1. Introduction
Foamed polymers are widely used in almost all industrial sectors, due to their interesting value-added properties, such as low density, low thermal conductivity, and adaptable mechanical properties depending on their relative density. Polyurethane (PU) is the polymer par excellence for foaming mainly because of the high applicability of PU foams, which is based on the possibility of tailoring the final properties of the material by adequately changing the types or quantities of the initial components (isocyanate, polyol, surfactants, catalysts, and blowing agents). Moreover, the simplicity of the PU foaming technology makes these materials very competitive from an economic perspective. PU foams represent 50% of the global foam market [
1]. Although most of the PU foams are thermoset, the study of foams based on thermoplastic polyurethanes (TPU) is gaining importance in recent years, because they are elastic thermoplastically processable materials, and also because they can be reprocessed or recycled. Thus, TPU foams are nowadays very interesting materials for several industries, such as furniture, automotive, sportswear, and packaging.
TPUs are block copolymers composed of alternating soft segments (SS) and hard segments (HS), which are produced by the polyaddition reaction of a diisocyanate with a medium molecular weight macrodiol and with a low molecular weight diol, which acts as a chain extender. The presence of SS and HS gives rise to a two-phase microstructure, in which the HS from the isocyanate and the chain extender segregate into semicrystalline domains, whereas the SS from the polyol chains form amorphous domains in which the HS are dispersed [
2]. Therefore, HS offers rigidity, and SS provides flexibility to the TPU material. Their ratio and distribution, which are key for the final properties, are determined by a large number of chemical and structural factors, including the characteristics of the raw materials and the polymerization procedure and processing conditions used for their synthesis.
The gas dissolution foaming process is one of the most reliable methods developed in order to produce thermoplastics foams, where carbon dioxide (CO
2) is normally used as a blowing agent. This technology allows manufacturing foamed samples by using a green solvent (CO
2), which can be removed leaving no residues nor pollutant compounds. Many studies have been published on the use of this method, and different cellular polymers have been obtained, such as poly(methylmethacrylate) (PMMA), polystyrene (PS), poly(lactic acid) (PLA), polycarbonate (PC), polyethylene (PE), or polypropylene (PP) among others [
3,
4,
5]. Nevertheless, the number of reports dealing with TPU foams obtained by the gas dissolution foaming process using CO
2 as a blowing agent is still rather limited [
6,
7,
8,
9,
10,
11,
12,
13,
14]. These previous papers clearly show that foaming TPU is complicated, not all TPUs can be foamed, and in addition, the processing window for those materials is narrow. As a result, it is important to understand the foamability behavior of the different types of TPU in which the chemical characteristics changes (chemical structure of the components, soft and hard segments contents, etc.). For that, it is necessary to investigate TPUs in which the chemical features are known since until now, the majority of studies are carried out using commercial TPUs.
Therefore, few studies have focused on the effect of chemical characteristics in the fabrication of TPU foams by the gas dissolution foaming process using CO
2 [
12,
13,
14,
15,
16]. Ito et al. [
12] studied the effect of the chain length of the SS from 1000 to 3000 g/mol of number-average molecular weight (Mn) and the SS contents from 50 to 80 wt.% (which correspond to HS from 50 to 20 wt.%) on the cellular structure of TPU foams obtained by two-step foaming. They concluded that the solubility of CO
2 in the SS is considerably higher than that in the HS, and thus, the nucleation and growth of the nuclei occurs in the SS area. Yet et al. [
13] used three different approaches to investigate the foaming behavior of TPU by a one-step foaming from previously synthesized TPU: increasing the HS content (37 wt.%, 48 wt.%, and 56 wt.%), replacing the SS type (polytetramethylene ether glycol and poly (1,4-butylene adipate) of molecular weight 1000 g/mol), and adding a graphene nucleation agent. All three approaches were able to produce cell sizes of less than 1 μm. The results of the TPUs with different HS and with different SS types indicate that
Tg is not the only factor that determines whether the sample can be foamed; both hardness and crystallinity significantly affect the foaming capability of the samples. Ge et al. [
14] foamed synthesized TPUs with very low HS contents (26 wt.%, 32 wt.%, and 36 wt.%) by two-step foaming. The cell structure evolution of TPU foams with the foaming temperature and the foaming time showed that the low HS content was beneficial for the expansion ratio because it reduced the unfoamed region, whereas the high HS content generated the microcrystalline region acting as the nucleating site to improve the cell density. Recently, Nofar et al. [
15] investigated the microcellular foaming behavior of three different commercial TPU grades that possessed similar SS molecular weight of around 1000 g/mol and contained different HS contents of 38.6 wt.%, 48.9 wt.%, and 56.9 wt.% using one-step pressure drop batch based bead foaming. It was observed that the increase in HS content expanded and shifted the processing windows to higher temperatures and also limited TPU foam expansion but at same time minimized the shrinkage. The same group later [
16] continued exploring the foaming behavior of three commercial TPU grades with a given HS content (39 wt.%) but with various SS molecular weights (1000, 2000, and 3500 g/mol) using the previous bead foaming method using supercritical CO
2. The results showed that the increase in SS length could not only enhance the foam expansion behavior but could also enhance the heterogeneous cell nucleation and more importantly minimize the shrinkage of the TPU foams as a result of broadened HS crystallization.
Considering the previous reports about the effect of the chemical characteristics of TPU on the foaming behaviour in which in many of the TPUs are provided by chemical companies, we have synthesized new TPUs to evaluate in more detail the effect of HS content on the foaming window of TPU. Mainly, the study aims to analysis the relation between the main characteristics of synthesized TPUs with their foamability. In order to fulfil this objective, a new series of TPUs with different characteristics have been synthesized. In particular, we herein report the synthesis of TPUs based on 4,4-diphenylmethanediisocyanate (MDI) and, poly(ethyleneglycol)-block-poly(propyleneglycol-block-poly(ethylen-glycol) (PEG-PPG-PEG) with an intermediate average Mn of ≈2000 g/mol and 1,5-pentanediol (1,5-PDO) components containing different HS contents (40 wt.%, 50 wt.%, and 60 wt.%) by using the pre-polymer method. These TPUs are herein characterized by Shore hardness, GPC, DSC, WAXD, DMTA and rheology. The foaming behavior of these materials is also extensively analyzed at different saturation and foaming conditions by a one-step gas dissolution foaming process. The resultant foams are characterized by measuring the density and cellular structure.
2. Materials and Methods
2.1. Materials
The reactants used to obtain TPU are as follows: MDI with a molecular weight of 250.25 g/mol and a functionality of 2, PEG-PPG-PEG as a macrodiol with an average Mn of ≈2000 g/mol and OH index of 56–59 mg KOH/g, 1,5-PDO as a chain extender with a molecular weight of 104.15 g/mol and a functionality of 2, 1,4-diazabicyclo[2.2.2]octane (DABCO) as catalyst and N,N-dimethylacetamide (DMAc) as solvent. All of them were supplied by Sigma Aldrich (St. Louis, MO, USA) and used as received, except for PEG-PPG-PEG and 1,5-PDO, which were dried in a vacuum oven overnight at 80 °C and then stored in sealed glass jars with molecular sieves, and DABCO, which was deoxygenated by bubbling nitrogen before use. Medical grade CO2 (99.9% purity) was used as a blowing agent for the gas dissolution foaming experiments.
2.2. TPU Synthesis
Three samples of TPU were synthesized with different weight fractions of HS: 40% HS, 50% HS and 60% HS (percentage indicated is by weight). The synthesis of TPUs was carried out in a reaction flask by a two-step, pre-polymer method under dry nitrogen atmosphere. Firstly, PEG-PPG-PEG was added drop wise from a funnel to an excess of MDI and this mixture was heated in an oil bath to 80 °C for 2 h with stirring at 400 rpm. After this step, a pre-polymer that contains a polyol end capped by MDI is obtained in the presence of an excess of MDI. In the second step, the required amount of the pre-polymer, MDI, and DMAc (240 mL) was stirred vigorously with a magnetic stirrer until the MDI granules were dissolved. Then, the above mixture was poured into a funnel and then added drop wise to a preheated mixture of chain extender (1,5-PDO) and DABCO catalyst (0.3 g, 0.003 mmol, 0.3%) in DMAc (60 mL). This reaction mixture was stirred at 400 rpm for 2 h in an oil bath at 80 °C. At the end of this stage, hard segments were created by the reaction of the MDI with the chain extender. Finally, the solution containing the TPU polymer was poured into silicone moulds and maintained in an oven at 80 °C for 3 days to obtain TPU casts.
The amount of each component used for the different TPUs is collected in
Table 1. The molar ratio of NCO/OH was adjusted as 1.02. The pre-polymer formulation (first step of synthesis) used is calculated having a molar ratio of the number of moles of MDI and PEG-PPG-PEG polyol as 6:1 for TPU 50% HS and TPU 60% HS, and as 4:1 for TPU 40% HS. In total, 100 g of TPU material were produced in each synthesis.
2.3. Samples Roduction of Solid TPU
Compression molded samples were prepared by using a hot plate press. The material was first heated at a temperature above melting temperature (between 165 and 180 °C) for 3 min, raising the pressure to 100 bar at 0.5 bar/s. Then, the samples were pressed under a constant pressure of 100 bar for 7 min, lowering the temperature to 60 °C at 25 °C/min. These compression molded samples were used to characterize their Shore hardness, WAXD, DMTA, and rheology.
Extruded samples with 1.5 ± 0.5 mm of diameter were also prepared by a TWELVindex extrusion plastometer from ATS Faar (Milan, Italy) using a temperature above melting temperature (between 165 and 180 °C). These extruded samples were prepared to carry out the foaming tests.
All the samples were dried in a vacuum oven overnight at 80 °C before any type of characterization and before the foaming process.
2.4. Solid TPU Characterization
2.4.1. Density
The density of the solid TPUs (ρs) (extruded samples) was measured with a gas pycnometer Accupyc II 1340 from Micromeritics (Norcross, GA, USA).
2.4.2. Shore Hardness
Shore A and B hardness tenting was measured with a Bareiss U72 durometer (Bareiss, Baiersbronn, Germany) following the “ISO 868:2003: Plastics and ebonite. Determination of indentation hardness by means of a duromer (Shore hardness)” procedure. The measurements were taken for 1 s at the temperature of 23 °C. Three samples of each material from the compression molded sheet with dimensions 10 × 10 × 2 mm3 were used.
2.4.3. Gel Permeation Chromatography (GPC)
Number-averaged molecular weights (Mn), weigh-averaged molecular weights (Mw) and polydispersity index (PDI) were determined by GPC. A dilute solution of 1 mg/mL of synthesized TPU was prepared in tetrahydrofuran (THF) and stirred for 2 h. The solutions were filtered through a 0.2 μm polyamide filter. Three measurements of each synthesized TPU were carried out in order to obtain an average.
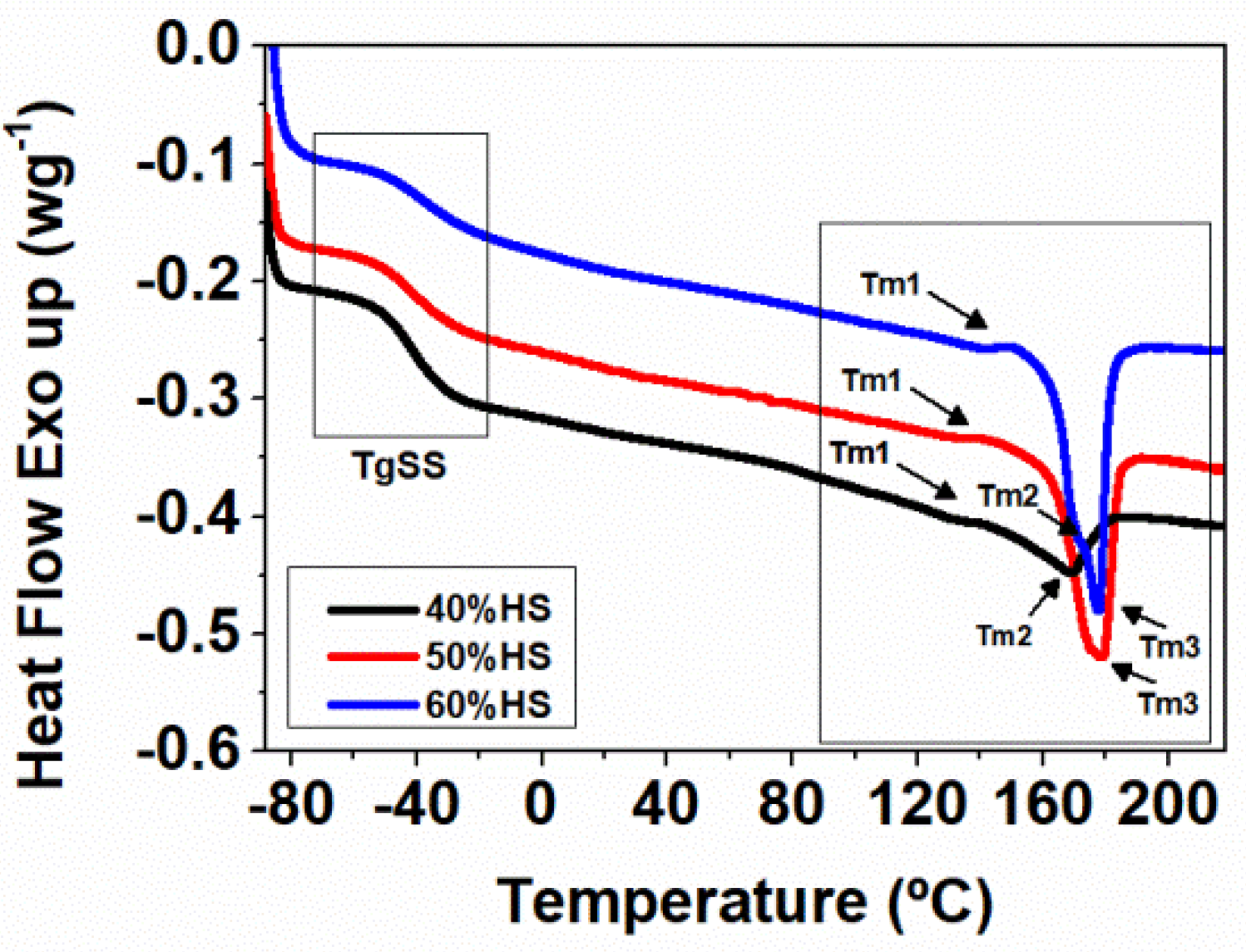
2.4.4. Differential Scanning Calorimetry (DSC)
DSC was performed using a DSC30 Mettler Toledo Instrument (Mettler-Toledo, Columbus, OH, USA). Tests were carried out under nitrogen atmosphere at a heating rate of 10 °C min
−1 from −90 to 220 °C. All DSC measurements were done in aluminum pans with 8 mg of extruded TPU. DSC experiments are carried out by performing a first heating, a cooling, and a second heating to eliminate the thermal history. The HS crystallinity of the TPUs (
Xc) has been estimated according to Equation (1), where Δ
Hm is the melting enthalpy measured for each TPU and Δ
Hom is the melting enthalpy for 100% crystalline TPU, which was taken as 155 Jg
−1 [
17].
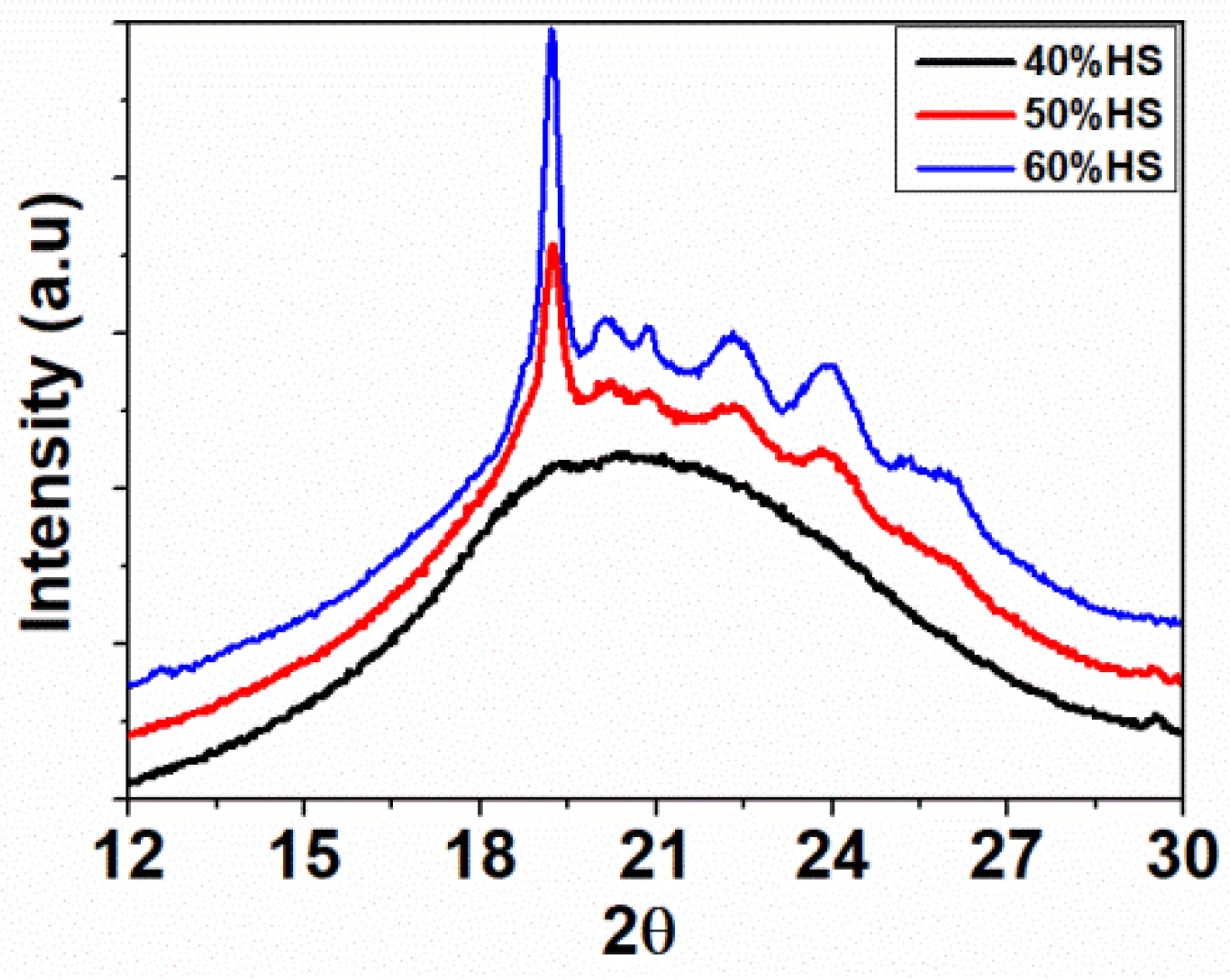
2.4.5. Wide Angle X-ray Diffraction (WAXD)
WAXD patterns were obtained using a PANalytical X’Pert Pro (XRD 5) instrument (Malvert Panalytical, Malvern, UK) employing Cu Kα radiation (λ = 1.54 Å) as the X-ray source, and a voltage of 40 KV and 40 mA of current. The diffraction angle was scanned from 5° to 90°. The samples were cut from the compressed sheet with dimensions 10 × 10 × 2 mm3.
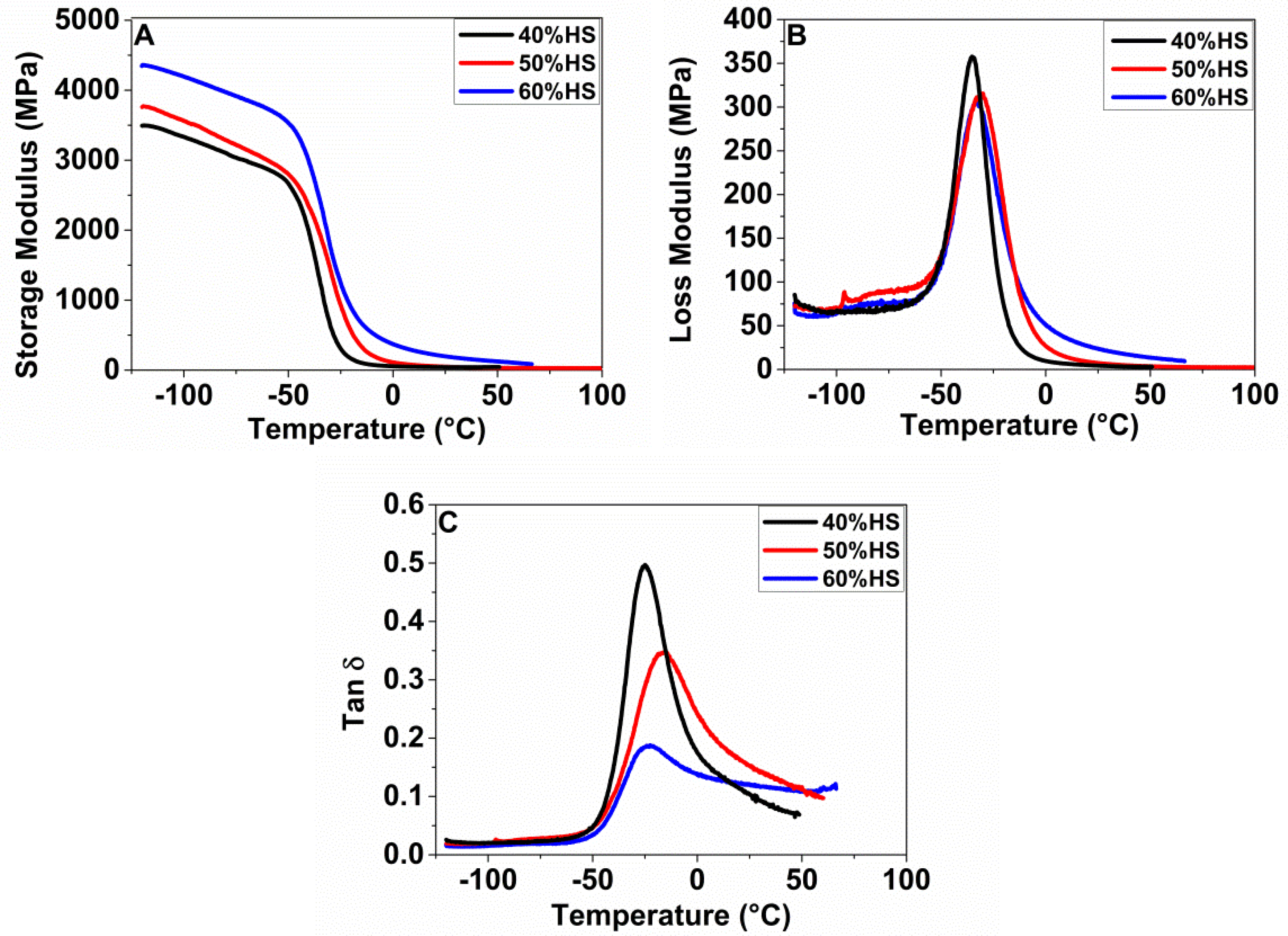
2.4.6. Dynamic Mechanical Thermal Analysis (DTMA)
DTMA tests were carried out using a Q800 DMA instrument (TA) in single cantilever mode at a heating rate of 3 °C/min, an amplitude of 15 µm and a frequency of 1 Hz. The samples were cut from the compressed sheet, with dimensions 17 × 6 × 2 mm3. Three samples of each sample were measured to obtain an average. The storage modulus (E’), loss modulus (E’’), and tan δ were recorded, and the glass transition temperature (Tg) of hard segments (HS) and soft segments (SS) was obtained from the peak tan δ.
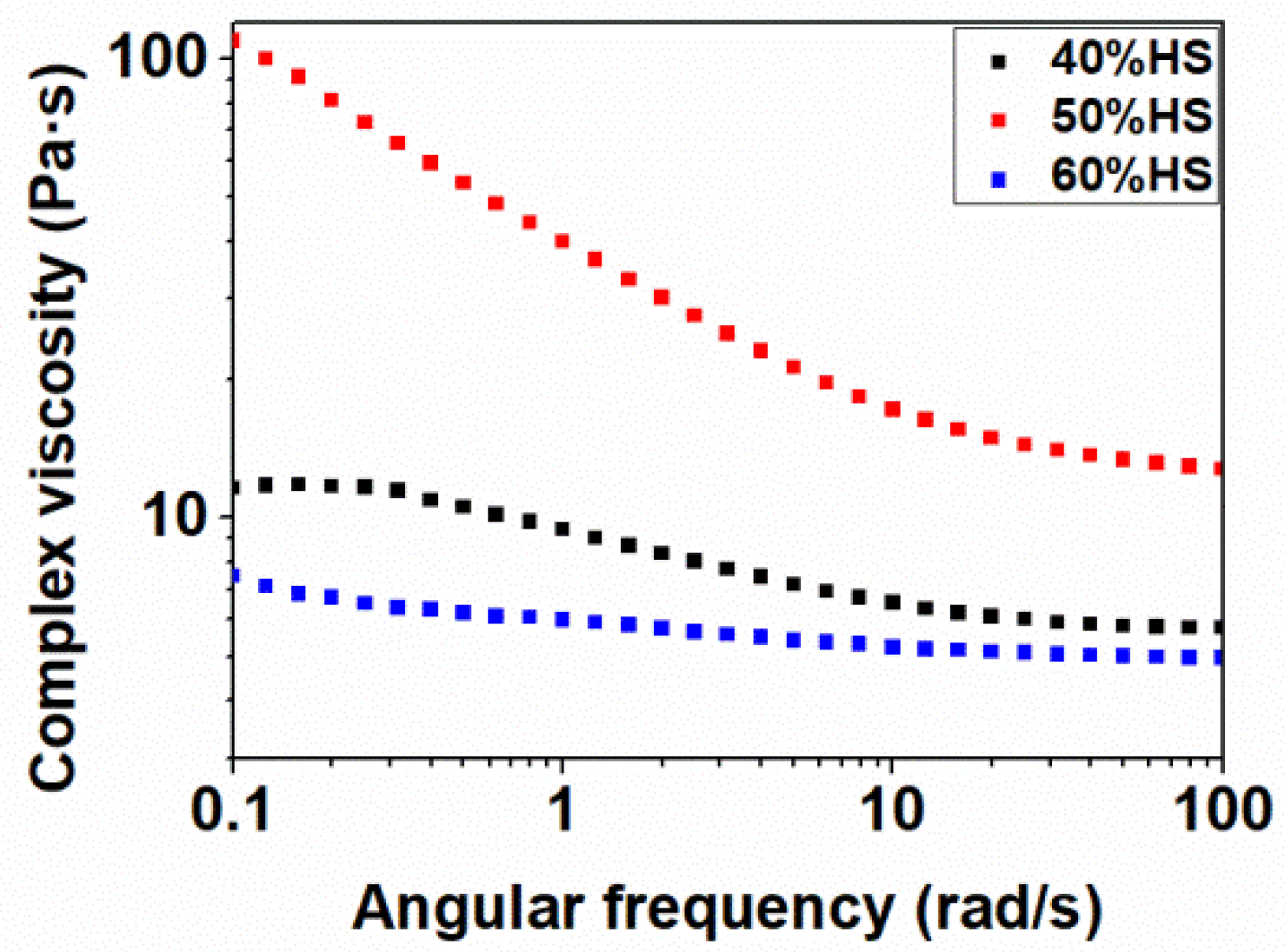
2.4.7. Shear Rheology
Cylindrical samples obtained by compression molding with a thickness of 1 mm and a diameter of 25 mm were used for the rheological tests. Rheological measurements were carried out on a strain-controlled rheometer (ARES-G2, TA Instruments, New Castle, DE, USA) with parallel plate geometry (diameter 25 mm, gap 1 mm) under a nitrogen atmosphere. The rheological characterization was performed in frequency sweep tests at 200 °C, a constant strain of 10% and an angular frequency range from 0.1 to 100 rad s−1. Strain was chosen in order to measure all the samples in the linear viscoelastic region (LVR), and it was estimated by an initial study through an amplitude sweep test at strains ranging from 0.01% to 1000%, an angular frequency of 1 rad/s and 200 °C.
2.5. Gas Dissolution Foaming Experiments
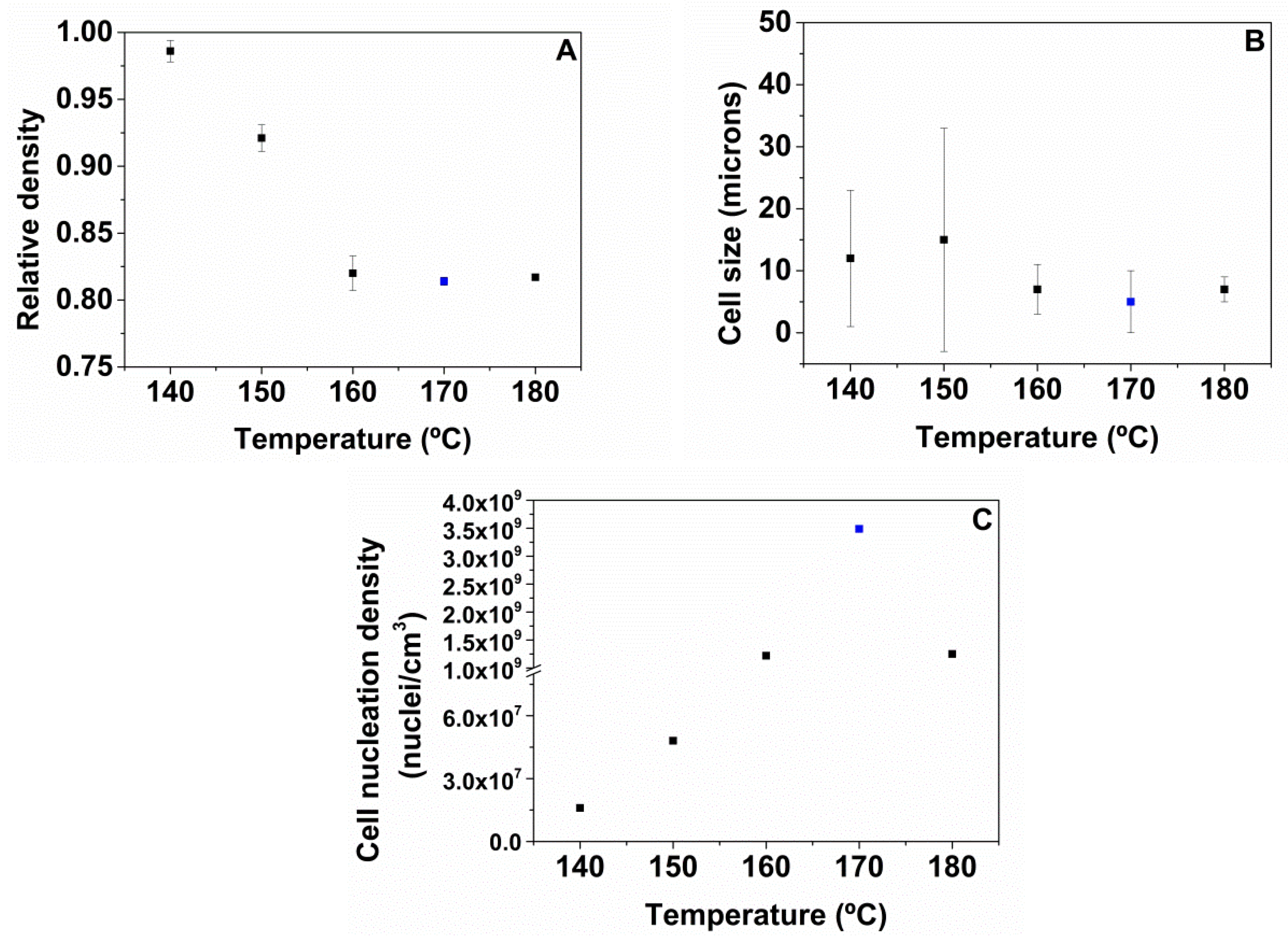
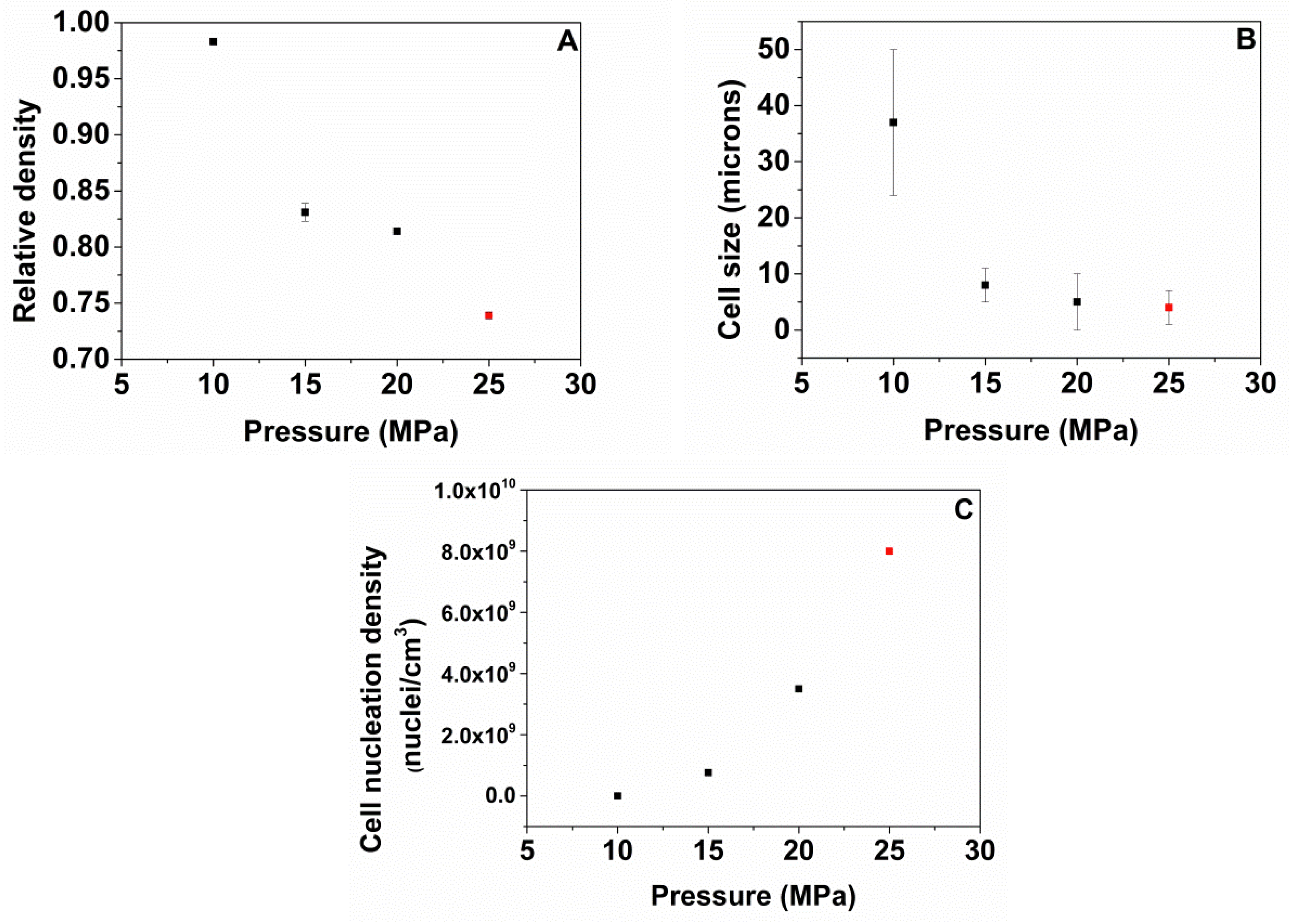
A high pressure vessel (model PARR 4681) provided by Parr Instrument Company with a capacity of 1 L was used for the foaming tests. The maximum temperature and pressure reached by this device are 350 °C and 41 MPa, respectively. The pressure was automatically controlled by an accurate pressure pump controller (model SFT-10) provided by Supercritical Fluid Technologies Inc. (Newark, USA, DE) The vessel is equipped with a clamp heater of 1200 W, and its temperature is controlled by a CAL 3300 temperature controller(CAL control, Hertfordshire, UK). This set up has been used to perform a set of experiments by using a one-step foaming process. Firstly, samples were introduced in the pressure vessel under certain pressure and temperature conditions for the saturation stage. After saturation, pressure was rapidly released and the samples expanded in the pressure vessel after depressurization. Two sets of experiments were performed for the extruded samples. Firstly, the effect of the foaming temperature was analyzed by fixing the saturation pressure to 20 MPa and the saturation time to 1 h, and varying the foaming temperature between 140, 150, 160, 170 and 180 °C. Secondly, the influence of the saturation pressure was evaluated by choosing four different saturation pressures: 10, 15, 20 and 25 MPa at 170 °C for 1 h. Extruded TPU samples were foamed and then were left to desorb the remaining CO2 before characterizing their density and cellular structure.
2.6. Characterization of TPU Foams
2.6.1. Density
Density of the TPU foams was determined with the water-displacement method based on Archimedes’ principle using a density determination kit with an AT261 Mettler-Toledo balance (Mettler-Toledo, Columbus, USA, OH). The density of TPU foams was measured when all CO2 was desorbed. Relative density (ρr) has been calculated by the ratio between the density of TPU foam (ρf) and the density of the solid TPU (extruded samples) with the same chemical composition (ρs).
2.6.2. Cellular Structure
With the aim of maintaining the cellular structure for the microscopic visualization, samples were cooled in liquid nitrogen and fractured. The cellular morphology of the extruded samples was observed by Scanning Electron Microscopy (SEM) with a JEOL JSM-820 microscope (JEOL Ltd., Tokio, Japan). The perpendicular plane of the extrusion direction was examined by SEM after vacuum coating with a gold monolayer. An image analysis technique [
18] of SEM micrographs was used to determine the average cell size (Ф) of the cellular structure of the TPU foamed. More than 100 cells of different areas of each cellular material have been used for this analysis.
Cell nucleation density (
N0) [
18] has been determined according to Equation (2) where
Nv is the cell density, Ф is the average cell size, and ρr is the relative density.
4. Conclusions
A series of TPU based on MDI, PEG-PPG-PEG and 1,5-PDO components with low contents of HS (40, 50 and 60 wt.%) are synthesized using the pre-polymer method. These TPUs have been characterized in detail by measuring their densities, Shore hardness, GPC, DSC, WAXD, DMTA, and rheology.
The foaming window for these materials in the one-step gas dissolution process has been evaluated at different saturation and foaming conditions. Initially, the effect of the foaming temperatures from 140 to 180 °C maintaining a saturation pressure of 20 MPa and a foaming time of 1 h has been evaluated for all the TPU grades. The results show that the optimum HS content is 50 wt.% because higher or lower HS ratios do not lead to TPU foams. In addition, the foaming conditions to obtain TPU foams with 50 wt.% HS have been optimized by varying the saturation pressure from 10 to 25 MPa, while maintaining the optimum foaming temperature (170 °C), and the foaming time (1 h). The best foaming conditions are 170 °C, 25 MPa and 1 h, since a TPU foam with the lowest relative density (0.74), the smallest average cell size (4 microns), and an improved cell nucleation density of 8.0 × 109 cells/cm3 is obtained.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}