
Chemical Feedback in Templated Reaction-Assembly of Polyelectrolyte Complex Micelles: A Molecular Simulation Study of the Kinetics and Clustering




Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Model
2.1. Coarse-Grained Molecular Dynamics Simulation Details
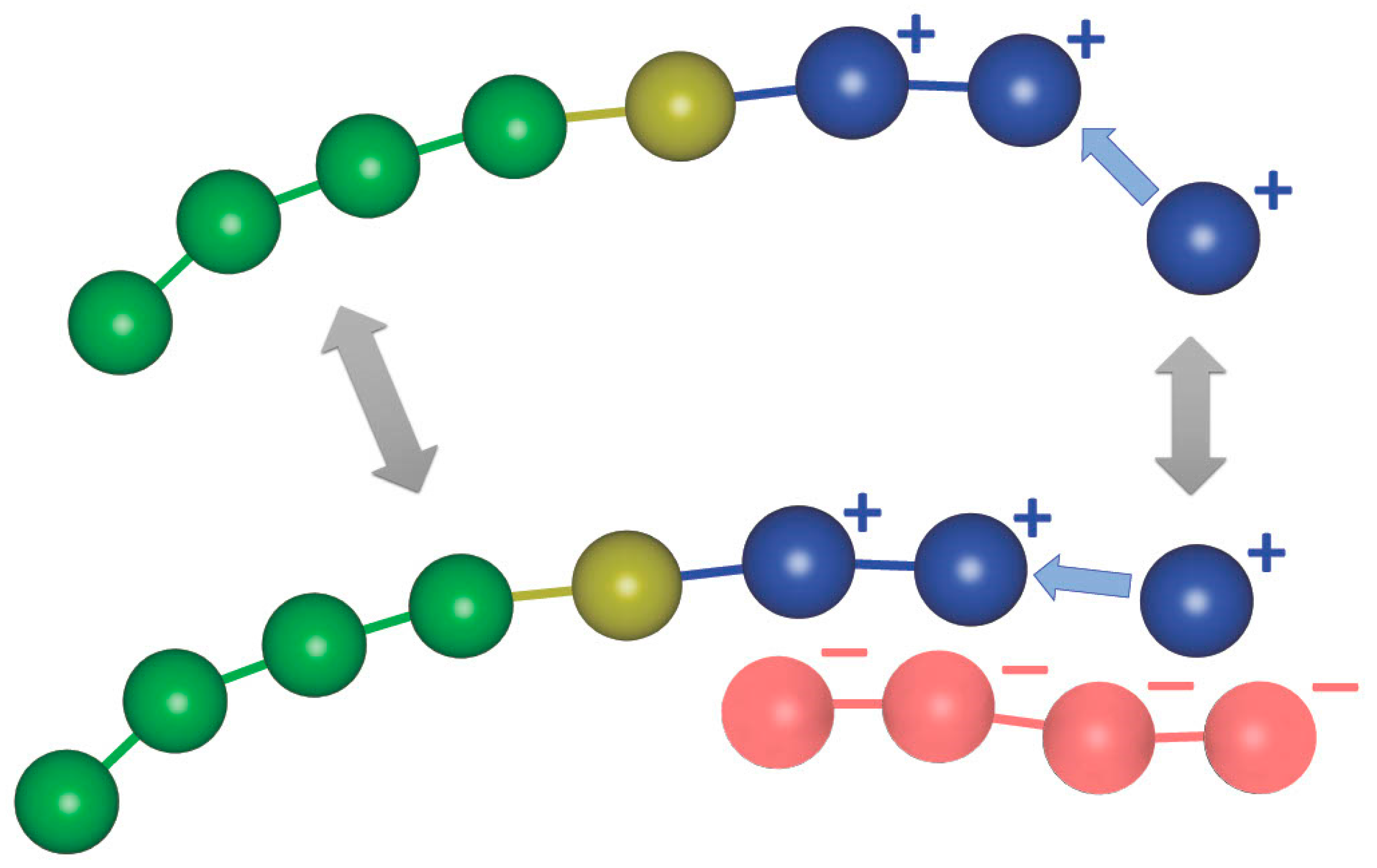
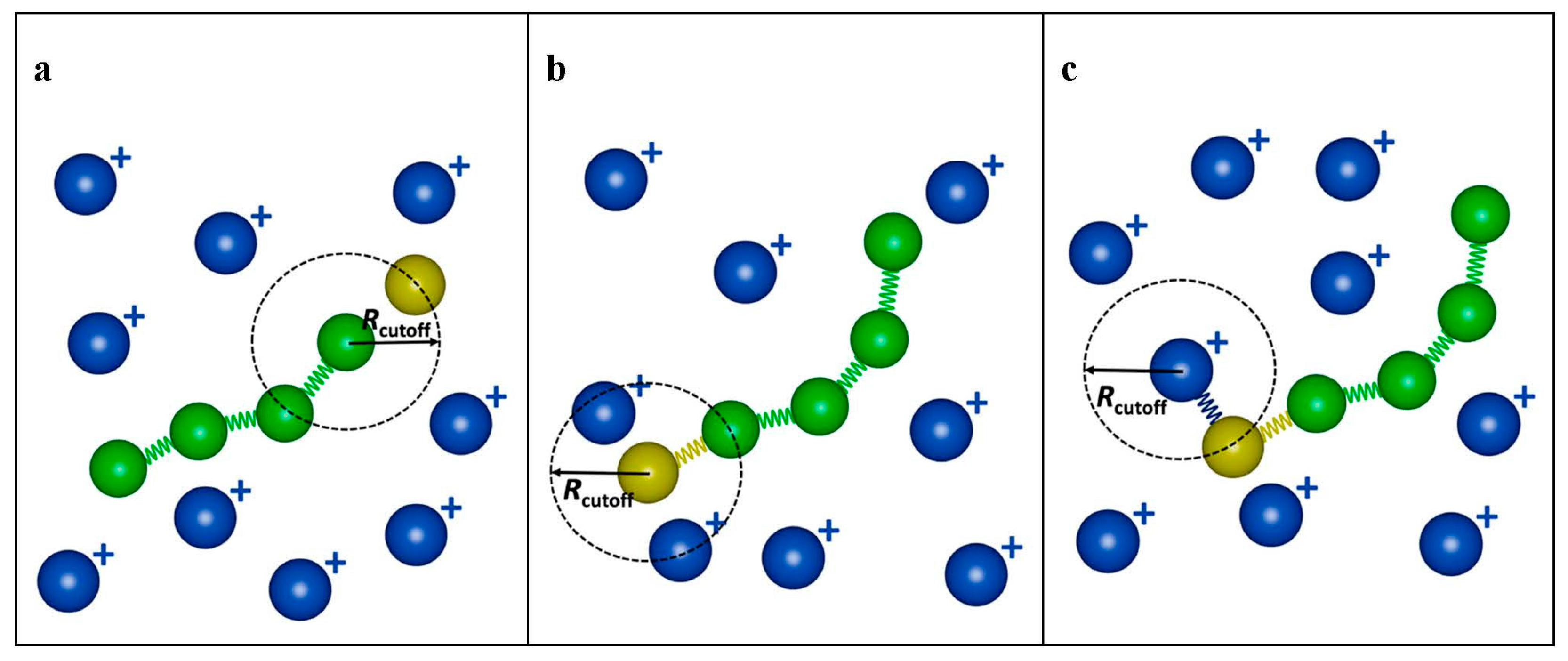
2.2. Modeling the Polymerization
2.3. Clustering Analysis
3. Results and Discussion
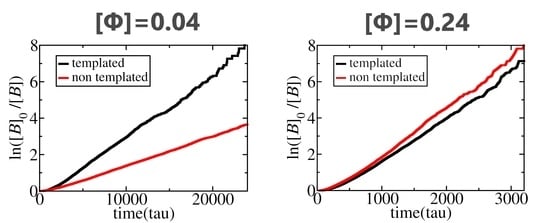
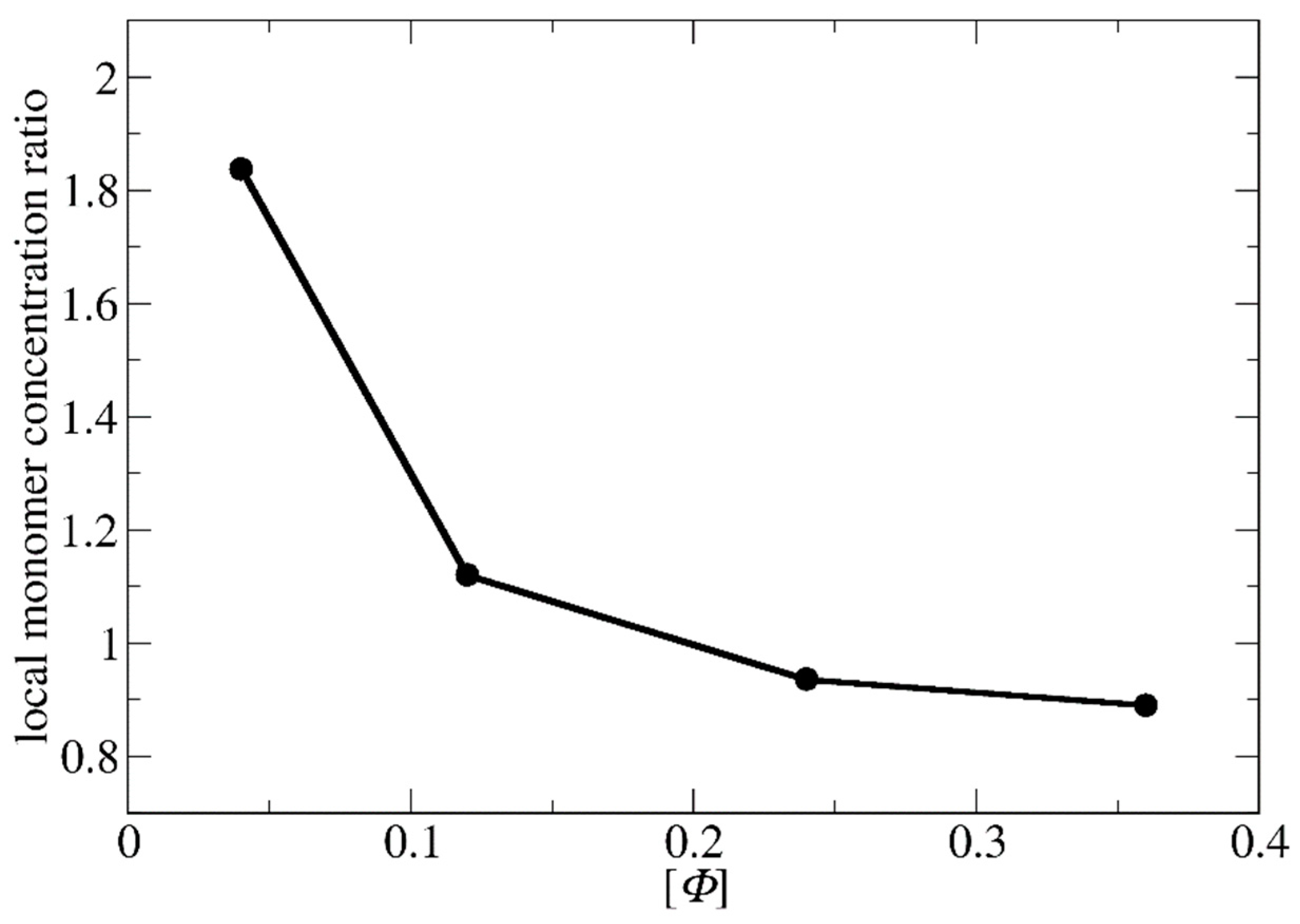
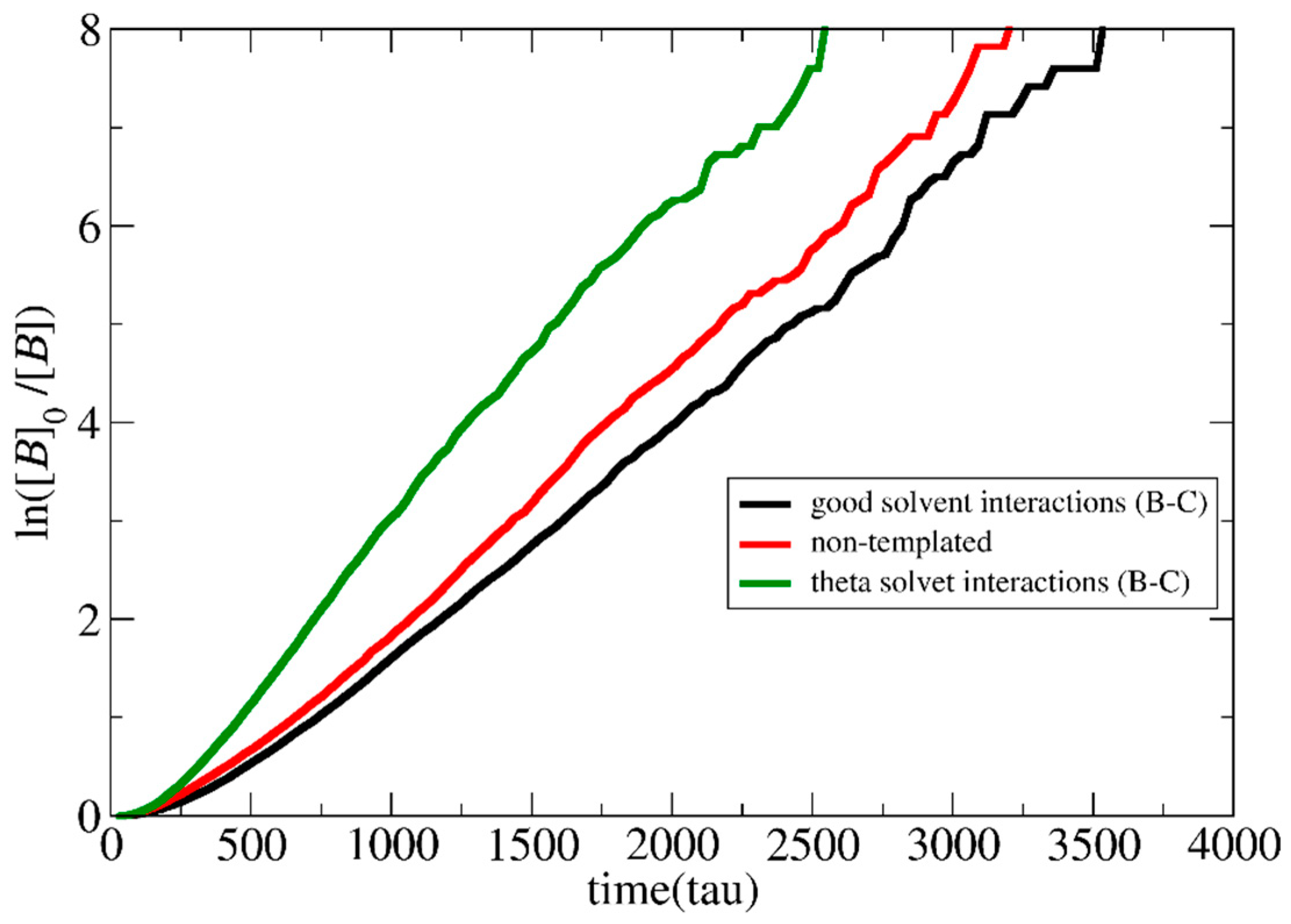
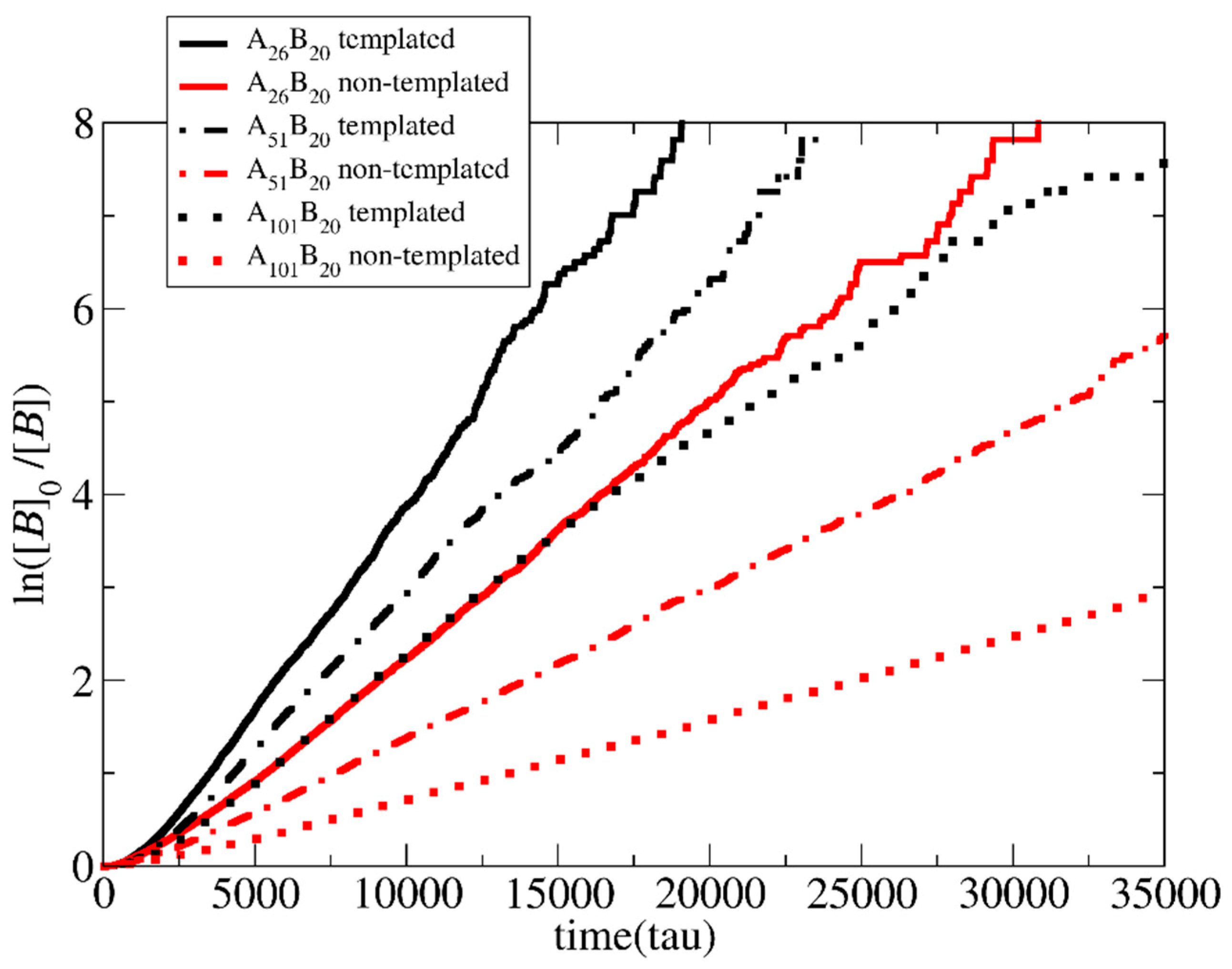
3.1. Polymerization Rate
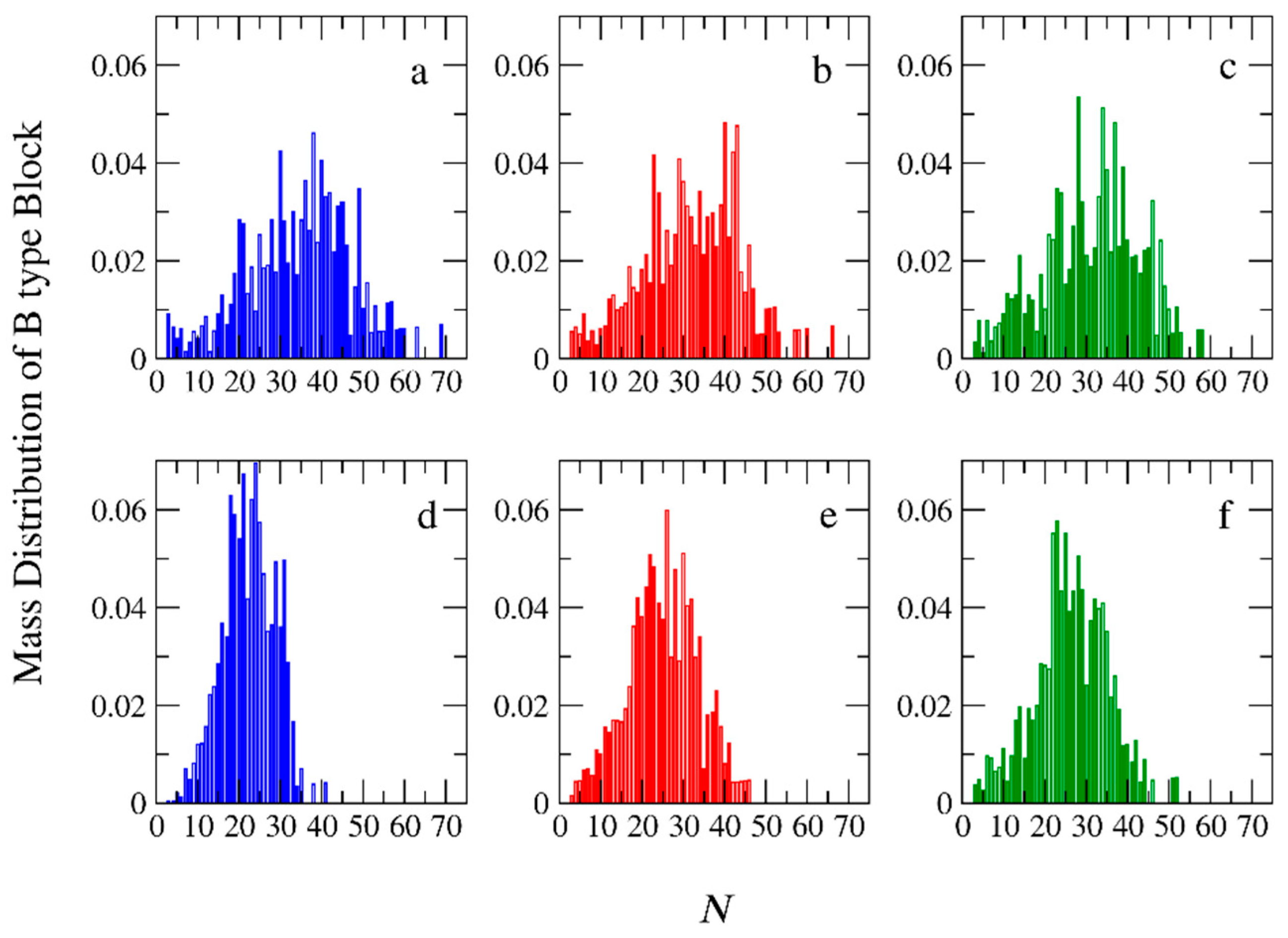
3.2. Molecular Weights and Polydispersity
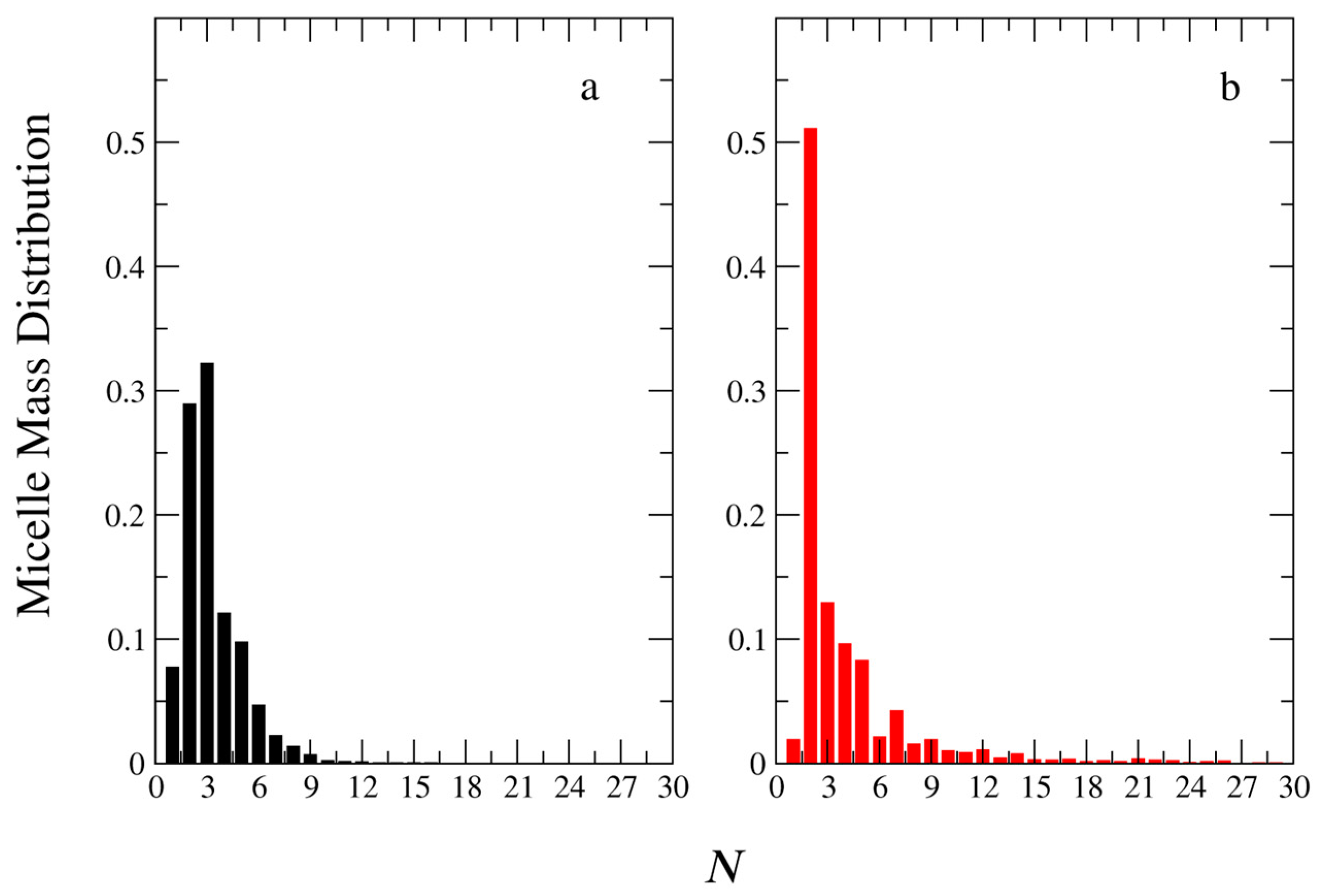
3.3. Micelle Size and Shape
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magana, J.R.; Sproncken, C.C.M.; Voets, I.K. On Complex Coacervate Core Micelles: Structure-Function Perspectives. Polymers 2020, 12, 1953. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Kataoka, K. Polyion complex micelle formation from double-hydrophilic block copolymers composed of charged and non-charged segments in aqueous media. Polym. J. 2018, 50, 95–100. [Google Scholar] [CrossRef]
- Van der Kooij, H.M.; Spruijt, E.; Voets, I.K.; Fokkink, R.; Stuart, M.A.C.; van der Gucht, J. On the Stability and Morphology of Complex Coacervate Core Micelles: From Spherical to Wormlike Micelles. Langmuir 2012, 28, 14180–14191. [Google Scholar] [CrossRef]
- Shah, S.; Leon, L. Structural dynamics, phase behavior, and applications of polyelectrolyte complex micelles. Curr. Opin. Colloid Interface Sci. 2021, 53, 101424. [Google Scholar] [CrossRef]
- Marras, A.E.; Ting, J.M.; Stevens, K.C.; Tirrell, M.V. Advances in the Structural Design of Polyelectrolyte Complex Micelles. J. Phys. Chem. B 2021, 125, 7076–7089. [Google Scholar] [CrossRef]
- Wu, H.; Ting, J.M.; Yu, B.; Jackson, N.E.; Meng, S.; de Pablo, J.J.; Tirrell, M.V. Spatiotemporal Formation and Growth Kinetics of Polyelectrolyte Complex Micelles with Millisecond Resolution. ACS Macro Lett. 2020, 9, 1674–1680. [Google Scholar] [CrossRef]
- Anraku, Y.; Kishimura, A.; Kamiya, M.; Tanaka, S.; Nomoto, T.; Toh, K.; Matsumoto, Y.; Fukushima, S.; Sueyoshi, D.; Kano, M.R.; et al. Systemically Injectable Enzyme-Loaded Polyion Complex Vesicles as In Vivo Nanoreactors Functioning in Tumors. Angew. Chem. Int. Ed. 2016, 55, 560–565. [Google Scholar] [CrossRef]
- Yoon, H.; Dell, E.J.; Freyer, J.L.; Campos, L.M.; Jang, W.-D. Polymeric supramolecular assemblies based on multivalent ionic interactions for biomedical applications. Polymer 2014, 55, 453–464. [Google Scholar] [CrossRef]
- Gioldasis, C.; Gergidis, L.N.; Vlahos, C. Micellization through complexation of oppositely charged diblock copolymers: Effects of composition, polymer architecture, salt of different valency, and thermoresponsive block. J. Polym. Sci. 2021, 59, 191–204. [Google Scholar] [CrossRef]
- van der Gucht, J.; Spruijt, E.; Lemmers, M.; Stuart, M.A.C. Polyelectrolyte complexes: Bulk phases and colloidal systems. J. Colloid Interface Sci. 2011, 361, 407–422. [Google Scholar] [CrossRef]
- Li, C.; Magana, J.R.; Sobotta, F.; Wang, J.; Stuart, M.A.C.; van Ravensteijn, B.G.P.; Voets, I.K. Switchable Electrostatically Templated Polymerization. Angew. Chem. Int. Ed. 2022, 61, e202206780. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wan, Y.; Cai, Y.; Ding, P.; Stuart, M.A.C.; Wang, J. Synthesis of Anionic Nanogels for Selective and Efficient Enzyme Encapsulation. Langmuir 2022, 38, 3234–3243. [Google Scholar] [CrossRef] [PubMed]
- Bos, I.; Terenzi, C.; Sprakel, J. Chemical Feedback in Templated Reaction-Assembly Networks. Macromolecules 2020, 53, 10675–10685. [Google Scholar] [CrossRef]
- Ding, Y.; Zhao, Q.; Wang, L.; Huang, L.; Liu, Q.; Lu, X.; Cai, Y. Polymerization-Induced Self-Assembly Promoted by Liquid–Liquid Phase Separation. ACS Macro Lett. 2019, 8, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Gavrilov, A.A.; Chertovich, A.V. Simulation of the RAFT polymerization in 3D: Steric restrictions and incompatibility between species. Polym. Chem. 2022, 13, 2143–2154. [Google Scholar] [CrossRef]
- Genzer, J. In Silico Polymerization: Computer Simulation of Controlled Radical Polymerization in Bulk and on Flat Surfaces. Macromolecules 2006, 39, 7157–7169. [Google Scholar] [CrossRef]
- Turgman-Cohen, S.; Genzer, J. Computer Simulation of Concurrent Bulk- and Surface-Initiated Living Polymerization. Macromolecules 2012, 45, 2128–2137. [Google Scholar] [CrossRef]
- Cheng, L.; Cao, D. Effect of Tail Architecture on Self-Assembly of Amphiphiles for Polymeric Micelles. Langmuir 2009, 25, 2749–2756. [Google Scholar] [CrossRef]
- Suek, N.W.; Lamm, M.H. Computer Simulation of Architectural and Molecular Weight Effects on the Assembly of Amphiphilic Linear−Dendritic Block Copolymers in Solution. Langmuir 2008, 24, 3030–3036. [Google Scholar] [CrossRef]
- Miliou, K.; Gergidis, L.N.; Vlahos, C. Polyelectrolyte micelles in salt-free solutions: Micelle size and electrostatic potential. J. Polym. Sci. Part B Polym. Phys. 2018, 56, 924–934. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Murat, M.; Grest, G.S. Molecular Dynamics Study of Dendrimer Molecules in Solvents of Varying Quality. Macromolecules 1996, 29, 1278–1285. [Google Scholar] [CrossRef]
- Georgiadis, C.; Moultos, O.; Gergidis, L.N.; Vlahos, C. Brownian Dynamics Simulations on the Self-Assembly Behavior of AB Hybrid Dendritic−Star Copolymers. Langmuir 2011, 27, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Moultos, O.; Gergidis, L.N.; Vlahos, C. Self-Assembly Behavior of Thermoresponsive Bis-Solvophilic Linear Block Terpolymers: A Simulation Study. Macromolecules 2012, 45, 2570–2579. [Google Scholar] [CrossRef]
- Kalogirou, A.; Gergidis, L.N.; Moultos, O.; Vlahos, C. Entropic effects, shape, and size of mixed micelles formed by copolymers with complex architectures. Phys. Rev. E 2015, 92, 052601. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Cao, D.; Wang, W.; Jusufi, A. Conformation of a Spherical Polyelectrolyte Brush in the Presence of Oppositely Charged Linear Polyelectrolytes. Macromolecules 2008, 41, 5477–5484. [Google Scholar] [CrossRef]
- Kalogirou, A.; Moultos, O.A.; Gergidis, L.N.; Vlahos, C. Micellization Properties of Θ-Shaped, Figure-Eight-Shaped and Linked Rings Copolymers. Macromolecules 2014, 47, 5851–5859. [Google Scholar] [CrossRef]
- Kalogirou, A.; Gergidis, L.N.; Miliou, K.; Vlahos, C. Complexation of Polyelectrolyte Micelles with Oppositely Charged Linear Chains. J. Phys. Chem. B 2017, 121, 1982–1991. [Google Scholar] [CrossRef]
- Stillinger, F.H. Rigorous Basis of the Frenkel-Band Theory of Association Equilibrium. J. Chem. Phys. 1963, 38, 1486–1494. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Du-bourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0 Contributors. SciPy 1.0 Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring Network Structure, Dynamics, and Function Using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy 2008), Pasadena, CA, USA, 19–24 August 2008. [Google Scholar]
- Ayoubi, M.A.; Zhu, K.; Nyström, B.; Olsson, U.; Almdal, K.; Khokhlov, A.R.; Piculell, L. Morphological investigation of polydisperse asymmetric block copolymer systems of poly(styrene) and poly(methacrylic acid) in the strong segregation regime. J. Polym. Sci. Part B Polym. Phys. 2013, 51, 1657–1671. [Google Scholar] [CrossRef]
- Petrov, A.; Chertovich, A.V.; Gavrilov, A.A. Phase Diagrams of Polymerization-Induced Self-Assembly Are Largely Determined by Polymer Recombination. Polymers 2022, 14, 5331. [Google Scholar] [CrossRef]
- Theodorou, D.N.; Suter, U.W. Shape of Unperturbed Linear Polymers: Polypropylene. Macromolecules 1985, 18, 1206–1214. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gioldasis, C.; Gkamas, A.; Moultos, O.A.; Vlahos, C.H. Chemical Feedback in Templated Reaction-Assembly of Polyelectrolyte Complex Micelles: A Molecular Simulation Study of the Kinetics and Clustering. Polymers 2023, 15, 3024. https://doi.org/10.3390/polym15143024
Gioldasis C, Gkamas A, Moultos OA, Vlahos CH. Chemical Feedback in Templated Reaction-Assembly of Polyelectrolyte Complex Micelles: A Molecular Simulation Study of the Kinetics and Clustering. Polymers. 2023; 15(14):3024. https://doi.org/10.3390/polym15143024
Chicago/Turabian StyleGioldasis, Christos, Apostolos Gkamas, Othonas A. Moultos, and Costas Hristos Vlahos. 2023. "Chemical Feedback in Templated Reaction-Assembly of Polyelectrolyte Complex Micelles: A Molecular Simulation Study of the Kinetics and Clustering" Polymers 15, no. 14: 3024. https://doi.org/10.3390/polym15143024
APA StyleGioldasis, C., Gkamas, A., Moultos, O. A., & Vlahos, C. H. (2023). Chemical Feedback in Templated Reaction-Assembly of Polyelectrolyte Complex Micelles: A Molecular Simulation Study of the Kinetics and Clustering. Polymers, 15(14), 3024. https://doi.org/10.3390/polym15143024