




Biomass-Based Shape-Stabilized Composite Phase-Change Materials with High Solar–Thermal Conversion Efficiency for Thermal Energy Storage


Abstract
:1. Introduction
2. Experimental Methods
2.1. Materials
2.2. Preparation of Pine Cone Porous Biomass Carbon
2.3. Preparation of Composite Form-Stable PCMs
2.4. Preparation of Rigid Polyurethane Foam for Energy-Saving Building
2.5. Characterizations
3. Results and Discussion
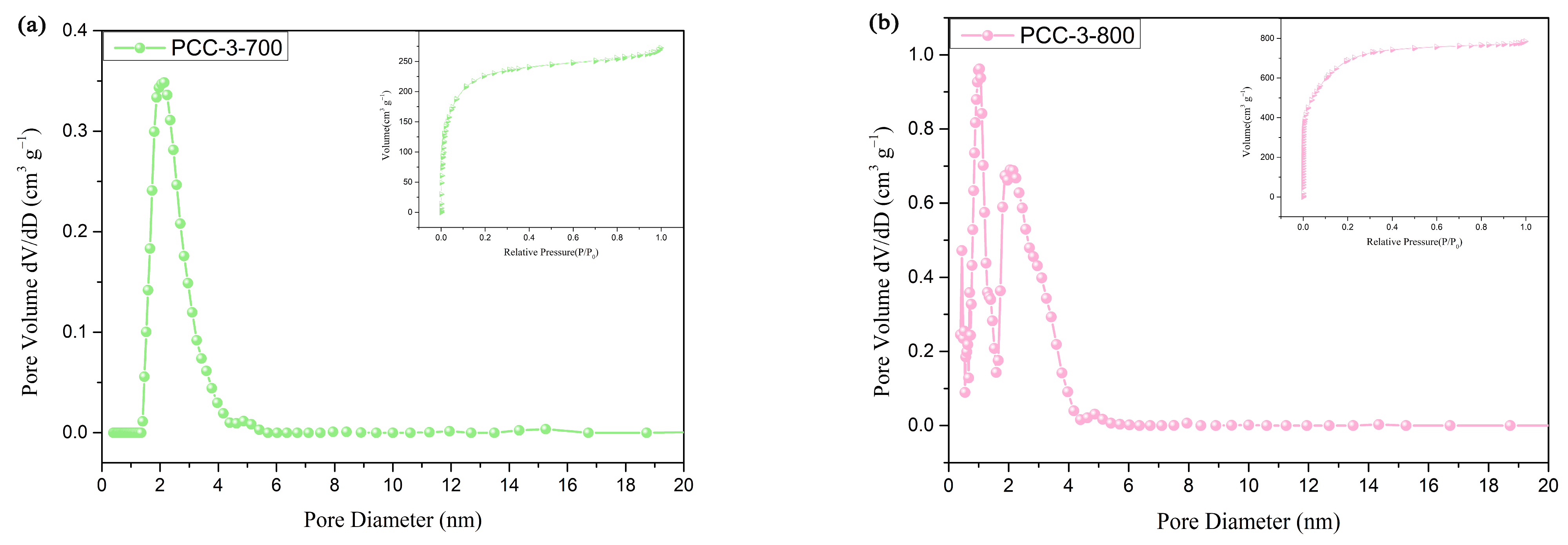
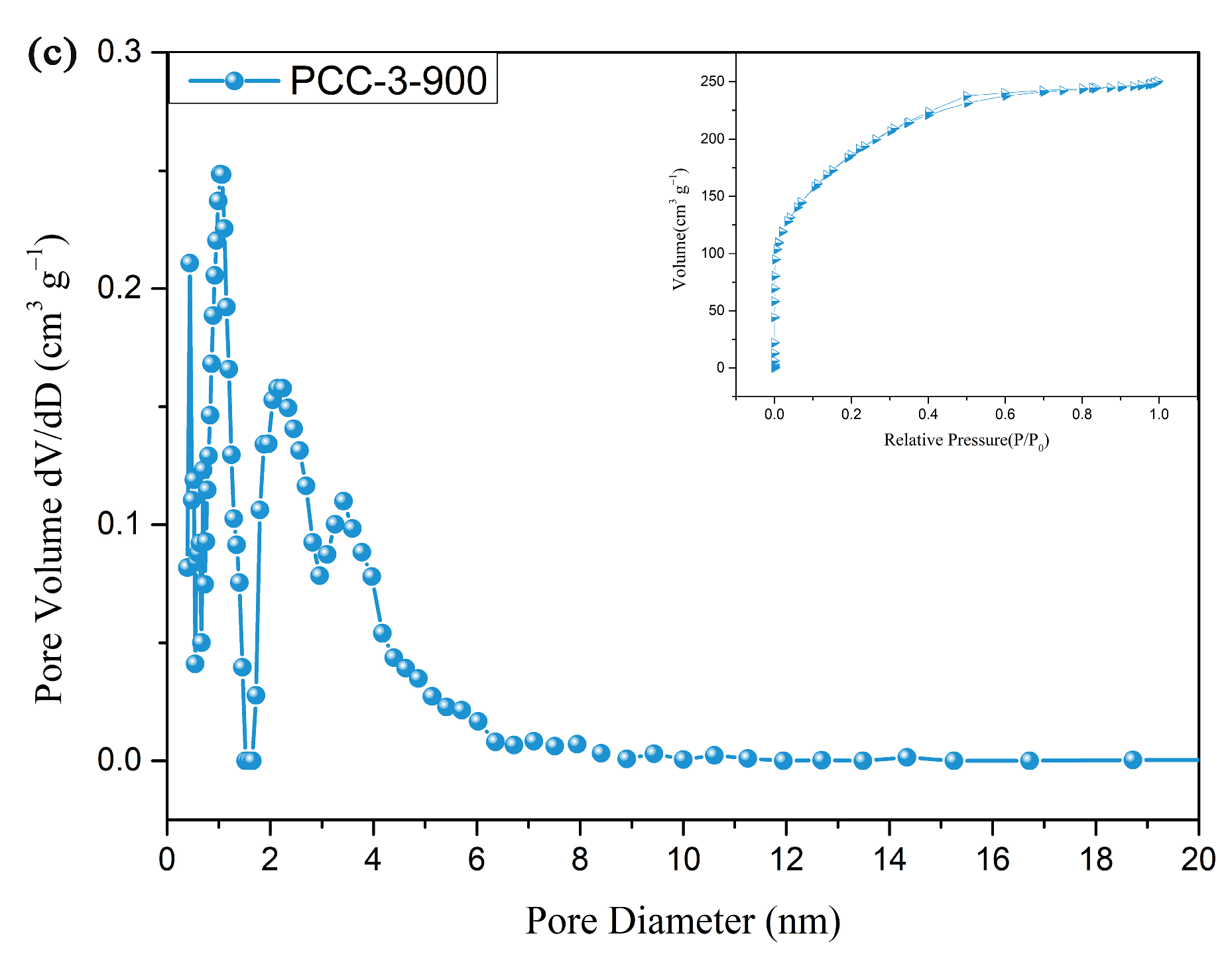
3.1. Structure of PCCs
3.2. Formation Mechanism
3.3. Structures of Composite PCMs
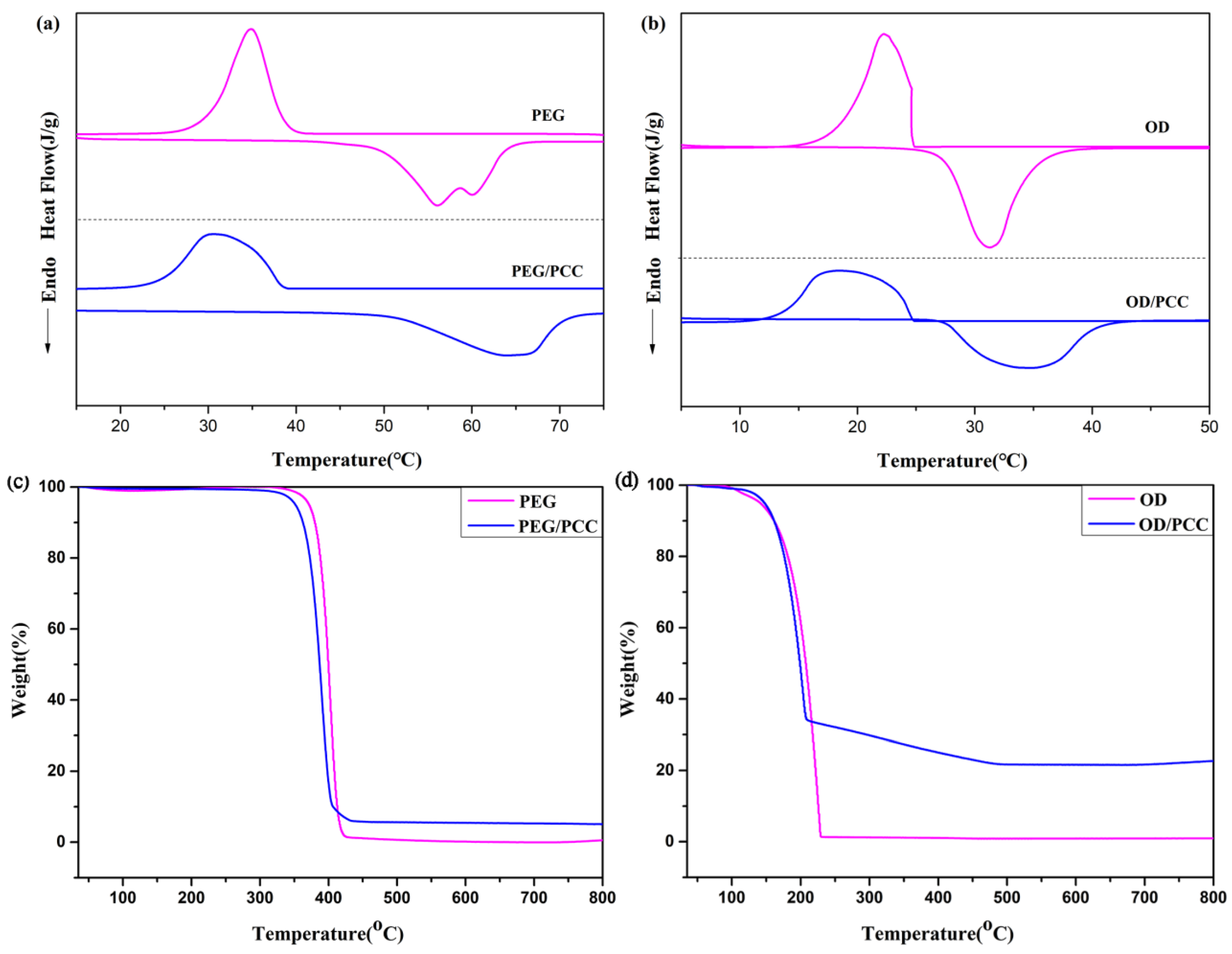
3.4. Thermal Properties and Stability of Composite PCMs
3.5. Thermal Conductivity and Solar–Thermal Energy Conversion
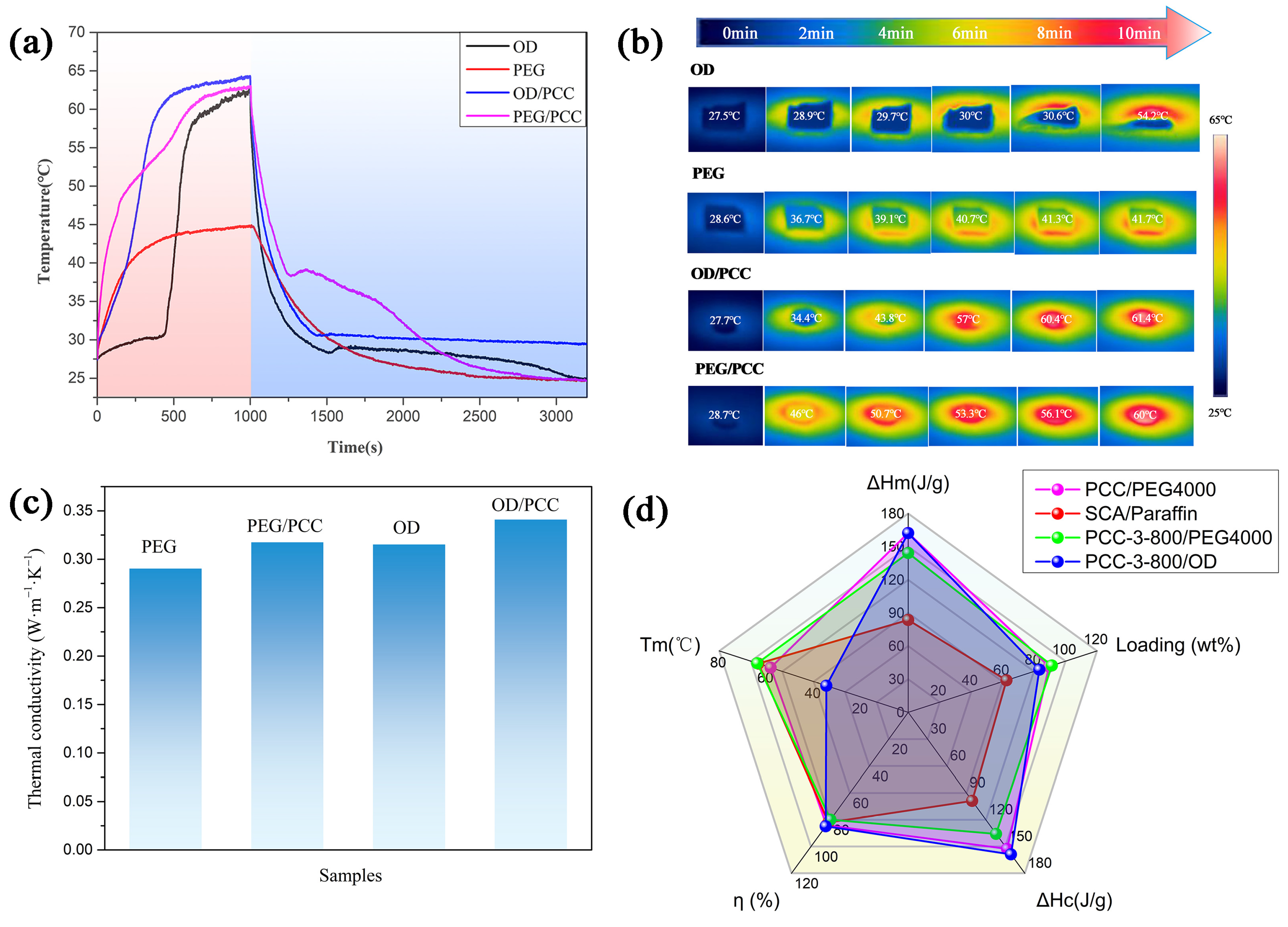
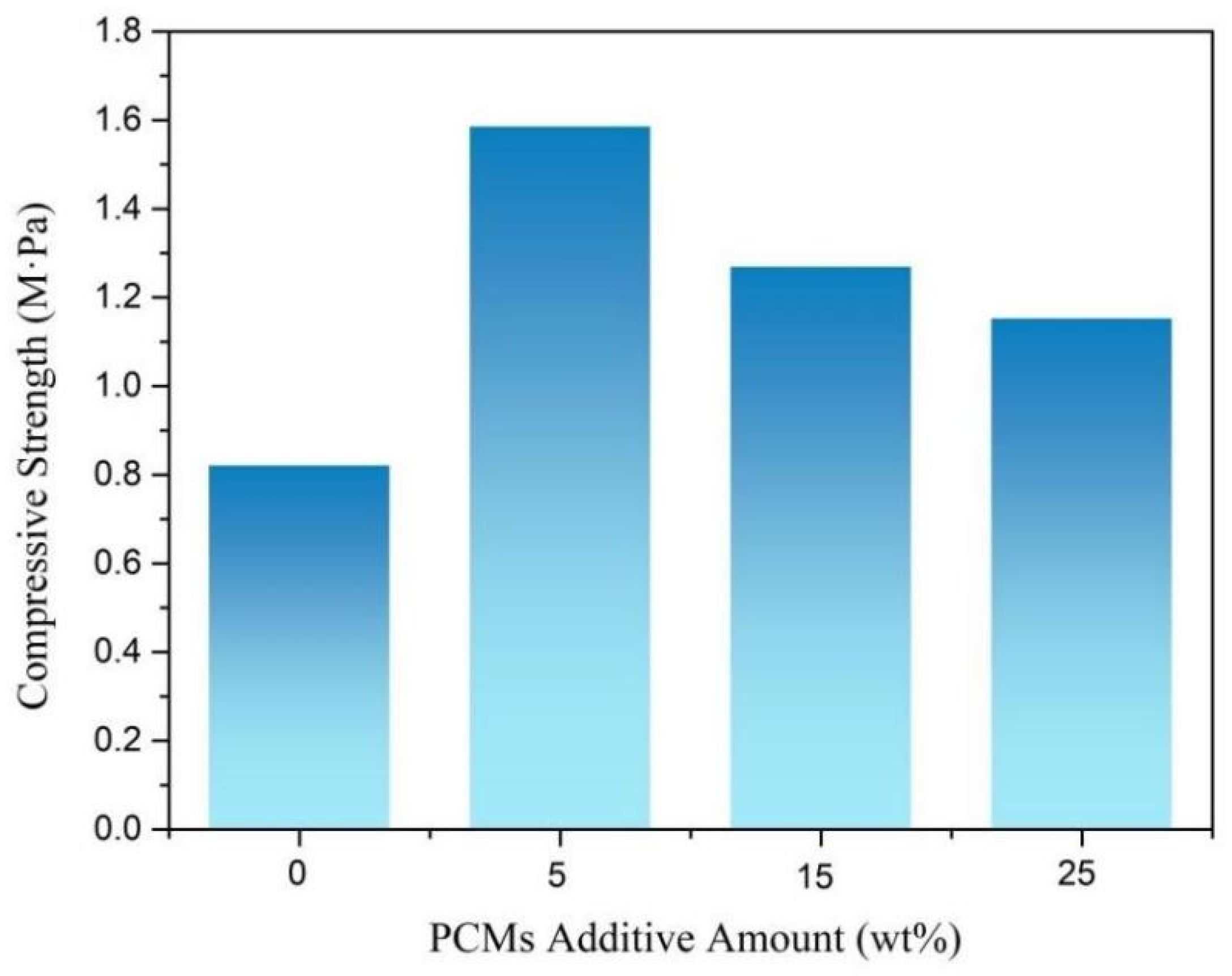
3.6. Compression Performance of Rigid Polyurethane Foam for Energy Storage
3.7. Thermal Conductivity of Rigid Polyurethane Foam for Energy Storage
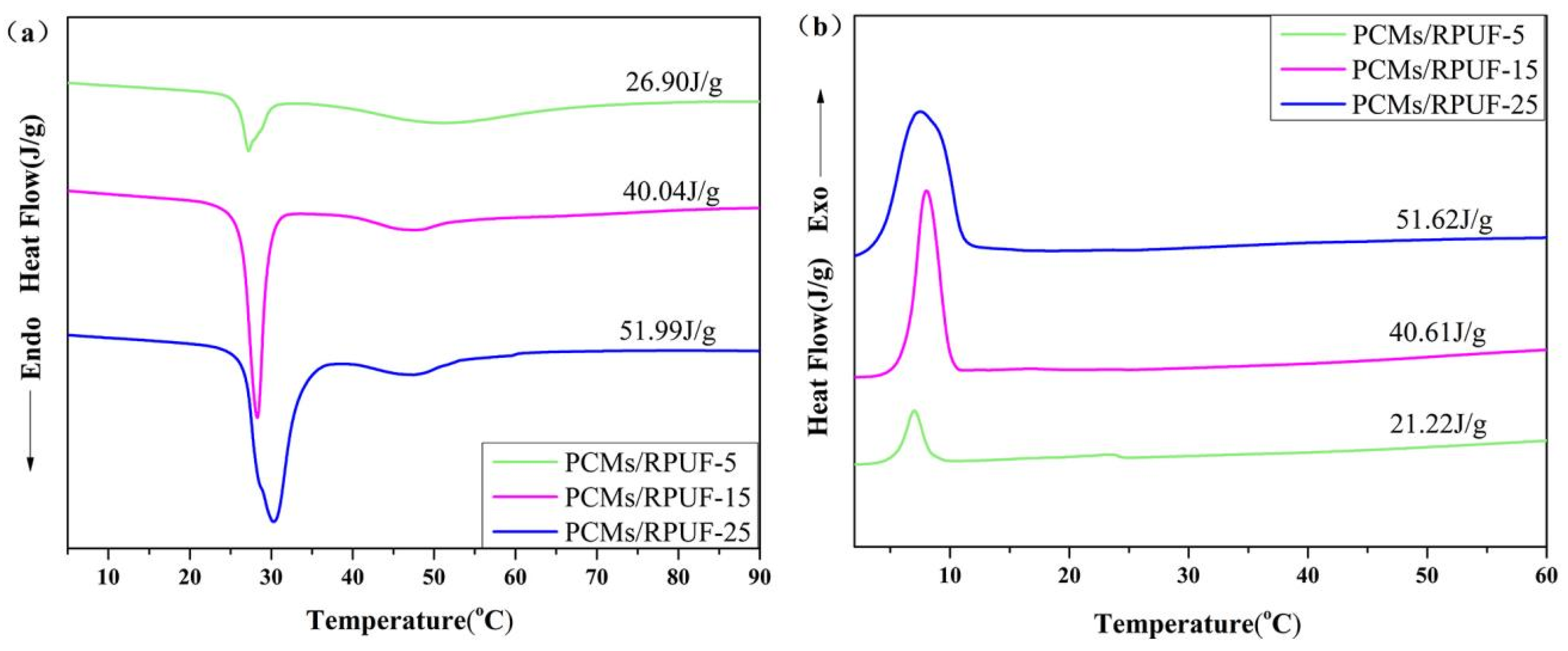
3.8. Thermal Performance of Rigid Polyurethane Foam for Energy Storage
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yin, L.; Zhao, M.; Yang, R. Preparation and thermal properties of propyl palmitate-based phase change composites with enhanced thermal conductivity for thermal energy storage. Polymers 2023, 15, 3192. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Xu, Y.; Cai, X.; Zheng, Z.J. Effect of the circumferential and radial graded metal foam on horizontal shell-and-tube latent heat thermal energy storage unit. Sol. Energy 2021, 226, 225–235. [Google Scholar] [CrossRef]
- Luo, Y.; Xie, Y.; Jiang, H.; Chen, Y.; Zhang, L.; Sheng, X.; Xie, D.; Wu, H.; Mei, Y. Flame-retardant and form-stable phase change composites based on MXene with high thermostability and thermal conductivity for thermal energy storage. Chem. Eng. J. 2021, 420, 130466. [Google Scholar] [CrossRef]
- Gao, N.; Tang, T.; Xiang, H.; Zhang, W.; Li, Y.; Yang, C.; Xia, T.; Liu, X. Preparation and structure-properties of crosslinking organic montmorillonite/polyurethane as solid-solid phase change materials for thermal energy storage. Sol. Energy Mater. Sol. Cells 2022, 244, 111831. [Google Scholar] [CrossRef]
- Zheng, Y.; Oguzlu, H.; Baldelli, A.; Zhu, Y.; Song, M.; Pratap-Singh, A.; Jiang, F. Sprayable cellulose nanofibrils stabilized phase change material pickering emulsion for spray coating application. Carbohydr. Polym. 2022, 291, 119583. [Google Scholar] [CrossRef]
- Nejman, A.; Cieslak, M. The impact of the heating/cooling rate on the thermoregulating properties of textile materials modified with PCM microcapsules. Appl. Therm. Eng. 2017, 127, 212–223. [Google Scholar] [CrossRef]
- Salgueiro, T.; Samagaio, A.; Gonçalves, M.; Figueiredo, A.; Labrincha, J.; Silva, L. Incorporation of phase change materials in an expanded clay containing mortar for indoor thermal regulation of buildings. J. Energy Storage 2021, 36, 102385. [Google Scholar] [CrossRef]
- Wu, M.; Liu, C.; Rao, Z. Experimental study on lauryl alcohol/expanded graphite composite phase change materials for thermal regulation in building. Constr. Build. Mater. 2022, 335, 127400. [Google Scholar] [CrossRef]
- Liu, X.; Su, H.; Huang, Z.; Lin, P.; Yin, T.; Sheng, X.; Chen, Y. Biomass-based phase change material gels demonstrating solar-thermal conversion and thermal energy storage for thermoelectric power generation and personal thermal management. Sol. Energy 2022, 239, 307–318. [Google Scholar] [CrossRef]
- Yan, D.; Li, M. Stearic acid-modified MOF-based composite phase change materials for solar-thermal energy conversion and storage. Sol. Energy 2023, 262, 111843. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, X.; Ji, J.; Liu, L.; Wu, Y.; Yang, M.; Lu, D.; Zheng, H. Development of flexible form-stable phase change material with enhanced electrical resistance for thermal management. J. Clean. Prod. 2021, 311, 127517. [Google Scholar] [CrossRef]
- Rakshamuthu, S.; Jegan, S.; Benyameen, J.J.; Selvakumar, V.; Anandeeswaran, K.; Iyahraja, S. Experimental analysis of small size solar dryer with phase change materials for food preservation. J. Energy Storage 2021, 33, 102095. [Google Scholar] [CrossRef]
- Gao, H.; Wang, J.; Chen, X.; Wang, G.; Huang, X.; Li, A.; Dong, W. Nanoconfinement effects on thermal properties of nanoporous shape-stabilized composite PCMs: A review. Nano Energy 2018, 53, 769–797. [Google Scholar] [CrossRef]
- Chen, X.; Gao, H.; Yang, M.; Dong, W.; Huang, X.; Li, A.; Dong, C.; Wang, G. Highly graphitized 3D network carbon for shape-stabilized composite PCMs with superior thermal energy harvesting. Nano Energy 2018, 49, 86–94. [Google Scholar] [CrossRef]
- Zhao, P.P.; Deng, C.; Zhao, Z.Y.; Lu, P.; He, S.; Wang, Y.Z. Hypophosphite tailored graphitized hierarchical porous biochar toward highly efficient solar thermal energy harvesting and stable storage/release. Chem. Eng. J. 2021, 420, 129942. [Google Scholar] [CrossRef]
- Liu, Y.; Xia, Y.; An, K.; Huang, C.; Cui, W.; Wei, S.; Ji, R.; Xu, F.; Zhang, H.; Sun, L. Fabrication and characterization of novel meso-porous carbon/n-octadecane as form-stable phase change materials for enhancement of phase-change behavior. J. Mater. Sci. Technol. 2019, 35, 939–945. [Google Scholar] [CrossRef]
- Xu, Y.; Ren, Q.; Zheng, Z.J.; He, Y.L. Evaluation and optimization of melting performance for a latent heat thermal energy storage unit partially filled with porous media. Appl. Energy 2017, 193, 84–95. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, Q.; Kang, S.; Li, J.; Jia, X. Coal-based ultrathin-wall graphitic porous carbon for high-performance form-stable phase change materials with enhanced thermal conductivity. Chem. Eng. J. 2020, 395, 125112. [Google Scholar] [CrossRef]
- Alshaer, W.G.; Rady, M.A.; Nada, S.A.; Palomo Del Barrio, E.; Sommier, A. An experimental investigation of using carbon foam-PCM-MWCNTs composite materials for thermal management of electronic devices under pulsed power modes. Heat Mass Transf. 2017, 53, 569–579. [Google Scholar] [CrossRef]
- Khadiran, T.; Hussein, M.Z.; Zainal, Z.; Rusli, R. Activated carbon derived from peat soil as a framework for the preparation of shape-stabilized phase change material. Energy 2015, 82, 468–478. [Google Scholar] [CrossRef]
- Atinafu, D.G.; Chang, S.J.; Kim, K.H.; Kim, S. Tuning surface functionality of standard biochars and the resulting uplift capacity of loading/energy storage for organic phase change materials. Chem. Eng. J. 2020, 394, 125049. [Google Scholar] [CrossRef]
- Gao, M.; Wang, W.K.; Zheng, Y.M.; Zhao, Q.B.; Yu, H.Q. Hierarchically porous biochar for supercapacitor and electrochemical H2O2 production. Chem. Eng. J. 2020, 402, 126171. [Google Scholar] [CrossRef]
- Negi, P.; Chhantyal, A.K.; Dixit, A.K.; Kumar, S.; Kumar, A. Activated carbon derived from mango leaves as an enhanced microwave absorbing material. Sustain. Mater. Technol. 2021, 27, e00244. [Google Scholar] [CrossRef]
- Kamali, M.; Appels, L.; Kwon, E.E.; Aminabhavi, T.M.; Dewil, R. Biochar in water and wastewater treatment-a sustainability assessment. Chem. Eng. J. 2021, 420, 129946. [Google Scholar] [CrossRef]
- Atinafu, D.G.; Wi, S.; Yun, B.Y.; Kim, S. Engineering biochar with multiwalled carbon nanotube for efficient phase change material encapsulation and thermal energy storage. Energy 2021, 216, 119294. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, X.; Zhang, X.; Yin, Z.; Liu, Y.; Fang, M.; Wu, X.; Min, X.; Huang, Z. Lauric-stearic acid eutectic mixture/carbonized biomass waste corn cob composite phase change materials: Preparation and thermal characterization. Thermochim. Acta 2019, 674, 21–27. [Google Scholar] [CrossRef]
- Hekimoğlu, G.; Sarı, A.; Kar, T.; Keleş, S.; Kaygusuz, K.; Yıldırım, N.; Tyagi, V.V.; Sharma, R.K.; Saleh, T.A. Carbonized waste hazelnut wood-based shape-stable composite phase change materials for thermal management implementations. Int. J. Energy Res. 2021, 45, 10271–10284. [Google Scholar] [CrossRef]
- Wen, R.; Liu, Y.; Yang, C.; Zhu, X.; Huang, Z.; Zhang, X.; Gao, W. Enhanced thermal properties of stearic acid/carbonized maize straw composite phase change material for thermal energy storage in buildings. J. Energy Storage 2021, 36, 102420. [Google Scholar] [CrossRef]
- Atinafu, D.G.; Yun, B.Y.; Kim, Y.U.; Wi, S.; Kim, S. Introduction of eicosane into biochar derived from softwood and wheat straw: Influence of porous structure and surface chemistry. Chem. Eng. J. 2021, 415, 128887. [Google Scholar] [CrossRef]
- Martinez, M.L.; Torres, M.M.; Guzman, C.A.; Maestri, D.M. Preparation and characteristics of activated carbon from olive stones and walnut shells. Ind. Crop Prod. 2006, 23, 23–28. [Google Scholar] [CrossRef]
- Hekimoğlu, G.; Sarı, A.; Kar, T.; Keleş, S.; Kaygusuz, K.; Tyagi, V.V.; Sharma, R.; Al-Ahmed, A.; Al-Sulaiman, F.A.; Saleh, T.A. Walnut shell derived bio-carbon/methyl palmitate as novel composite phase change material with enhanced thermal energy storage properties. J. Energy Storage 2021, 35, 102288. [Google Scholar] [CrossRef]
- Li, W.; Peng, J.; Zhang, L.; Yang, K.; Xia, H.; Zhang, S.; Guo, S.H. Preparation of activated carbon from coconut shell chars in pilot-scale microwave heating equipment at 60 kW. Waste Manag. 2009, 29, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Muchtar, A.R.; Hassam, C.L.; Srinivasan, B.; Berthebaud, D.; Mori, T.; Soelami, N.; Yuliarto, B. Shape-stabilized phase change materials: Performance of simple physical blending synthesis and the potential of coconut based materials. J. Energy Storage 2022, 52, 104974. [Google Scholar] [CrossRef]
- Tham, Y.J.; Latif, P.A.; Abdullah, A.M.; Shamala-Devi, A.; Taufiq-Yap, Y.H. Performances of toluene removal by activated carbon derived from durian shell. Bioresour. Technol. 2011, 102, 724–728. [Google Scholar] [CrossRef]
- Maldhure, A.V.; Ekhe, J.D. Preparation and characterization of microwave assisted AC form industrial waste lignin for Cu (II) sorption. Chem. Eng. J. 2011, 168, 1103–1111. [Google Scholar] [CrossRef]
- Braghiroli, F.L.; Bouafif, H.; Neculita, C.M.; Koubaa, A. Influence of pyro-gasification and activation conditions on the porosity of activated biochars: A literature review. Waste Biomass Valor. 2020, 11, 5079–5098. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, F.; Li, C.; Cai, J. Biomass-based shape-stable phase change materials supported by garlic peel-derived porous carbon for thermal energy storage. J. Energy Storage 2022, 46, 103929. [Google Scholar] [CrossRef]
- Zhou, X.M.; Liu, Y.F. Study on the preparation of high adsorption activated carbon material and its application as phase change energy storage carrier material. J. Therm. Anal. Calorim. 2022, 147, 8169–8176. [Google Scholar] [CrossRef]
- Wang, X.; Liang, X.; Wang, Y.; Wang, X.; Liu, M.; Yin, D.; Xia, S.; Zhao, J.; Zhang, Y. Adsorption of Copper (II) onto activated carbons from sewage sludge by microwave-induced phosphoric acid and zinc chloride activation. Desalination 2011, 278, 231–237. [Google Scholar] [CrossRef]
- Gao, Y.; Yue, Q.; Gao, B.; Sun, Y.; Wang, W.; Li, Q.; Wang, Y. Comparisons of porous, surface chemistry and adsorption properties of carbon derived from enteromorpha prolifera activated by H4P2O7 and KOH. Chem. Eng. J. 2013, 232, 582–590. [Google Scholar] [CrossRef]
- Zhao, B.; O’Connor, D.; Zhang, J.; Peng, T.; Shen, Z.; Tsang, D.C.; Hou, D. Effect of pyrolysis temperature, heating rate, and residence time on rapeseed stem derived biochar. J. Clean. Prod. 2018, 174, 977–987. [Google Scholar] [CrossRef]
- Taran, O.P.; Miroshnikova, A.V.; Baryshnikov, S.V.; Kazachenko, A.S.; Skripnikov, A.M.; Sychev, V.V.; Malyar, Y.N.; Kuznetsov, B.N. Reductive catalytic fractionation of spruce wood over Ru/C bifunctional catalyst in the medium of ethanol and molecular hydrogen. Catalysts 2022, 12, 1384. [Google Scholar] [CrossRef]
- Wang, F.; Zheng, Y.; Qiu, J.; Liu, S.; Tong, Y.; Zhu, F.; Ouyang, G. Graphene-based metal and nitrogen-doped carbon composites as adsorbents for highly sensitive solid phase microextraction of polycyclic aromatic hydrocarbons. Nanoscale 2018, 10, 10073–10078. [Google Scholar] [CrossRef] [PubMed]
- Atinafu, D.G.; Dong, W.; Wang, C.; Wang, G. Synthesis of porous carbon from cotton using an Mg(OH)2 template for form-stabilized phase change materials with high encapsulation capacity, transition enthalpy and reliability. J. Mater. Chem. A 2018, 6, 8969–8977. [Google Scholar] [CrossRef]
- Liu, S.; Peng, S.; Zhang, B.; Xue, B.; Yang, Z.; Wang, S.; Xu, G. Effects of biochar pyrolysis temperature on thermal properties of polyethylene glycol/biochar composites as shape-stable biocomposite phase change materials. RSC Adv. 2022, 12, 9587–9598. [Google Scholar] [CrossRef] [PubMed]
- Quosai, P.; Anstey, A.; Mohanty, A.K.; Misra, M. Characterization of biocarbon generated by high-and low-temperature pyrolysis of soy hulls and coffee chaff: For polymer composite applications. R. Soc. Open Sci. 2018, 5, 171970. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, X.; Wang, L.; Bei, K.; Wang, J.; Chou, I.M.; Pan, Z. Progress of Raman spectroscopic investigations on the structure and properties of coal. J. Raman Spectrosc. 2020, 51, 1874–1884. [Google Scholar] [CrossRef]
- Gao, Y.; Yue, Q.; Gao, B.; Sun, Y.; Wang, W.; Li, Q.; Wang, Y. Preparation of high surface area-activated carbon from lignin of papermaking black liquor by KOH activation for Ni (II) adsorption. Chem. Eng. J. 2013, 217, 345–353. [Google Scholar] [CrossRef]
- Om Prakash, M.; Gujjala, R.; Panchal, M.; Ojha, S. Mechanical characterization of arhar biomass based porous nano activated carbon polymer composites. Polym. Compos. 2020, 41, 3113–3123. [Google Scholar] [CrossRef]
- Zhang, G.H.; Bon, S.A.F.; Zhao, C.Y. Synthesis, characterization and thermal properties of novel nanoencapsulated phase change materials for thermal energy storage. Sol. Energy 2012, 86, 1149–1154. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, J.; Xu, L.; Xie, H.; Guo, Z. Preparation and thermal characterization of n-octadecane/pentafluorostyrene nanocapsules for phase-change energy storage. J. Energy Storage 2021, 35, 102327. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, G.; Wang, S.; Fang, X.; Liu, X. Thermal energy storage cement mortar containing n-octadecane/expanded graphite composite phase change material. Renew. Energy 2013, 50, 670–675. [Google Scholar]
- Hua, J.S.; Yuan, C.; Zhao, X.; Zhang, J.; Du, J. Structure and thermal properties of expanded graphite/paraffin composite phase change material. Energy Sources Part A Recovery Util. Environ. Eff. 2019, 41, 86–93. [Google Scholar]
- Tang, J.; Chen, X.; Zhang, L.; Yang, M.; Wang, P.; Dong, W.; Wang, G.; Yu, F.; Tao, J. Alkylated meso-macroporous metal-organic framework hollow tubes as nanocontainers of octadecane for energy storage and thermal regulation. Small 2018, 14, 1801970. [Google Scholar] [CrossRef]
- Jiang, L.; Lei, Y.; Liu, Q.; Lei, J. Polyethylene glycol based self-luminous phase change materials for both thermal and light energy storage. Energy 2020, 193, 116802. [Google Scholar] [CrossRef]
- Kong, W.; Lei, Y.; Jiang, Y.; Lei, J. Preparation and thermal performance of polyurethane/PEG as novel form-stable phase change materials for thermal energy storage. J. Therm. Anal. Calorim. 2017, 130, 1011–1019. [Google Scholar] [CrossRef]
- Jing, R.; Zhang, H.; Huang, C.; Su, F.; Wu, B.; Sun, Z.; Xu, F.; Sun, L.; Xia, Y.; Peng, H.; et al. Construction of double cross-linking PEG/h-BN@ GO polymeric energy-storage composites with high structural stability and excellent thermal performances. Colloids Surf. A Physicochem. Eng. Asp. 2022, 638, 128193. [Google Scholar] [CrossRef]
- Zhang, D.; Tian, S.; Xiao, D. Experimental study on the phase change behavior of phase change material confined in pores. Sol. Energy 2007, 81, 653–660. [Google Scholar] [CrossRef]
- Wang, C.; Feng, L.; Li, W.; Zheng, J.; Tian, W.; Li, X. Shape-stabilized phase change materials based on polyethylene glycol/porous carbon composite: The influence of the pore structure of the carbon materials. Sol. Energy Mater. Sol. Cells 2012, 105, 21–26. [Google Scholar] [CrossRef]
- Paneliya, S.; Khanna, S.; Singh, A.P.; Patel, Y.K.; Vanpariya, A.; Makani, N.H.; Banerjee, R.; Mukhopadhyay, I. Core shell paraffin/silica nanocomposite: A promising phase change material for thermal energy storage. Renew. Energy 2021, 167, 591–599. [Google Scholar] [CrossRef]
- Mandal, S.; Ishak, S.; Lee, D.E.; Park, T. Optimization of eco-friendly pinus resinosa biochar-dodecanoic acid phase change composite for the cleaner environment. J. Energy Storage 2022, 55, 105414. [Google Scholar] [CrossRef]
- Sarabandi, D.; Roudini, G.; Barahuie, F. Activated carbon derived from pine cone as a framework for the preparation of n-heptadecane nanocomposite for thermal energy storage. J. Energy Storage 2019, 24, 100795. [Google Scholar] [CrossRef]
- Wang, C.; Liang, W.; Yang, Y.; Liu, F.; Sun, H.; Zhu, Z.; Li, A. Biomass carbon aerogels based shape-stable phase change composites with high light-to-thermal efficiency for energy storage. Renew. Energy 2020, 153, 182–192. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Geng, Y.; Liao, Y.; Chen, S.; Sun, K.; Li, M. Shape-stable phase change composites based on carbonized waste pomelo peel for low-grade thermal energy storage. J. Energy Storage 2022, 47, 103556. [Google Scholar] [CrossRef]
- Wei, Y.; Li, J.; Sun, F.; Wu, J.; Zhao, L. Leakage-proof phase change composites supported by biomass carbon aerogels from succulents. Green Chem. 2018, 20, 1858–1865. [Google Scholar] [CrossRef]
- Borreguero, A.M.; Rodríguez, J.F.; Valverde, J.L.; Peijs, T.; Carmona, M. Characterization of rigid polyurethane foams containing microencapsulted phase change materials: Microcapsules type effect. J. Appl. Polym. Sci. 2013, 128, 582–590. [Google Scholar] [CrossRef]
- Tinti, A.; Tarzia, A.; Passaro, A.; Angiuli, R. Thermographic analysis of polyurethane foams integrated with phase change materials designed for dynamic thermal insulation in refrigerated transport. Appl. Therm. Eng. 2014, 70, 201–210. [Google Scholar] [CrossRef]
- Lu, M.; Huang, Y.; Chen, C. Cedarwood bark-derived hard carbon as an anode for high-performance sodium-ion batteries. Energy Fuels 2020, 34, 11489–11497. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, A.; Izquierdo, M.T.; Mathieu, S.; González-Álvarez, J.; Celzard, A.; Fierro, V. Outstanding electrochemical performance of highly N-and O-doped carbons derived from pine tannin. Green Chem. 2017, 19, 2653–2665. [Google Scholar] [CrossRef]
- Murali, G.; Kesavan, T.; Ponnusamy, S.; Harish, S.; Navaneethan, M. Improved supercapacitor performance based on sustainable synthesis using chemically activated porous carbon. J. Alloys Compd. 2022, 906, 164287. [Google Scholar] [CrossRef]
- He, R.; Neupane, M.; Zia, A.; Huang, X.; Bowers, C.; Wang, M.; Lu, J.; Yang, Y.; Dong, P. Binder-free wood converted carbon for enhanced water desalination performance. Adv. Funct. Mater. 2022, 32, 2208040. [Google Scholar] [CrossRef]
- Leng, C.; Sun, K.; Li, J.; Jiang, J. From dead pine needles to O, N codoped activated carbons by a one-step carbonization for high rate performance supercapacitors. ACS Sustain. Chem. Eng. 2017, 5, 10474–10482. [Google Scholar] [CrossRef]
- Wan, Y.; Chen, Y.; Cui, Z.; Ding, H.; Gao, S.; Han, Z.; Gao, J. A promising form-stable phase change material prepared using cost effective pinecone biochar as the matrix of palmitic acid for thermal energy storage. Sci. Rep. 2019, 9, 11535. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Huang, Z.; Yin, Z.; Min, X.; Liu, Y.G.; Wu, X.; Fang, M. Preparation and thermal properties of shape-stabilized composite phase change materials based on polyethylene glycol and porous carbon prepared from potato. RSC Adv. 2016, 6, 15821–15830. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass Ratio of Pine Cones to KOH | Calcination Temperature (°C) | ||
|---|---|---|---|
| 700 | 800 | 900 | |
| 1:1 | - | PCC-1-800 | - |
| 1:2 | PCC-2-700 | PCC-2-800 | PCC-2-900 |
| 1:3 | PCC-3-700 | PCC-3-800 | PCC-3-900 |
| 1:4 | - | PCC-4-800 | - |
| Samples | RPUF | PCMs/RPUF-5 | PCMs/RPUF-15 | PCMs/RPUF-25 |
|---|---|---|---|---|
| OD/PCC wt% | 0 | 2.5 | 7.5 | 12.5 |
| PEG/PCC wt% | 0 | 2.5 | 7.5 | 12.5 |
| Samples | BET Surface Area (m2/g) | Total Pore Volume (cm3/g) | Mean Pore Size (nm) | Ref. |
|---|---|---|---|---|
| Rapeseed stem biochar | 316.90 | 0.11 | 2.20 | [41] |
| Activated garlic peel biomass carbon (AGP) | 1309.00 | 0.54 | 2.20 | [37] |
| Synthetic graphite-like carbon (Sib-4-ox-450 c) | 380.00 | 0.53 | 5.66 | [42] |
| Synthetic graphite-like carbon (3%Ru/Sib-4-ox-450 c) | 341.00 | 0.50 | 5.88 | [42] |
| Graphene-based metal and nitrogen-doped carbon (GNC-Co) | 123.10 | - | 5.70 | [43] |
| PCC-2-800 | 1405.40 | 0.55 | 1.58 | This work |
| PCC-3-700 | 862.04 | 0.42 | 1.93 | This work |
| PCC-3-800 | 1758.60 | 1.21 | 2.74 | This work |
| PCC-3-900 | 679.73 | 0.38 | 2.26 | This work |
| Samples | Tm (°C) | ΔHm (J/g) | Tc (°C) | ΔHc (J/g) | R(%) |
|---|---|---|---|---|---|
| OD | 27.5 | 194.5 | 24.6 | 193.3 | - |
| PEG | 56.1 | 157.8 | 34.9 | 151.9 | - |
| OD/PCC | 34.8 | 162.3 | 18.5 | 158.7 | 83.4 |
| PEG/PCC | 64.0 | 144.3 | 30.7 | 135.8 | 91.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, N.; Du, J.; Yang, W.; Li, Y.; Chen, N. Biomass-Based Shape-Stabilized Composite Phase-Change Materials with High Solar–Thermal Conversion Efficiency for Thermal Energy Storage. Polymers 2023, 15, 3747. https://doi.org/10.3390/polym15183747
Gao N, Du J, Yang W, Li Y, Chen N. Biomass-Based Shape-Stabilized Composite Phase-Change Materials with High Solar–Thermal Conversion Efficiency for Thermal Energy Storage. Polymers. 2023; 15(18):3747. https://doi.org/10.3390/polym15183747
Chicago/Turabian StyleGao, Ning, Jiaoli Du, Wenbo Yang, Youbing Li, and Ning Chen. 2023. "Biomass-Based Shape-Stabilized Composite Phase-Change Materials with High Solar–Thermal Conversion Efficiency for Thermal Energy Storage" Polymers 15, no. 18: 3747. https://doi.org/10.3390/polym15183747
APA StyleGao, N., Du, J., Yang, W., Li, Y., & Chen, N. (2023). Biomass-Based Shape-Stabilized Composite Phase-Change Materials with High Solar–Thermal Conversion Efficiency for Thermal Energy Storage. Polymers, 15(18), 3747. https://doi.org/10.3390/polym15183747