: A High-Throughput Approach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Materials
2.1.1. Calibration and Control Standards
2.1.2. Reaction Reagent Preparation
2.1.3. Commercial Beer Samples
2.2. Quantification of WEAX by Acid Hydrolysis and Colorimetry
2.2.1. Basic Method According to Douglas and Kiszonas
2.2.2. Measurement of Absorbance Using a Spectrophotometer
2.2.3. Measurement of Absorbance Using a Multi-Mode Microplate Reader
2.2.4. Absorbance Loss over Time of the Colored Reaction Product
2.2.5. Validation of the High-Throughput Approach
2.3. Elimination of Interfering Sugars with Saccharomyces diastaticus Fermentation
2.4. Investigation of the Red Color Complex by Liquid Chromatography
2.4.1. High-Performance Liquid Chromatography (HPLC)
2.4.2. Liquid Chromatography-Mass Spectrometry (LC-MS)
2.5. Statistical Analysis
3. Results and Discussion
3.1. Improvement of the Colorimetric Quantification of WEAX
3.1.1. Spectrophotometer vs. Multi-Mode Microplate Reader
3.1.2. Determination of the Optimum Measurement Time Point Using Multi-Mode Microplate Reader
3.1.3. Comparison of Standard Material for External Calibration Using Multi-Mode Microplate Reader
3.1.4. Reproducibility of the Method Using Multi-Mode Microplate Reader
3.2. Influence of S. diastaticus Fermentation on the Quantification of AX in Samples with Higher Amounts of Interfering Sugars
3.3. Investigation of the Red Color Complex by LC-MS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bettge, A.D.; Morris, C.F. Oxidative gelation measurement and influence on soft wheat batter viscosity and end-use quality. Cereal Chem. 2007, 84, 237–242. [Google Scholar] [CrossRef]
- Ramseyer, D.D.; Bettge, A.D.; Morris, C.F. Endogenous and Enhanced Oxidative Cross-Linking in Wheat Flour Mill Streams. Cereal Chem. J. 2011, 88, 217–222. [Google Scholar] [CrossRef]
- Ramseyer, D.D.; Bettge, A.D.; Morris, C.F. Flour mill stream blending affects sugar snap cookie and Japanese sponge cake quality and oxidative cross-linking potential of soft white wheat. J. Food Sci. 2011, 76, C1300–C1306. [Google Scholar] [CrossRef]
- Salovaara, H.; Sontag-Strohm, T.; Anttila, H. Viscosity of beta-glucan in oat products. Agric. Food Sci. 2008, 13, 80–87. [Google Scholar] [CrossRef]
- Sahan, N.; Yasar, K.; Hayaloglu, A.A. Physical, chemical and flavour quality of non-fat yogurt as affected by a β-glucan hydrocolloidal composite during storage. Food Hydrocoll. 2008, 22, 1291–1297. [Google Scholar] [CrossRef]
- Hoffmann, R.A.; Leeflang, B.R.; De Barse, M.M.J.; Kamerling, J.P.; Vliegenthart, J.F.G. Characterisation by 1H-n.m.r. spectroscopy of oligosaccharides, derived from arabinoxylans of white endosperm of wheat, that contain the elements →4)[α-l-Araf-(1-ar3)]-β-d-Xylp-(1→ or →4)[α-l-Araf-(1→2)][α-lAraf-(1→3)]-β-d-Xylp-(1→. Carbohydr. Res. 1991, 221, 63–81. [Google Scholar] [CrossRef]
- Perlin, A.S. Structure of the soluble pentosans of wheat flours. Cerreal Chem. 1951, 28, 382–393. [Google Scholar]
- Renard, C.M.G.C.; Rouau, X.; Thibault, J.F. Structure and properties of water-soluble pentosans from wheat flour. Journ. D’etudes Sci. Aliment. 1990, 10, 283–292. [Google Scholar]
- Gruppen, H.; Hamer, R.J.; Voragen, A.G.J. Barium hydroxide as a tool to extract pure arabinoxylans from water-insoluble cell wall material of wheat flour. J. Cereal Sci. 1991, 13, 275–290. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; Biliaderis, C.G. Studies on the structure of wheat-endosperm arabinoxylans. Carbohydr. Polym. 1994, 24, 61–71. [Google Scholar] [CrossRef]
- Pellny, T.K.; Patil, A.; Wood, A.J.; Freeman, J.; Halsey, K.; Plummer, A.; Kosik, O.; Temple, H.; Collins, J.D.; Dupree, P.; et al. Loss of TaIRX9b gene function in wheat decreases chain length and amount of arabinoxylan in grain but increases cross-linking. Plant. Biotechnol. J. 2020, 18, 2316–2327. [Google Scholar] [CrossRef] [PubMed]
- Kupetz, M.; Zeh, A.; Fischer, S.; Becker, T. Investigation of Filtration-inhibitory Substances in German Wheat Beer. Brew. Sci. 2017, 70, 1–8. [Google Scholar]
- Michiels, P.; Delputte, N.; Debyser, W.; Langenaeken, N.; Courtin, C. The occurrence and structural heterogeneity of arabinoxylan in commercial pilsner beers and their non-alcoholic counterparts. Carbohydr. Polym. 2023, 306, 100310. [Google Scholar] [CrossRef]
- Langenaeken, N.; De Schutter, D.; Courtin, C. Arabinoxylan from non-malted cereals can act as mouthfeel contributor in beer. Carbohydr. Polym. 2020, 239, 116257. [Google Scholar] [CrossRef] [PubMed]
- Gastl, M.; Kupetz, M.; Becker, T. Determination of Cytolytic Malt Modification—Part I: Influence of Variety Characteristics. J. Am. Soc. Brew. Chem. 2020, 79, 53–65. [Google Scholar] [CrossRef]
- Gastl, M.; Kupetz, M.; Becker, T. Determination of Cytolytic Malt Modification—Part II: Impact on Wort Separation. J. Am. Soc. Brew. Chem. 2020, 79, 66–79. [Google Scholar] [CrossRef]
- Rübsam, H.; Gastl, M.; Becker, T. Influence of the range of molecular weight distribution of beer components on the intensity of palate fullness. Eur. Food Res. Technol. 2013, 236, 65–75. [Google Scholar] [CrossRef]
- Englyst, H.N.; Cummings, J.H. Simplified method for the measurement of total non-starch polysaccharides by gas-liquid chromatography of constituent sugars as alditol acetates. Analyst 1984, 109, 937–942. [Google Scholar] [CrossRef]
- Cleemput, G.; van Oort, M.; Hessing, M.; Bergmans, M.E.F.; Gruppen, H.; Grobe, P.J.; Delcour, J.A. Variation in the degree of D-Xylose substitution in arabinoxylans extracted from a European wheat flour. J. Cereal Sci. 1995, 22, 73–84. [Google Scholar] [CrossRef]
- Houben, R.; De Ruijter, C.F.; Brunt, K. Determination of the Pentosan Content of Wheat Products by Hydrolysis, Glucose Oxidase Treatment and Analysis by HPAEC/PAD. J. Cereal Sci. 1997, 26, 37–46. [Google Scholar] [CrossRef]
- Saulnier, L.; Quemener, B. Enzymatic mapping of arabinoxylan structure. In HEALTHGRAIN Methods: Analysis of Bioactive Components in Small Grain Cereals; Shewry, W., Ed.; AACC International: St. Paul, MN, USA, 2009; pp. 191–201. [Google Scholar]
- Andersson, R.; Andersson, A.; Ǻman, P. Molecular weight distributions of water-extractable β-glucan and arabinoxylan. In HEALTHGRAIN Methods: Analysis of Bioactive Components in Small Grain Cereals; American Association of Cereal Chemists, Inc. (AACC): St. Paul, MN, USA, 2009; pp. 203–216. [Google Scholar]
- Toole, G.A.; Wellner, N.; Faulds, C.B.; Mills, E.N.C.; Barron, C.; Devaux, M.F.; Guillon, F. Spatial mapping of cell wall components in the cereal endosperm using spectroscopic, fluorescent and immunochemical methods. In HEALTHGRAIN Methods: Analysis of Bioactive Components in Small Grain Cereals; Shewry, W., Ed.; AACC International: St. Paul, MN, USA, 2009; pp. 217–246. [Google Scholar]
- Gebruers, K.; Courtin, C.M.; Delcour, J.A. Quantification of arabinoxylans and their degree of branching using gas chromatography. In HEALTHGRAIN Methods: Analysis of Bioactive Components in Small Grain Cereals; Shewry, W., Ed.; AACC International: St. Paul, MN, USA, 2009; pp. 177–189. [Google Scholar]
- Douglas, S.G. A rapid method for the determination of pentosans in wheat flour. Food Chem. 1981, 7, 139–145. [Google Scholar] [CrossRef]
- Wheeler, H.J.; Tollens, B.V. Ueber die Xylose oder den Holzzucker, eine zweite Penta-Glycose. Justus Liebigs Ann. Chem. 1889, 254, 304–320. [Google Scholar] [CrossRef]
- Kröber, E.; Rimbach, C.; Tollens, B. Anwendung der Pentosan-Bestimmungsmethode auf verschiedene vegetabilische Stoffe und die Materialien der Papierfabrikation. Angew. Chem. 1902, 15, 508–510. [Google Scholar] [CrossRef]
- Chase, E.F. The Phloroglucinol Furfural Reaction; University of Massachusetts Amherst: Amherst, MA, USA, 1925. [Google Scholar]
- Fraser, J.R.; Brandon-Bravo, M.; Holmes, D.C. The proximate analysis of wheat flour carbohydrates. I.—Methods and Scheme of Analysis. J. Sci. Food Agric. 1956, 7, 577–589. [Google Scholar] [CrossRef]
- Dische, Z.; Borenfreund, E. A new color reaction for the determination of aldopentose in presence of other saccharides. Biochim. Biophys. Acta 1957, 23, 639–642. [Google Scholar] [CrossRef]
- Tollens, B. Über Pentosanbestimmung. Ber. Dtsch. Chem. Ges. 1903, 36, 261–264. [Google Scholar] [CrossRef]
- Cracknell, R.L.; Moye, C.J. A colorimetric method for the determination of pentosan-cereal products. In Proceedings of the 20th Annual Conference R.A.C.I, Canberra, Australia, 17–20 August 1970; pp. 67–77. [Google Scholar]
- Bell, B.M. A Rapid Method of Dietary Fiber Estimation in Wheat Products. J. Sci. Food Agric. 1985, 36, 815–821. [Google Scholar] [CrossRef]
- Rouau, X.; Surget, A. A rapid semi-automated method for the determination of total and water-extractable pentosans in wheat flours. Carbohydr. Polym. 1994, 24, 123–132. [Google Scholar] [CrossRef]
- Hashimoto, S.; Shogren, M.D.; Pomeranz, Y. Cereal pentosans: Their estimation and significance. I. Pentosans in wheat and milled wheat products. Cereal Chem. 1987, 64, 30–34. [Google Scholar]
- Delcour, J.A.; Vanhamel, S.; Geest, C.d. Physico-chemical and functional properties of rye nonstarch polysaccharides. I. Colorimetric analysis of pentosans and their relative monosaccharide compositions in fractionated (milled) rye products. Cereal Chem. 1989, 66, 107–111. [Google Scholar]
- Bettge, A.D.; Morris, C.F. Relationships among grain hardness, pentosan fractions, and end-use quality of wheat. Cereal Chem. 2000, 77, 241–247. [Google Scholar] [CrossRef]
- Finnie, S.M.; Bettge, A.D.; Morris, C.F. Influence of cultivar and environment on water-soluble and water-insoluble arabinoxylans in soft wheat. Cereal Chem. 2006, 83, 617–623. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; Dexter, J.E. Barley β-glucans and arabinoxylans: Molecular structure, physicochemical properties, and uses in food products—A Review. Food Res. Int. 2008, 41, 850–868. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; Jacobs, M.; Dexter, J.E. Distribution and structural variation of nonstarch polysaccharides in milling fractions of hull-less barley with variable amylose content. Cereal Chem. 2003, 80, 645–653. [Google Scholar] [CrossRef]
- Kiszonas, A.M.; Courtin, C.M.; Morris, C.F. A Critical Assessment of the Quantification of Wheat Grain Arabinoxylans Using a Phloroglucinol Colorimetric Assay. Cereal Chem. J. 2012, 89, 143–150. [Google Scholar] [CrossRef]
- Herzig, J.; Kohn, R. Zur Kenntnis des Phloroglucides. Mon. Chem. 1908, 29, 677–688. [Google Scholar] [CrossRef]
- Klingstedt, F.W. Über die Bestimmung des Pentosans. Fresenius’ Z. Anal. Chem. 1925, 66, 129–160. [Google Scholar] [CrossRef]
- Goodwin, W.; Tollens, B. Über die Zusammensetzung des Furfurolphloroglucides. Ber. Dtsch. Chem. Ges. 1904, 37, 315–319. [Google Scholar] [CrossRef]
- Jäger, R.; Unger, E. Über Pentosanbestimmung. Ber. Dtsch. Chem. Ges. 1902, 35, 4440–4443. [Google Scholar] [CrossRef]
- Mann, F.; Krüger, M.; Tollens, B. Über die Bestimmung der Pentosen und Pentosane durch Furfuroldestillation. Z. Angew. Chem. 1896, 9, 33–46. [Google Scholar] [CrossRef]
- Kröber, E. Untersuchungen über die Pentosanbestimmungen mittelst der Salzsäure-Phloroglucinmethode nebst einigen Anwendungen. J. Landwirtsch. 1901, 48, 357–384. [Google Scholar]
- Councler, C. Ein neues Verfahren zur quantitativen Bestimmung von Furfurol bzw. den in den Vegetabilien enthaltenen Pentosanen. Chem. Ztg. 1894, 18, 966–968. [Google Scholar]
- Leach, A.E.; Winton, A.L. For the use of public analysts, health officers, sanitary chemists, and food economists. In Food Inspection and Analysis, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1913; p. 1001. [Google Scholar]
- Foo, L.Y.; Hemingway, R.W. Condensed Tannins: Reactions of Model Compounds with Furfuryl Alcohol and Furfuraldehyde. J. Wood Chem. Technol. 1985, 5, 135–158. [Google Scholar] [CrossRef]
 spectrophotometer,
spectrophotometer,  multi-mode microplate reader.
spectrophotometer, multi-mode microplate reader.
multi-mode microplate reader.
spectrophotometer, multi-mode microplate reader.
 (dark green) spectrophotometer,
(dark green) spectrophotometer,  (light green) multi-mode microplate reader. Beer samples: lager (A), lager (B), pilsner (C), pilsner (D), pilsner (E), wheat beer (F), non-alcoholic wheat beer (G).
(dark green) spectrophotometer, (light green) multi-mode microplate reader. Beer samples: lager (A), lager (B), pilsner (C), pilsner (D), pilsner (E), wheat beer (F), non-alcoholic wheat beer (G).
(light green) multi-mode microplate reader. Beer samples: lager (A), lager (B), pilsner (C), pilsner (D), pilsner (E), wheat beer (F), non-alcoholic wheat beer (G).
(dark green) spectrophotometer, (light green) multi-mode microplate reader. Beer samples: lager (A), lager (B), pilsner (C), pilsner (D), pilsner (E), wheat beer (F), non-alcoholic wheat beer (G).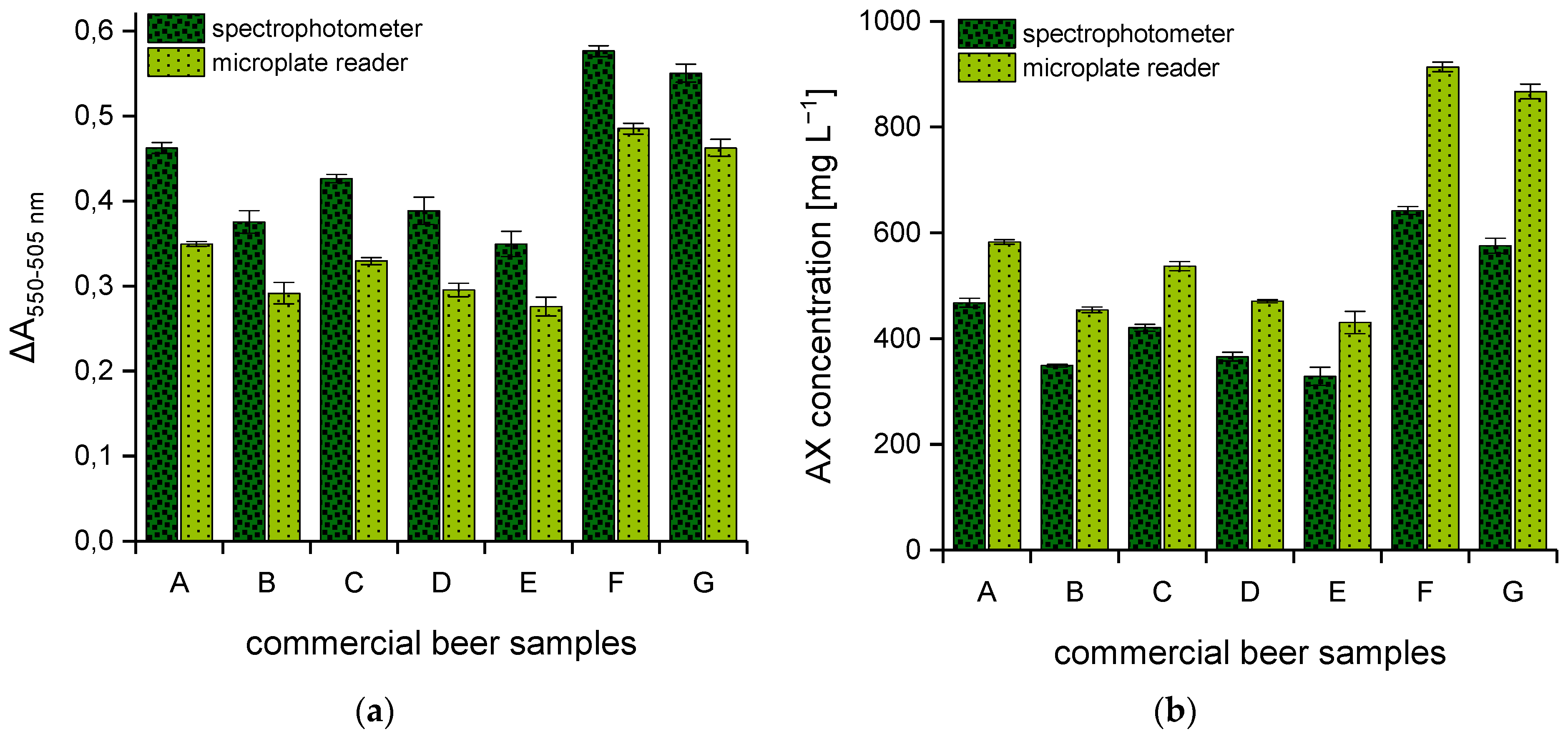
 X 50,
X 50,  X 150,
X 150,  X 250,
X 250,  AX250,
AX250,  AX500.
X 50, X 150, X 250, AX250, AX500.
AX500.
X 50, X 150, X 250, AX250, AX500.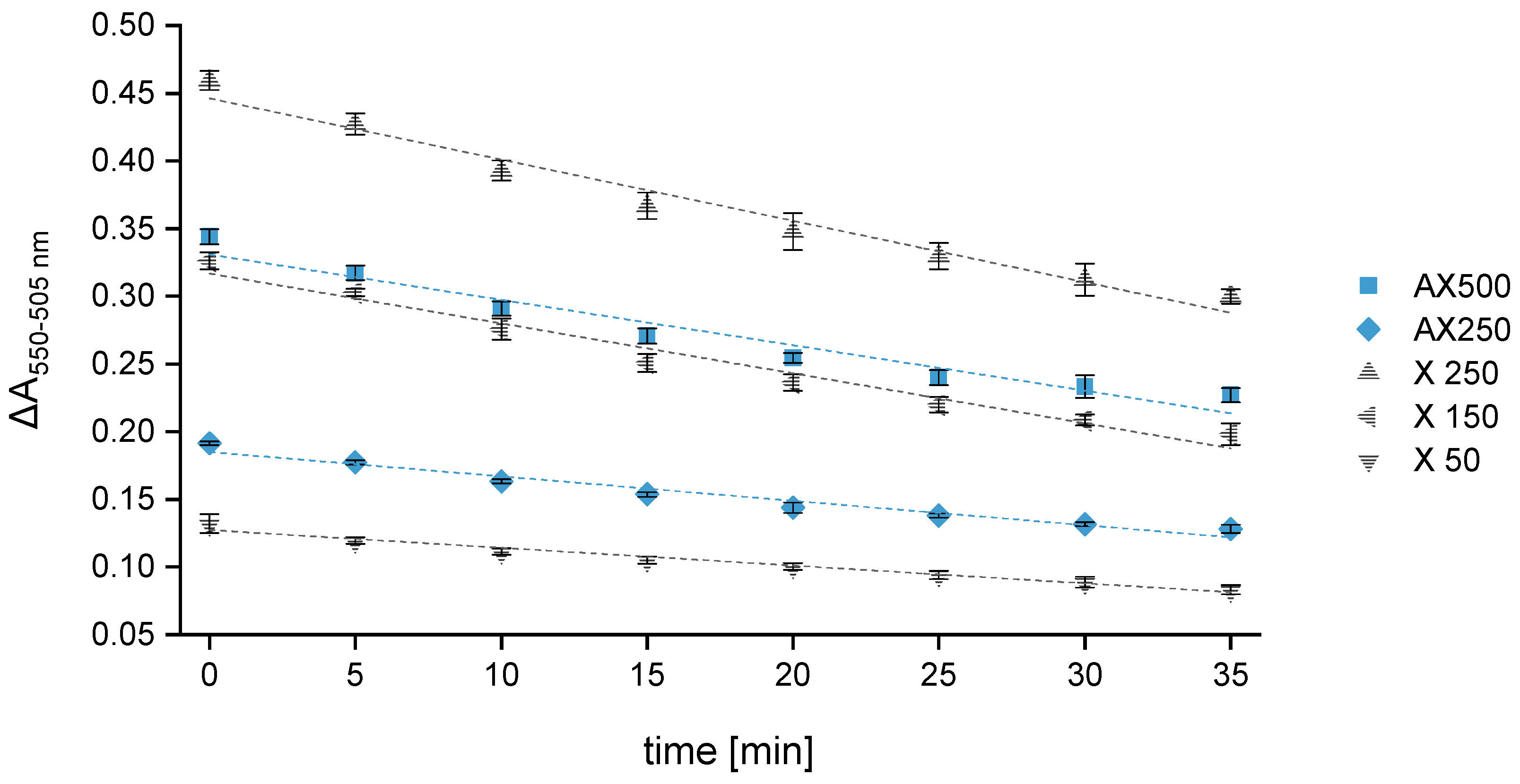

 reaction reagent as reference,
reaction reagent as reference,  reaction reagent with xylose,
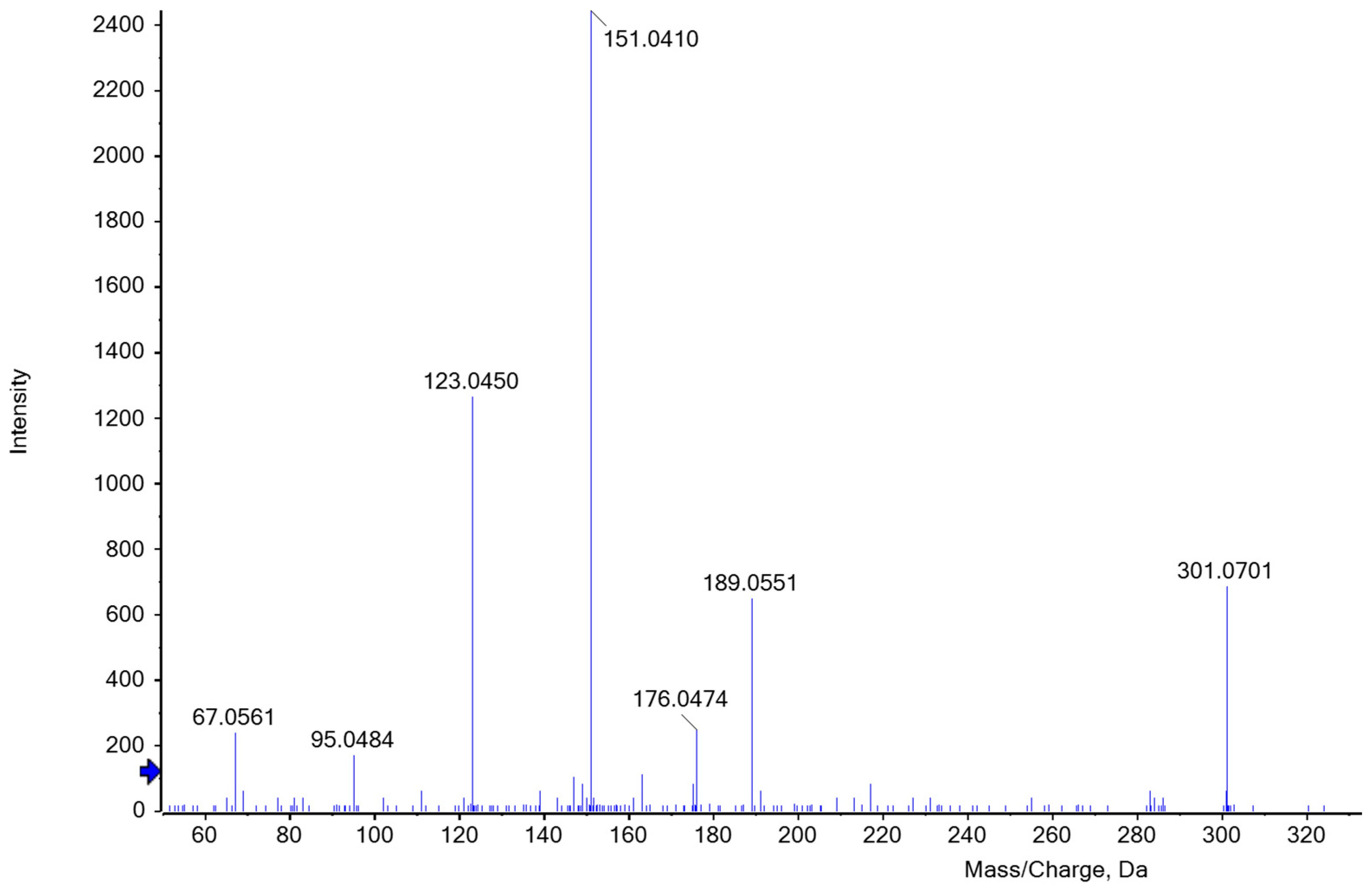
reaction reagent with xylose,  reaction reagent with AX. (b) ESI-MS/MS spectrum of [M+H]+ ion (m/z 235.06) of phloroglucide in the hydrolyzed reaction reagent.
reaction reagent as reference, reaction reagent with xylose, reaction reagent with AX. (b) ESI-MS/MS spectrum of [M+H]+ ion (m/z 235.06) of phloroglucide in the hydrolyzed reaction reagent.
reaction reagent with AX. (b) ESI-MS/MS spectrum of [M+H]+ ion (m/z 235.06) of phloroglucide in the hydrolyzed reaction reagent.
reaction reagent as reference, reaction reagent with xylose, reaction reagent with AX. (b) ESI-MS/MS spectrum of [M+H]+ ion (m/z 235.06) of phloroglucide in the hydrolyzed reaction reagent.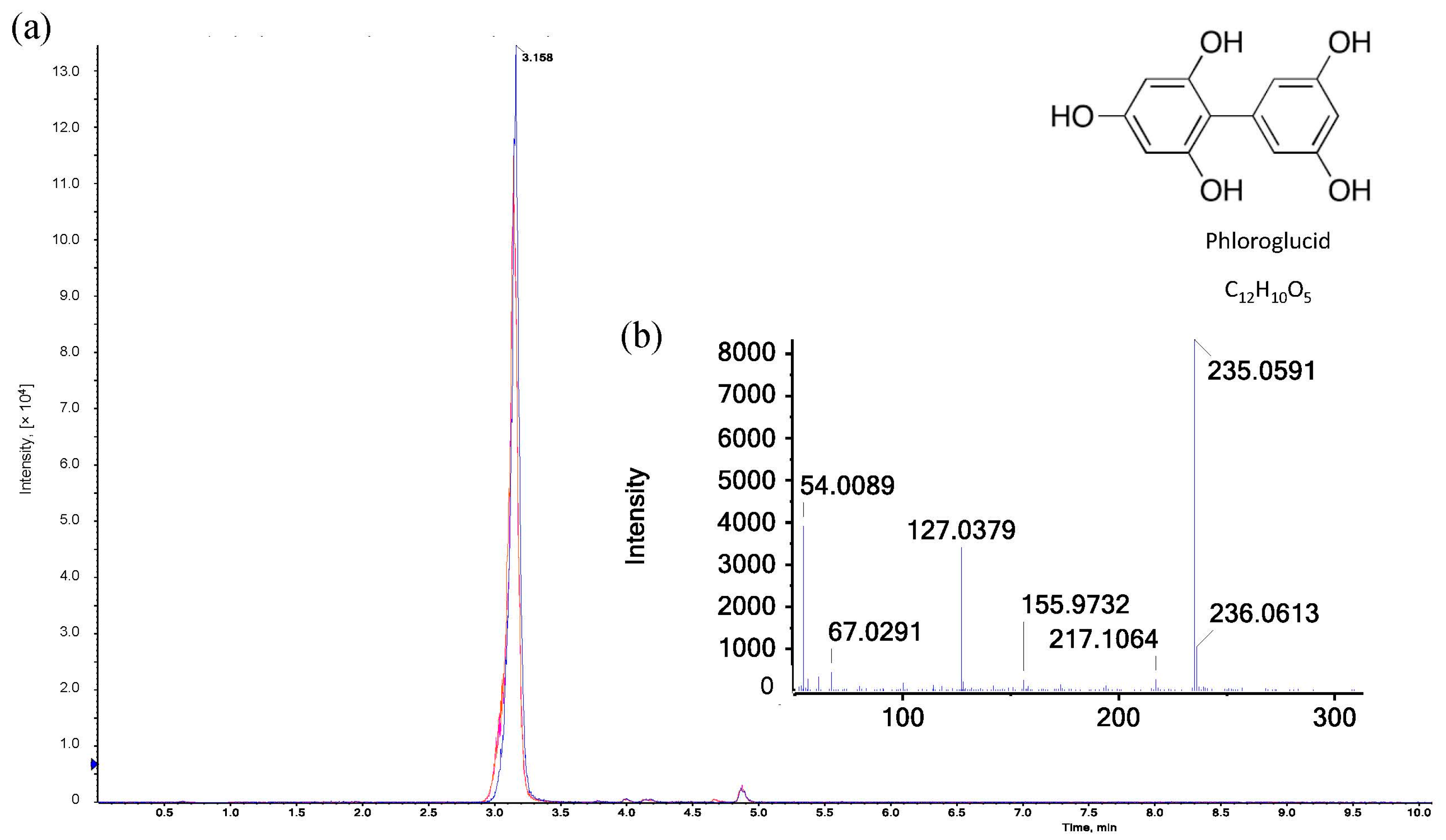

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Phloroglucide Derivatives | Molecular Weight | Reference |
|---|---|---|---|
| C22H16O8 | 2P + 2F − 2H2O | 409 Da | Goodwin and Tollens [44] |
| C11H8O4 | P + F − H2O | 205 Da | Jäger and Unger [45]; Goodwin and Tollens [44] |
| C11H6O3 | P + F − 2H2O | 187 Da | Mann et al. [46]; Kröber [47] |
| C16H12O6 | P + 2F − H2O | 301 Da | Councler [48]; Leach and Winton [49] |
| C17H14O7 | 2P + F − H2O | 331 Da | Foo and Hemingway [50] |
| C17H12O6 | 2P + F − 2H2O | 313 Da | Chase [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steiner, J.; Kupetz, M.; Becker, T. Advancing Quantification of Water-Extractable Arabinoxylan in Beer: A High-Throughput Approach. Polymers 2023, 15, 3959. https://doi.org/10.3390/polym15193959
Steiner J, Kupetz M, Becker T. Advancing Quantification of Water-Extractable Arabinoxylan in Beer: A High-Throughput Approach. Polymers. 2023; 15(19):3959. https://doi.org/10.3390/polym15193959
Chicago/Turabian StyleSteiner, Julia, Michael Kupetz, and Thomas Becker. 2023. "Advancing Quantification of Water-Extractable Arabinoxylan in Beer: A High-Throughput Approach" Polymers 15, no. 19: 3959. https://doi.org/10.3390/polym15193959
APA StyleSteiner, J., Kupetz, M., & Becker, T. (2023). Advancing Quantification of Water-Extractable Arabinoxylan in Beer: A High-Throughput Approach. Polymers, 15(19), 3959. https://doi.org/10.3390/polym15193959