
A Directly Compressible Pregelatinised Sago Starch: A New Excipient in the Pharmaceutical Tablet Formulations



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Pregelatinised Sago Starch
2.3. Spectroscopic Characterisations
2.3.1. FT-IR Analysis
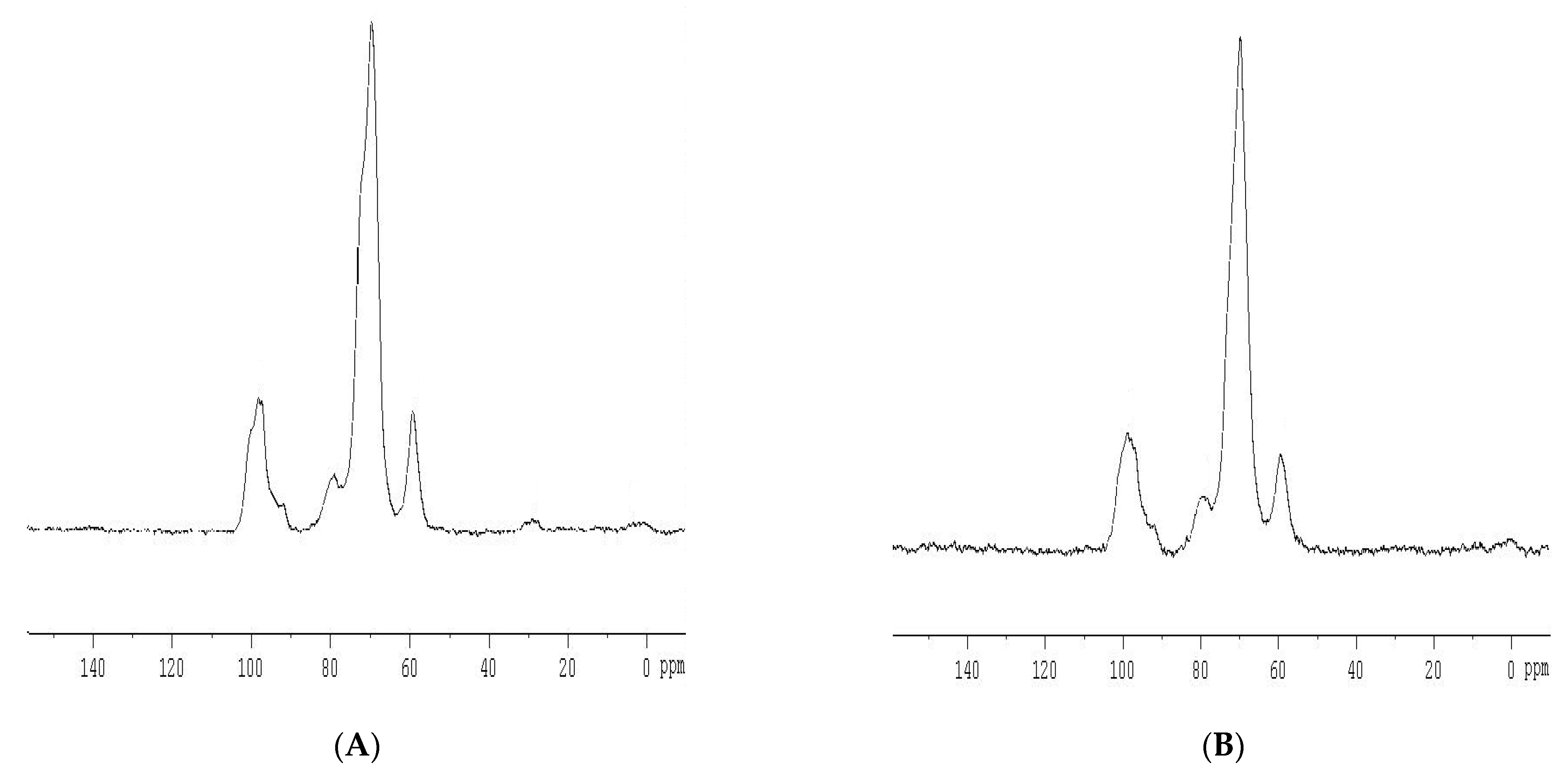
2.3.2. NMR Analysis
2.3.3. X-ray Diffraction
2.4. Evaluation of Excipient Bulk Powders
2.4.1. Particle Size Distribution, Shape and Surface Texture
2.4.2. Moisture Content
2.4.3. Bulk Density (ρ0) and Tapped Density (ρt)
2.4.4. Flow Properties
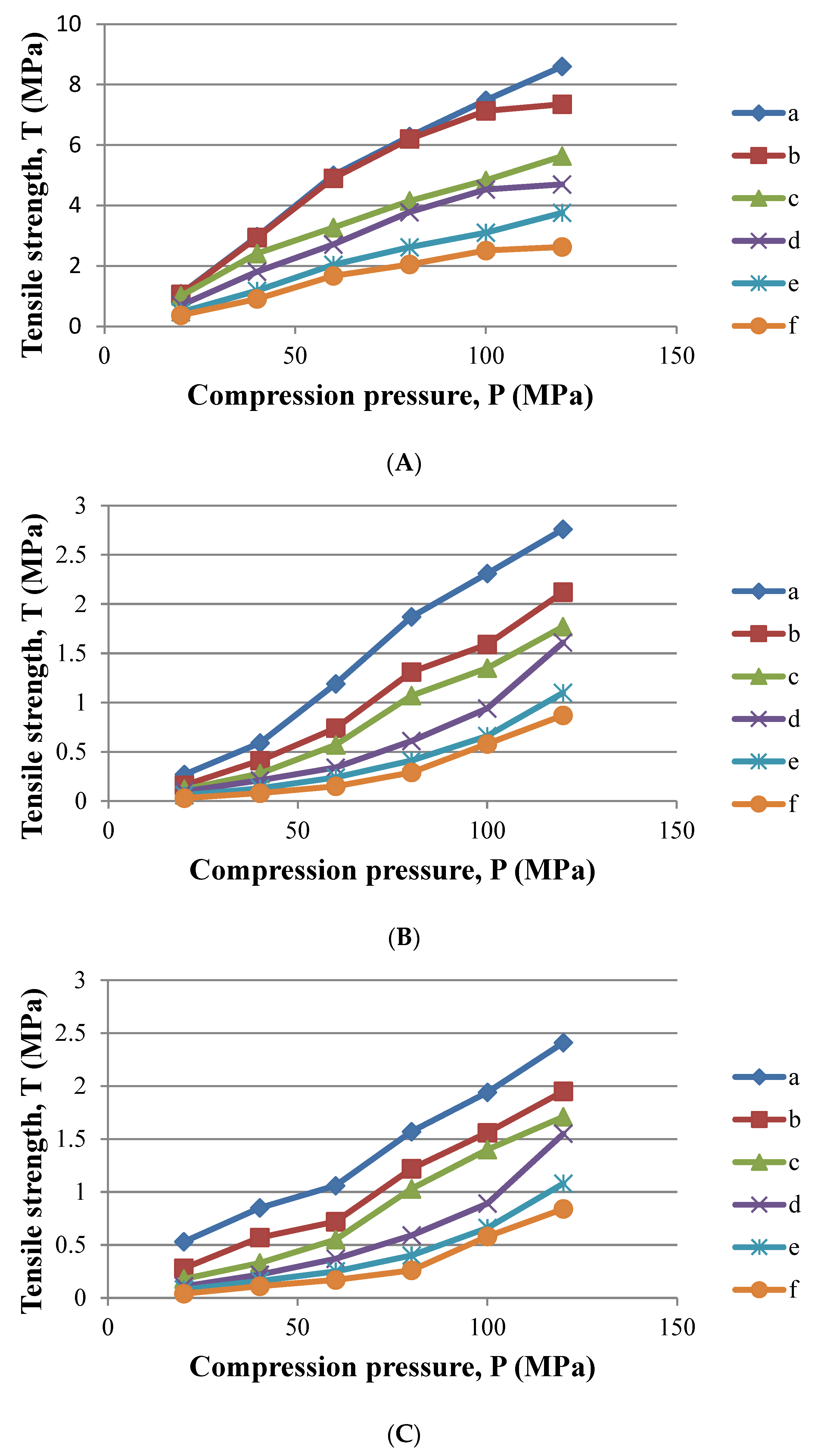
2.4.5. Compactibility
2.4.6. Loading Capacity
2.4.7. Development of Model Drug Formulation
2.4.8. Short-Term Accelerated Stability Study
2.4.9. Statistical Analysis
3. Results and Discussion
3.1. Spectroscopic Characterisation
3.1.1. FT-IR Analysis
3.1.2. C-NMR Analysis
3.1.3. X-ray Diffraction
3.2. Evaluation of Excipient Bulk Powders
3.2.1. Particle Analysis, Moisture Content, Bulk and Tap Densities and Flow Properties
3.2.2. Compactibility and Loading Capacity
3.3. In Vitro Evaluations of Developed Formulation of Paracetamol Tablets
3.3.1. Uniformity of Weight, Friability and Dimension
3.3.2. Hardness
3.3.3. Disintegration
3.3.4. Dissolution
3.3.5. Short-Term Accelerated Stability Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolhuis, G.K.; Chowhan, Z.T. Materials for direct compression. In Pharmaceutical Powder Compaction Technology; Alderborn, G., Nström, C., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1996; pp. 419–500. [Google Scholar]
- Shangraw, R.F. Compressed tablets by direct compression. In Pharmaceutical Dosage Forms: Tablets; Lieberman, H.A., Lachman, L., Schwartz, J.B., Eds.; Marcel Decker Inc.: New York, NY, USA; Basel, Switzerland, 1989; pp. 195–247. [Google Scholar]
- Zhang, Y.; Law, Y.; Chakrabarti, S. Physical properties and compact analysis of commonly used direct compression binders. A.A.P.S PharmSciTech. 2003, 4, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, R.S.; Kennedy, J.F.; Gopalakrishnan, S.M.; Kaczmarek, A.; Knill, C.J.; Akmar, P.F. Industrial production, processing and utilization of sago palm-derived products. Carbohyd Polym. 2008, 72, 1–20. [Google Scholar] [CrossRef]
- Widodo, R.T.; Hassan, A. Compression and mechanical properties of directly compressible pregelatinised sago starches. Powder Technol. 2015, 269, 15–21. [Google Scholar] [CrossRef]
- Nuwamanya, E.; Baguma, Y.; Emmambux, N.; Rubaihayo, P. Crystalline and pasting properties of cassava starch are influenced by its molecular properties. Afr. J. Food Sci. 2010, 4, 8–15. [Google Scholar]
- The United States of Pharmacopeia. 27/The National Formulary 22; Port City Press: Baltimore, MA, USA, 2004. [Google Scholar]
- Gonnissen, Y.; Remon, J.P.; Vervait, C. Development of directly compressible powders via co-spray drying. Eur. J. Pharm. Biopharm. 2007, 67, 220–226. [Google Scholar] [CrossRef]
- Busignies, V.; Leclerc, B.; Porion, P.; Evesque, P.; Couarraze, G.; Tchoreloff, P. Compaction behaviour and new prespective approach to the compressibility of binary mixtures of pharmaceutical excipients. Eur. J. Pharm. Biopharm. 2006, 64, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Habib, Y.; Augsberger, L.; Reier, G.; Wheatley, T.; Shangraw, R. Dilution Potential: A new prespective. Pharm. Dev. Technol. 1996, 1, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Ngwuluka, N.C.; Idiakhoa, B.A.; Nep, I.E.; Ogaji, I.; Okafor, I.S. Formulation and evaluation of paracetamol tablets manufactured using the dried fruit of Phoenic dactilyfera Linn as an excipient. Res. Pharm. Biotech. 2010, 2, 25–32. [Google Scholar]
- Anderson, N.H.; Bauer, M.; Boussac, N.; Khan-Malek, R.; Munden, P.; Sardaro, M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profile. J. Pharm. Biomed. Anal. 1998, 17, 811–822. [Google Scholar] [CrossRef]
- International Conference on Harmonization. Guideline on Stability Testing of New Drug Substances and Products; European Medicines Agency: London, UK, 2003. [Google Scholar]
- Sun, R.C.; Tomkinson, J.; Ma, P.L.; Liang, S.F. Comparative study of hemicelluloses from rice straw by alkali and hydrogen peroxyde treatments. Carbohyd. Polym. 2000, 42, 111–122. [Google Scholar] [CrossRef]
- Ummi-Shafiqah, M.S.; Fazilah, A.; Karim, A.A.; Kaur, B.; Yusuf, Y. The effects of UV treatment on the properties of sago and mung bean films. Int. Food Res. J. 2012, 19, 265–270. [Google Scholar]
- Hoover, R. Composition, molecular structure, and physicochemical properties of tuber and root starches: A review. Carbohyd. Polym. 2001, 45, 253–267. [Google Scholar] [CrossRef]
- Morgan, K.R.; Furneaux, R.H.; Larsen, N.G. Solid-state NMR studies on the structure of starch granules. Carbohydr. Res. 1995, 276, 387–399. [Google Scholar] [CrossRef]
- Ahmad, F.B.; Williams, P.A.; Doublier, J.L.; Durand, S.; Buleon, A. Physico-chemical characterisation of sago starch. Carbohyd. Polym. 1999, 38, 361–370. [Google Scholar] [CrossRef]
- Karim, A.A.; Pei-Ling, A.T.; Manan, D.M.A.; Zaidul, I.S.M. Starch from the sago (Metroxylan sagu) Palm Tree-Properties, Prospects, and Challenges as a New industrial Source for Food and Other Uses. Compr. Rev. Food Sci. Food Saf. 2008, 7, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Fazilah, A.; Karim, A.A. Alcoholic-alkaline treatment of sago starch and its effects on physicochemical properties. Food Bioprod. Process. 2011, 89, 463–471. [Google Scholar] [CrossRef]
- Carr, R.L. Evaluating flow properties of solids. Chem. Eng. 1965, 72, 163–168. [Google Scholar]
- Bhimte, N.A.; Tayade, P.T. Evaluation of microcrystalline cellulose prepared from sisal fibers as a tablet excipient: A technical note. A.A.P.S PharmSciTech. 2007, 8, E1–E7. [Google Scholar] [CrossRef]
- Mitrevej, A.; Sinchaipanid, N.; Faroongsarng, D. Spray-dried rice starch: Comparative evaluation of direct compression fillers. Drug Dev. Ind. Pharm. 1996, 22, 587–594. [Google Scholar] [CrossRef]
- Kibbe, A.H. Handbook of Pharmaceutical Excipients, 3rd ed.; American Pharmaceutical Assosiation and Pharmaceutical Press: London, UK, 2000. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | Formulation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |||||
| D1 | 3 M | 6 M | D1 | 3 M | 6 M | ||||
| Paracetamol | 120.0 | 120.0 | 120.0 | 120.0 | 120.0 | 120.0 | 120.0 | 120.0 | 120.0 |
| Avicel PH 101 | 229.1 | - | - | 70.0 | 70.0 | 70.0 | 70.0 | 70.0 | 70.0 |
| Spress® B820 | - | 229.1 | - | 145.1 | 145.1 | 145.1 | - | - | - |
| Pregelatinised sago starch | - | - | 229.1 | - | - | - | 145.1 | 145.1 | 145.1 |
| Sodium starch glycolate | - | - | - | 14.0 | 14.0 | 14.0 | 14.0 | 14.0 | 14.0 |
| Magnesium stearate | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 |
| Excipients | Avicel PH 101 | Spress® B820 | Pregelatinised Sago Starch |
|---|---|---|---|
| Mean particle diameter (µm) ± SD a | 56.70 ± 11.51 | 89.30 ± 20.29 | 88.00 ± 18.98 |
| Moisture content (%) ± SD b | 5.19 ± 0.06 | 9.91 ± 0.02 | 10.39 ± 0.41 |
| Bulk density (g/cm3) ± SD b | 0.35 ± 0.00 | 0.64 ± 0.00 | 0.52 ± 0.01 |
| Tap density (g/cm3) ± SD b | 0.44 ± 0.00 | 0.71 ± 0.00 | 0.61 ± 0.01 |
| Angle of repose, α (°) ± SD b | 41.87 ± 0.51 | 30.23 ± 0.46 | 30.37 ± 0.23 |
| AUC T vs P | 1270.6 | 446.8 | 392.7 |
| Evaluation | Formulation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |||||
| D1 | 3 M | 6 M | D1 | 3 M | 6 M | ||||
| Thickness (mm) a | 3.96 ± 0.05 | 3.55 ± 0.03 | 3.66 ± 0.03 | 3.59 ± 0.02 | 3.60 ± 0.01 | 3.61 ± 0.02 | 3.62 ± 0.01 | 3.62 ± 0.02 | 3.62 ± 0.03 |
| Diameter (mm) a | 9.90 ± 0.01 | 9.88 ± 0.01 | 9.90 ± 0.01 | 9.88 ± 0.00 | 9.90 ± 0.02 | 9.90 ± 0.02 | 9.90 ± 0.00 | 9.91 ± 0.02 | 9.92 ± 0.03 |
| Hardness (N) a | 92.91 ± 4.37 | 103.506. ± 72 | 103.50 ± 7.46 | 98.70 ± 5.17 | 100.00 ± 2.21 | 100.40 ± 3.60 | 109.10 ± 5.00 | 107.40 ± 2.55 | 106.50 ± 2.37 |
| Uniformity of weight (mg) b | 347.82 ± 1.00 | 348.79 ± 0.76 | 350.05 ± 1.29 | 350.06 ± 0.93 | 348.79 ± 0.66 | 348.68 ± 0.89 | 349.40 ± 1.05 | 348.97 ± 0.76 | 348.75 ± 0.55 |
| Friability (%) | 0.38 | 0.48 | 0.51 | 0.54 | 0.54 | 0.58 | 0.43 | 0.46 | 0.46 |
| Disintegration time (min) c | 0.23 ± 0.05 | 1.41 ± 0.16 | 1.33 ± 0.12 | 0.57 ± 0.37 | 0.56 ± 0.04 | 0.58 ± 0.06 | 0.44 ± 0.18 | 0.47 ± 0.03 | 0.45 ± 0.04 |
| Dissolution efficiency (%) | 40.14 | 69.50 | 71.25 | 86.34 | 87.76 | 85.75 | 86.36 | 86.23 | 85.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Widodo, R.T.; Hassan, A.; Liew, K.B.; Ming, L.C. A Directly Compressible Pregelatinised Sago Starch: A New Excipient in the Pharmaceutical Tablet Formulations. Polymers 2022, 14, 3050. https://doi.org/10.3390/polym14153050
Widodo RT, Hassan A, Liew KB, Ming LC. A Directly Compressible Pregelatinised Sago Starch: A New Excipient in the Pharmaceutical Tablet Formulations. Polymers. 2022; 14(15):3050. https://doi.org/10.3390/polym14153050
Chicago/Turabian StyleWidodo, Riyanto Teguh, Aziz Hassan, Kai Bin Liew, and Long Chiau Ming. 2022. "A Directly Compressible Pregelatinised Sago Starch: A New Excipient in the Pharmaceutical Tablet Formulations" Polymers 14, no. 15: 3050. https://doi.org/10.3390/polym14153050
APA StyleWidodo, R. T., Hassan, A., Liew, K. B., & Ming, L. C. (2022). A Directly Compressible Pregelatinised Sago Starch: A New Excipient in the Pharmaceutical Tablet Formulations. Polymers, 14(15), 3050. https://doi.org/10.3390/polym14153050