
Quantum Mechanical Investigation of the Oxidative Cleavage of the C–C Backbone Bonds in Polyethylene Model Molecules


 ,
, 

Abstract
:1. Introduction
2. Calculation Methods
2.1. Computer Programs for QM Calculations
2.2. Bond Strength Descriptors
3. Results and Discussion
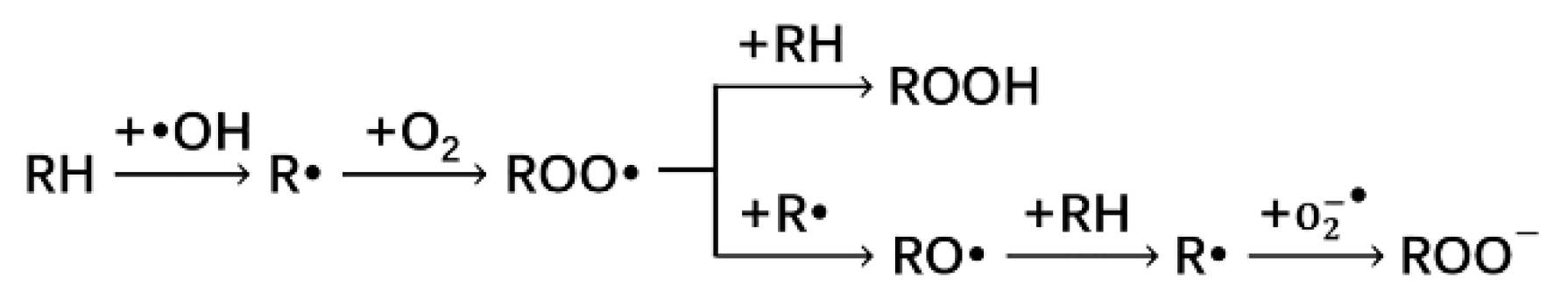
3.1. Oxidation of PE by Free Oxygen Radicals
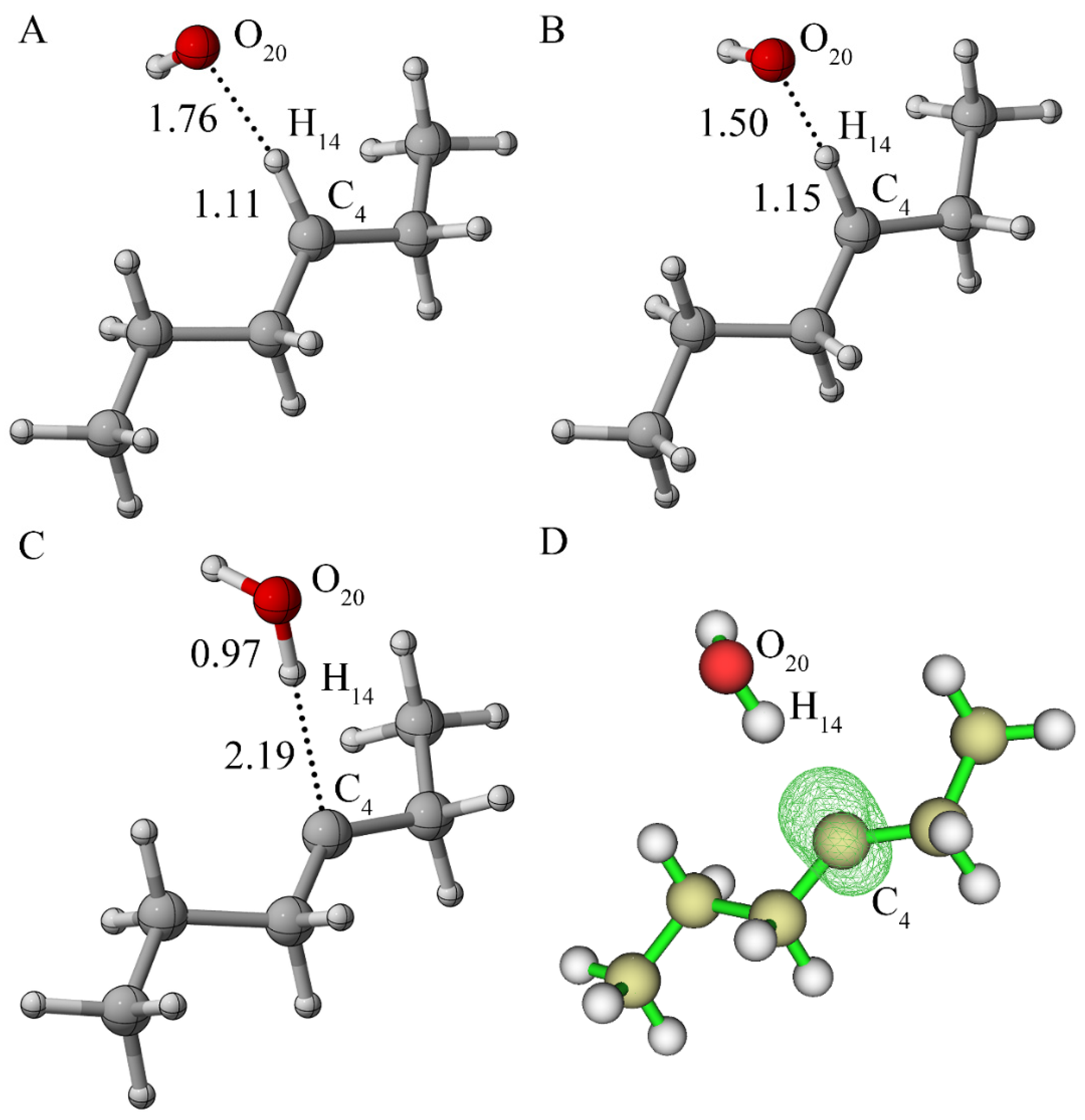
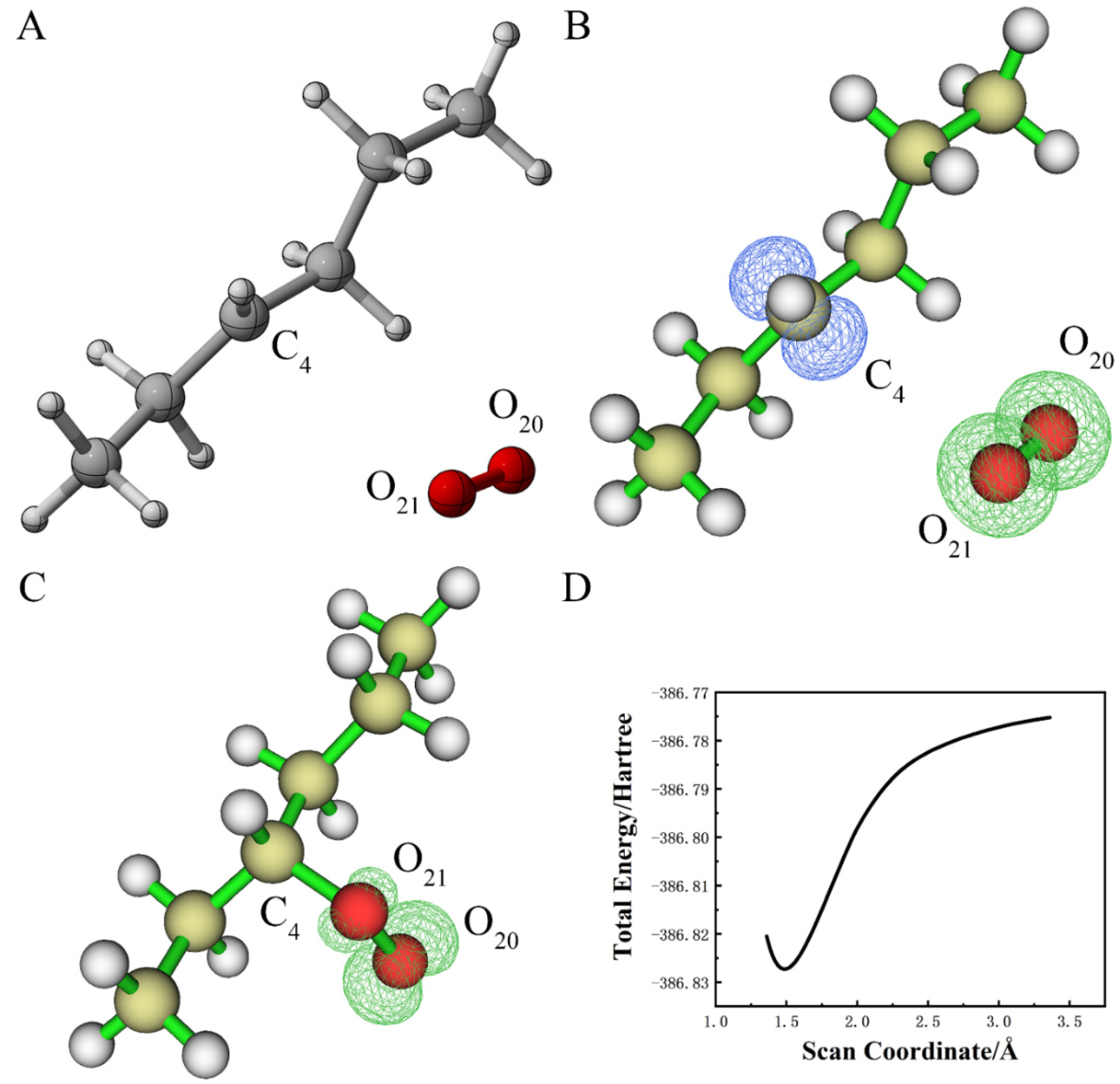
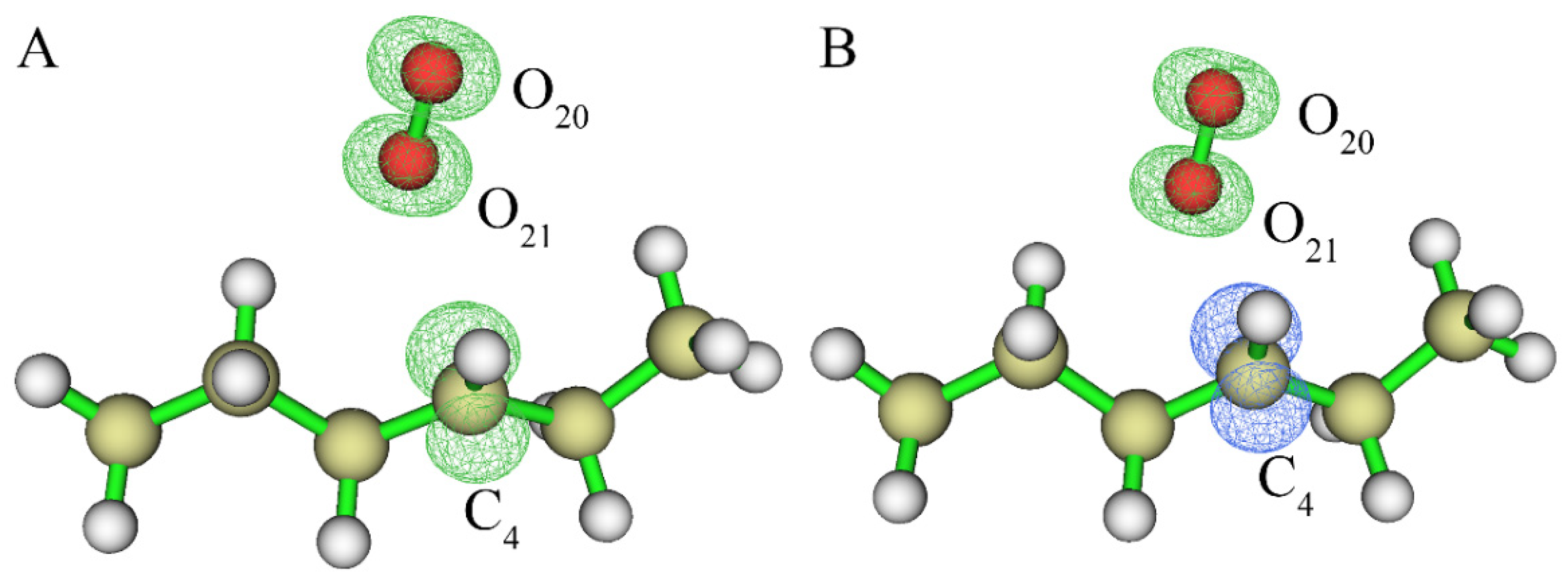
3.1.1. Reaction of Alkane with Hydroxyl Radical
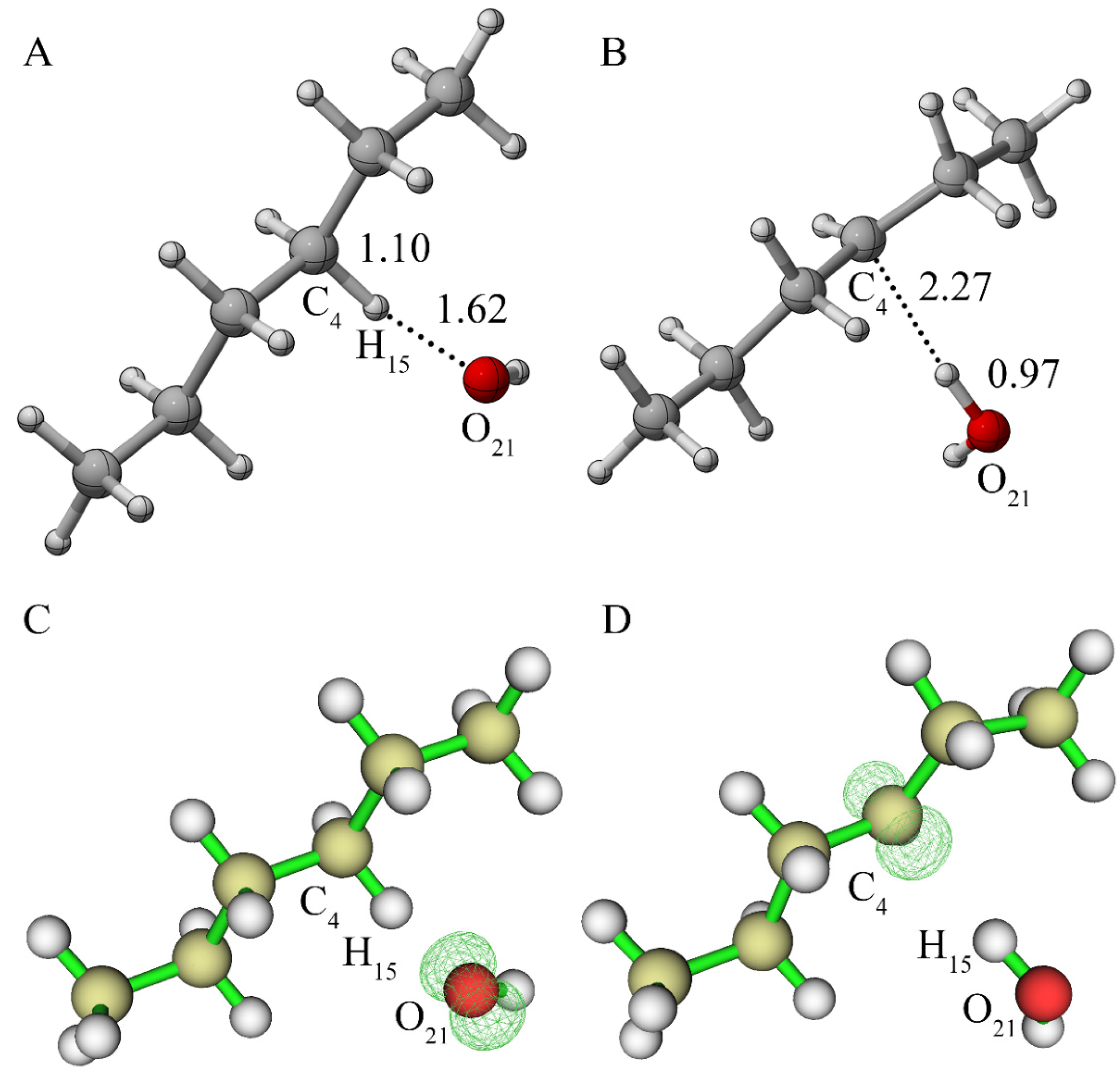
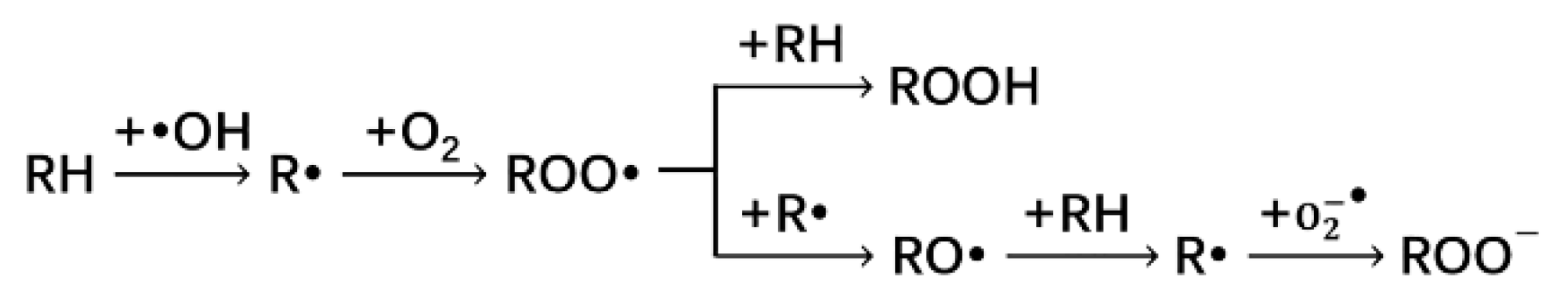
3.1.2. Reaction of Alkane Free Radical with Oxygen
3.1.3. Reaction of Peroxyl Radical (ROO·) with Alkane or Alkane Radical
ROO· + R·→2RO·
3.1.4. Reaction of Alkoxy Radical (RO·) with Alkane
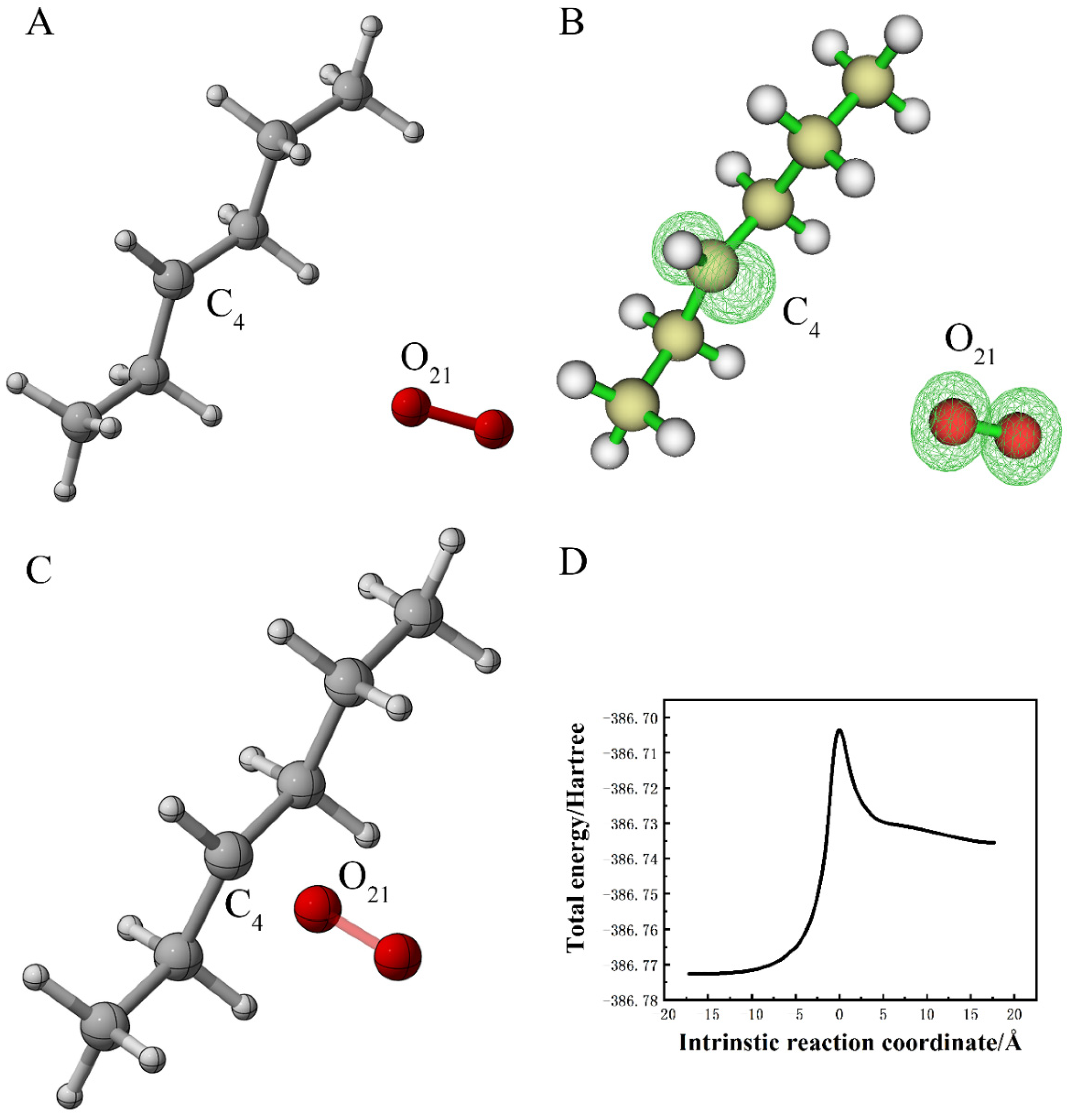
3.1.5. Reaction of Alkane Radical with Superoxide Anion Radical
3.2. The Influence of Carbocation on the Strength of PE Carbon Backbone
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [Green Version]
- Andrady, A.L. Plastics and Environmental Sustainability; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Facts, P.E.P.T. An Analysis of European Plastics Production, Demand and Waste Data; Plastics Europe: Brussels, Belgium, 2019. [Google Scholar]
- Miandad, R.; Barakat, M.; Aburiazaiza, A.S.; Rehan, M.; Nizami, A. Catalytic pyrolysis of plastic waste: A review. Process Saf. Environ. Prot. 2016, 102, 822–838. [Google Scholar] [CrossRef]
- North, E.J.; Halden, R.U. Plastics and environmental health: The road ahead. Rev. Environ. Health 2013, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Chatterjee, S. Microplastic pollution, a threat to marine ecosystem and human health: A short review. Environ. Sci. Pollut. Res. 2017, 24, 21530–21547. [Google Scholar] [CrossRef] [PubMed]
- Weinig, H.G. Science to Enable Sustainable Plastics. Nachr. Chem. 2020, 68, 82. [Google Scholar] [CrossRef]
- Fotopoulou, K.N.; Karapanagioti, H.K. Degradation of various plastics in the environment. In Hazardous Chemicals Associated with Plastics in the Marine Environment; Springer: Cham, Switzerland, 2017; pp. 71–92. [Google Scholar]
- Wei, R.; Zimmermann, W. Biocatalysis as a green route for recycling the recalcitrant plastic polyethylene terephthalate. Microb. Biotechnol. 2017, 10, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Devi, R.S.; Kannan, V.R.; Nivas, D.; Kannan, K.; Chandru, S.; Antony, A.R. Biodegradation of HDPE by Aspergillus spp. from marine ecosystem of Gulf of Mannar, India. Mar. Pollut. Bull. 2015, 96, 32–40. [Google Scholar] [CrossRef]
- Li, Z.; Wei, R.; Gao, M.; Ren, Y.; Yu, B.; Nie, K.; Xu, H.; Liu, L. Biodegradation of low-density polyethylene by Microbulbifer hydrolyticus IRE-31. J. Environ. Manag. 2020, 263, 110402. [Google Scholar] [CrossRef]
- Rojas-Parrales, A.; Orantes-Sibaja, T.; Redondo-Gómez, C.; Vega-Baudrit, J. Biological Degradation of Plastics: Polyethylene Biodegradation by Aspergillus and Streptomyces Species—A Review. In Integrated and Sustainable Environmental Remediation; ACS Publications: Washington, DC, USA, 2018; pp. 69–79. [Google Scholar]
- Santo, M.; Weitsman, R.; Sivan, A. The role of the copper-binding enzyme–laccase–in the biodegradation of polyethylene by the actinomycete Rhodococcus ruber. Int. Biodeterior. Biodegrad. 2013, 84, 204–210. [Google Scholar] [CrossRef]
- Jeon, H.J.; Kim, M.N. Functional analysis of alkane hydroxylase system derived from Pseudomonas aeruginosa E7 for low molecular weight polyethylene biodegradation. Int. Biodeterior. Biodegrad. 2015, 103, 141–146. [Google Scholar] [CrossRef]
- Yoon, M.G.; Jeon, H.J.; Kim, M.N. Biodegradation of polyethylene by a soil bacterium and AlkB cloned recombinant cell. J. Biorem. Biodegrad. 2012, 3, 1–8. [Google Scholar]
- Ru, J.; Huo, Y.; Yang, Y. Microbial degradation and valorization of plastic wastes. Front. Microbiol. 2020, 11, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Cui, Z.; Nie, K.; Cao, H.; Jiang, M.; Xu, H.; Tan, T.; Liu, L. A quantum mechanism study of the cc bond cleavage to predict the bio-catalytic polyethylene degradation. Front. Microbiol. 2019, 10, 489. [Google Scholar] [CrossRef] [PubMed]
- Inderthal, H.; Tai, S.L.; Harrison, S.T. Non-Hydrolyzable Plastics—An Interdisciplinary Look at Plastic Bio-Oxidation. Trends Biotechnol. 2020. [Google Scholar] [CrossRef]
- Gewert, B.; Plassmann, M.M.; MacLeod, M. Pathways for degradation of plastic polymers floating in the marine environment. Environ. Sci. Process. Impacts 2015, 17, 1513–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitroka, S.M.; Smiley, T.D.; Tanko, J.; Dietrich, A.M. Reaction mechanism for oxidation and degradation of high density polyethylene in chlorinated water. Polym. Degrad. Stab. 2013, 98, 1369–1377. [Google Scholar] [CrossRef]
- Wei, R.; Tiso, T.; Bertling, J.; O’Connor, K.; Blank, L.M.; Bornscheuer, U.T. Possibilities and limitations of biotechnological plastic degradation and recycling. Nat. Catal. 2020, 3, 867–871. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D. 01. 2009. Available online: http://www.rsc.org/suppdata/c5/sc/c5sc02423d/c5sc02423d1.pdf (accessed on 10 August 2021).
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Lu, T. Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Brandhorst, K.; Grunenberg, J. How strong is it? The interpretation of force and compliance constants as bond strength descriptors. Chem. Soc. Rev. 2008, 37, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Brandhorst, K.; Grunenberg, J. Efficient computation of compliance matrices in redundant internal coordinates from Cartesian Hessians for nonstationary points. J. Chem. Phys. 2010, 132, 184101. [Google Scholar] [CrossRef]
- Grunenberg, J. Ill-defined chemical concepts: The problem of quantification. Int. J. Quantum Chem. 2017, 117, e25359. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Bond order analysis based on the Laplacian of electron density in fuzzy overlap space. J. Phys. Chem. A 2013, 117, 3100–3108. [Google Scholar] [CrossRef]
- Matito, E.; Poater, J.; Solà, M.; Duran, M.; Salvador, P. Comparison of the AIM delocalization index and the mayer and fuzzy atom bond orders. J. Phys. Chem. A 2005, 109, 9904–9910. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.; Salvador, P. Overlap populations, bond orders and valences for ‘fuzzy’atoms. Chem. Phys. Lett. 2004, 383, 368–375. [Google Scholar] [CrossRef]
- Cremer, D.; Wu, A.; Larsson, A.; Kraka, E. Some thoughts about bond energies, bond lengths, and force constants. Mol. model. annu. 2000, 6, 396–412. [Google Scholar] [CrossRef]
- Paul, S.; Deka, J.; Deka, A.; Gour, N.K. Degradation mechanism of propylene carbonate initiated by hydroxyl radical and fate of its product radicals: A hybrid density functional study. Atmos. Environ. 2019, 216, 116952. [Google Scholar] [CrossRef]
- Ahn, Y.; Colin, X.; Roma, G. Atomic scale mechanisms controlling the oxidation of polyethylene: A first principles study. Polymers 2021, 13, 2143. [Google Scholar] [CrossRef]
- Shi, B.; Wang, W.; Zhou, L.; Li, J.; Wang, J.; Chen, Y.; Zhang, W.; Ge, M. Kinetics and mechanisms of the gas-phase reactions of OH radicals with three C15 alkanes. Atmos. Environ. 2019, 207, 75–81. [Google Scholar] [CrossRef]
- Bertin, D.; Leblanc, M.; Marque, S.R.; Siri, D. Polypropylene degradation: Theoretical and experimental investigations. Polym. Degrad. Stab. 2010, 95, 782–791. [Google Scholar] [CrossRef]
- Ward, C.P.; Armstrong, C.J.; Walsh, A.N.; Jackson, J.H.; Reddy, C.M. Sunlight converts polystyrene to carbon dioxide and dissolved organic carbon. Environ. Sci. Technol. Lett. 2019, 6, 669–674. [Google Scholar] [CrossRef]
- Xie, Y.; Jiao, X.; Zheng, K.; Chen, Q.; Li, X.; Li, Y.; Shao, W.; Xu, J.; Zhu, J.; Pan, Y. Photocatalyzing Waste Plastics into C2 Fuels under Simulated Natural Environments. Angew. Chem. Int. Engl. 2020, 59, 15497–15501. [Google Scholar]
- Tao, H.; Lin, K.C. Pathways, kinetics and thermochemistry of methyl-ester peroxy radical decomposition in the low-temperature oxidation of methyl butanoate: A computational study of a biodiesel fuel surrogate. Combust. Flame 2014, 161, 2270–2287. [Google Scholar] [CrossRef]
- Battin-Leclerc, F.; Herbinet, O.; Glaude, P.A.; Fournet, R.; Zhou, Z.; Deng, L.; Guo, H.; Xie, M.; Qi, F. Experimental confirmation of the low-temperature oxidation scheme of alkanes. Angew. Chem. 2010, 122, 3237–3240. [Google Scholar] [CrossRef] [Green Version]
- Maurya, P.K. Animal biotechnology as a tool to understand and fight aging. Anim. Biotechnol. 2014, 117–119. [Google Scholar]
- Belcher, J.; McLean, K.J.; Matthews, S.; Woodward, L.S.; Fisher, K.; Rigby, S.E.; Nelson, D.R.; Potts, D.; Baynham, M.T.; Parker, D.A. Structure and biochemical properties of the alkene producing cytochrome P450 OleTJE (CYP152L1) from the Jeotgalicoccus sp. 8456 bacterium. J. Biol. Chem. 2014, 289, 6535–6550. [Google Scholar] [CrossRef] [Green Version]
- Rude, M.A.; Baron, T.S.; Brubaker, S.; Alibhai, M.; Del Cardayre, S.B.; Schirmer, A. Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus species. Appl. Environ. Microbiol. 2011, 77, 1718–1727. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Sharma, N. Mechanistic implications of plastic degradation. Polym. Degrad. Stab. 2008, 93, 561–584. [Google Scholar] [CrossRef]
- Abbas-Abadi, M.S. The effect of process and structural parameters on the stability, thermo-mechanical and thermal degradation of polymers with hydrocarbon skeleton containing PE, PP, PS, PVC, NR, PBR and SBR. J. Therm. Anal. Calorim. 2021, 143, 2867–2882. [Google Scholar] [CrossRef]
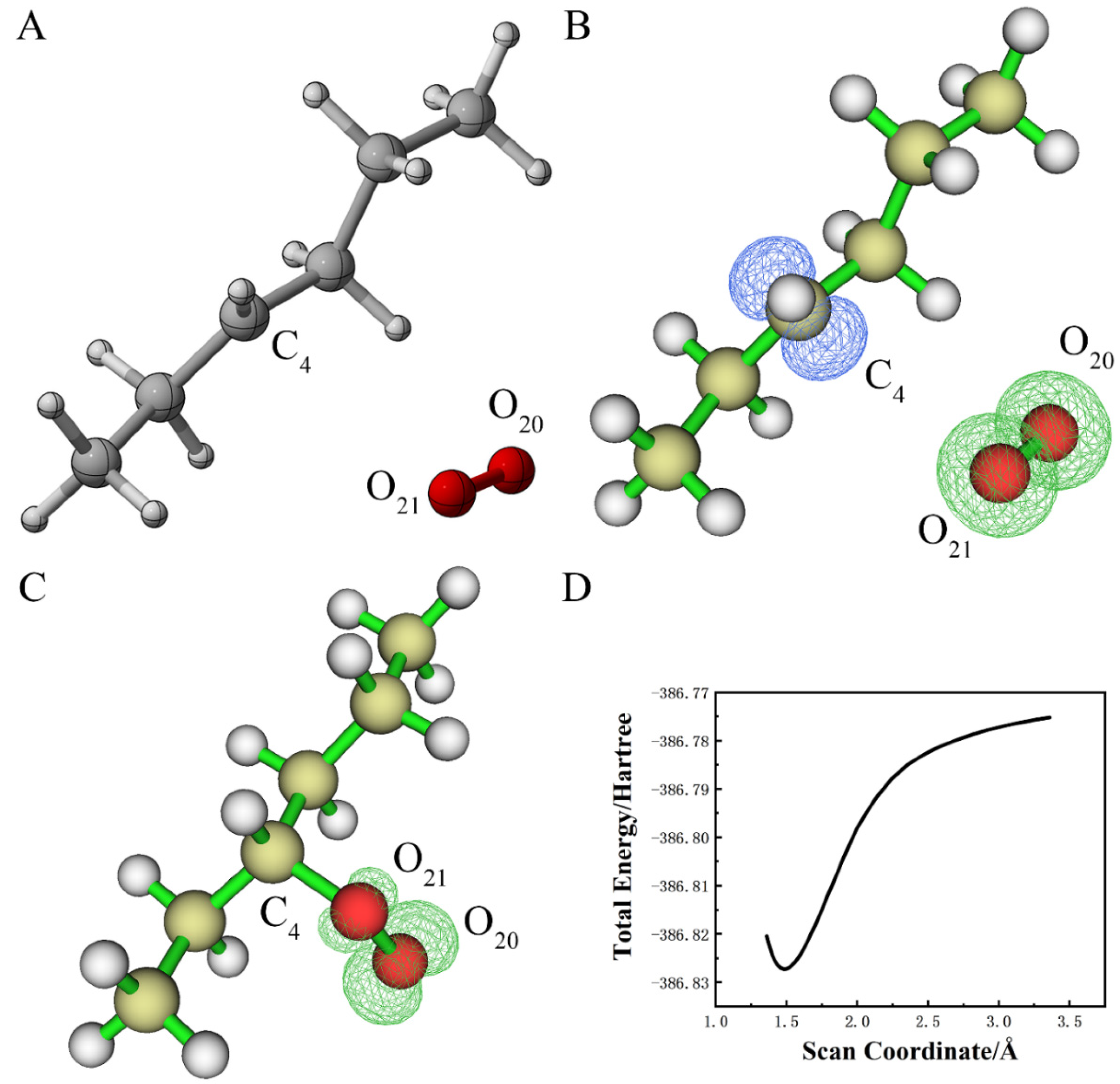


{kind=link}
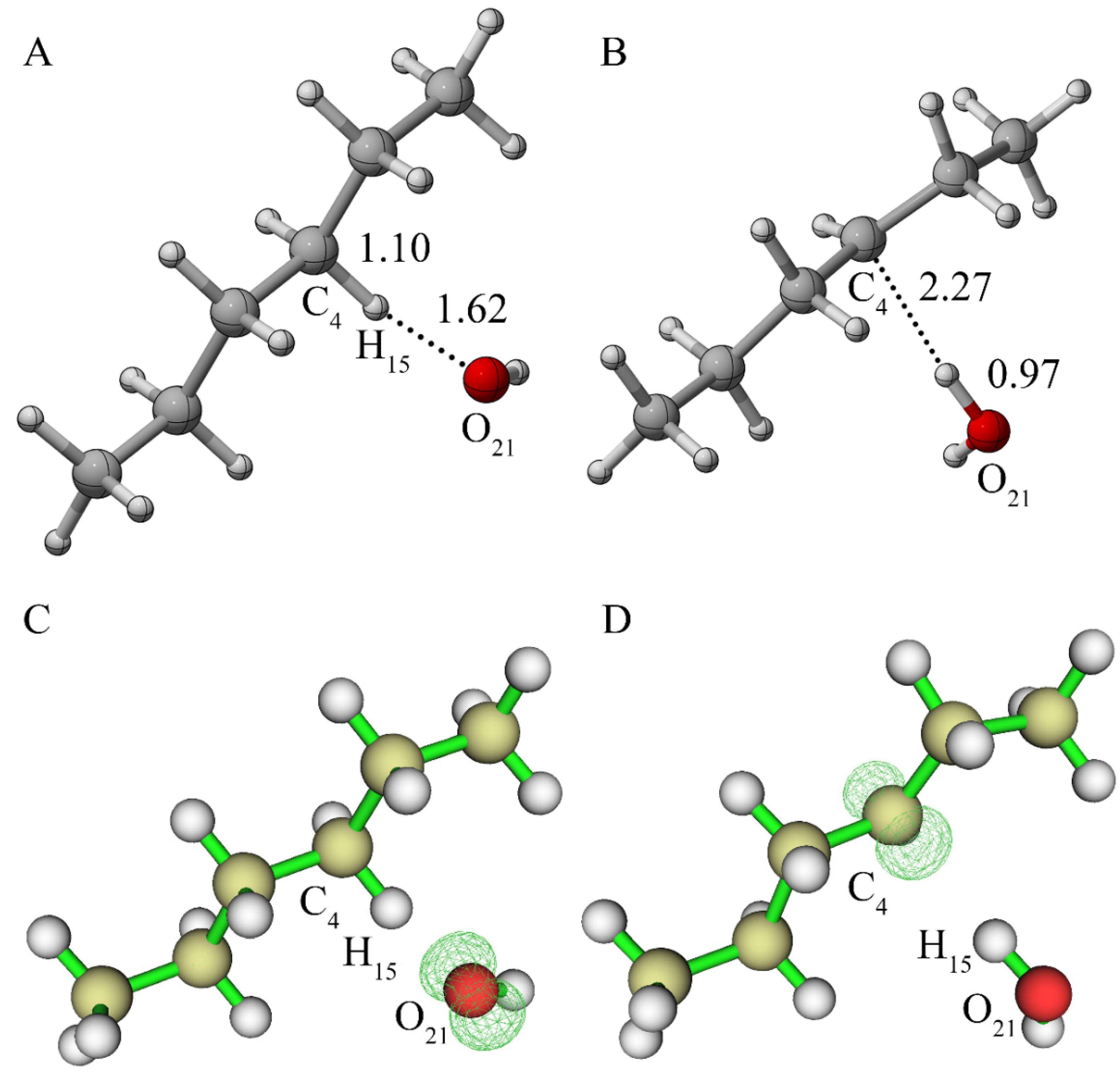
{kind=link}
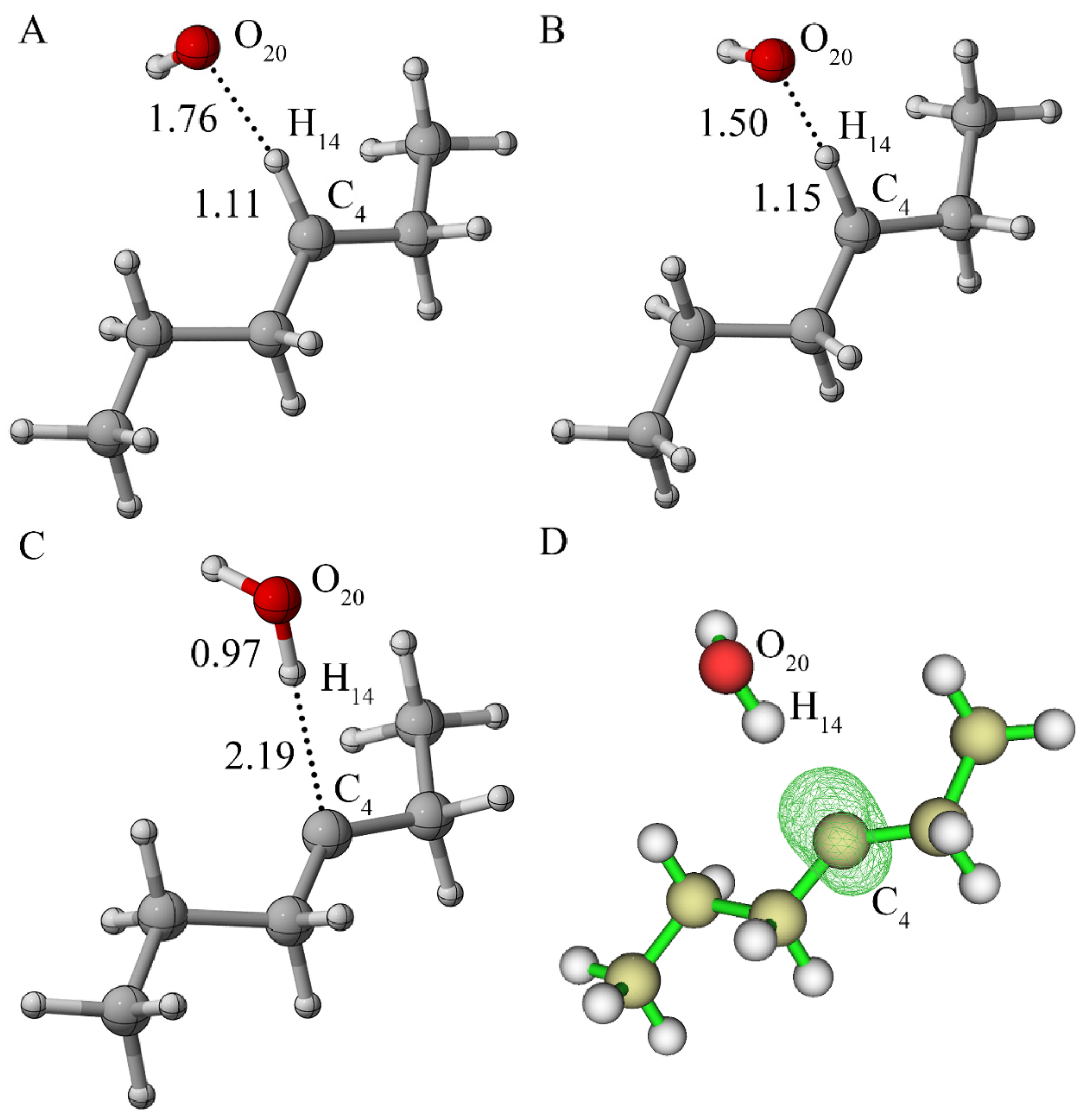
{kind=link}
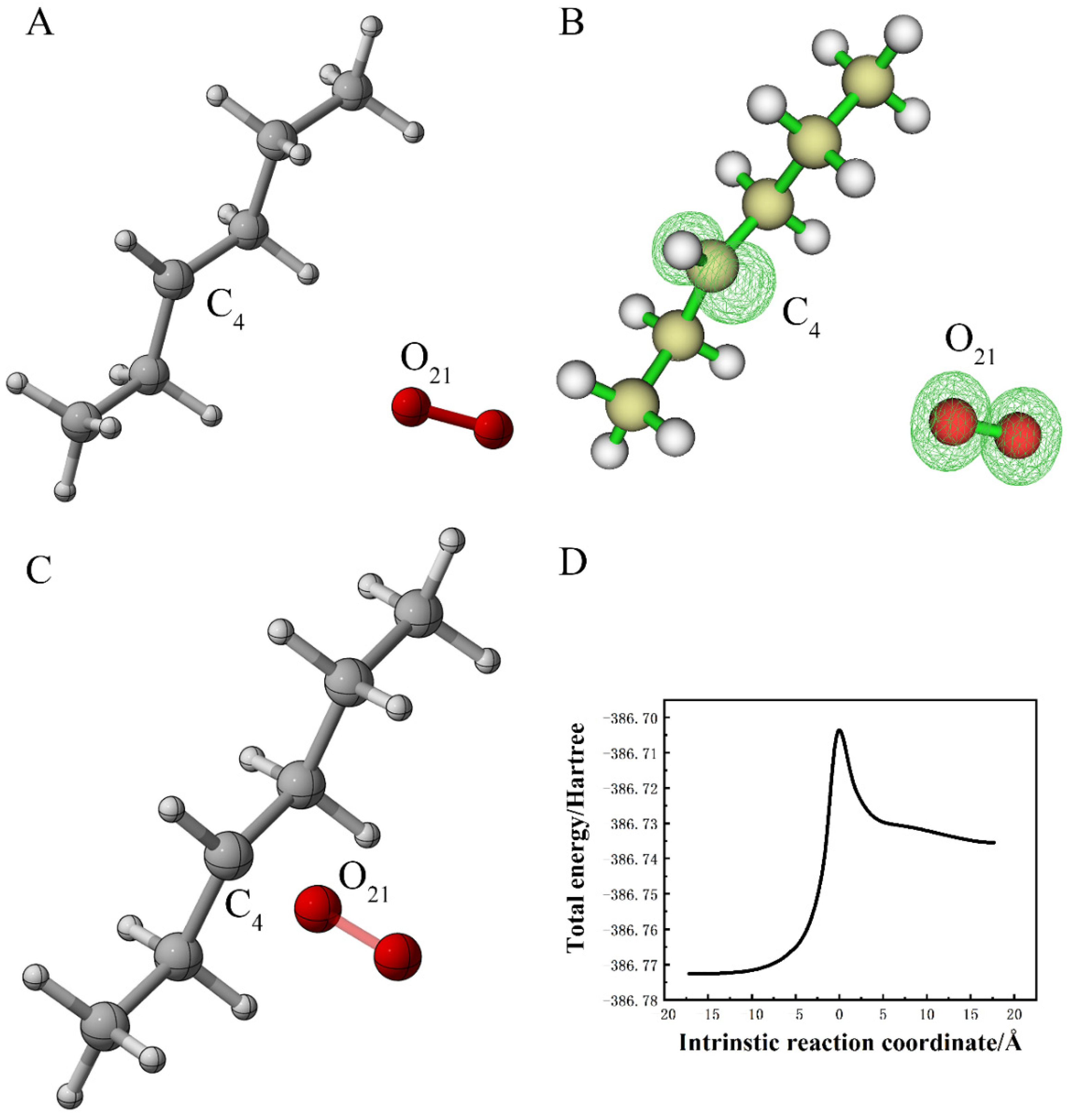
{kind=link}
{kind=link}
{kind=link}
| Bond | Alkane | C4 Position Losing One H Atom | C4 Position Losing Two H atoms | C3 and C4 Position Losing One H atom Respectively | C2 and C4 Position Losing One H atom Respectively |
|---|---|---|---|---|---|
| C1–C2 | 1.17 | 1.17 | 1.17 | 1.16 | 1.25 |
| C2–C3 | 1.12 | 1.11 | 1.10 | 1.18 | 1.19 |
| C3–C4 | 1.12 | 1.20 | 1.27 | 1.28 | 1.19 |
| C4–C5 | 1.12 | 1.20 | 1.27 | 1.19 | 1.20 |
| C5–C6 | 1.17 | 1.16 | 1.15 | 1.16 | 1.16 |
| Number | Schematic Diagram of the Carbocation Distribution (The Positions of Carbocation Are Shown in Bold and Italic) | Flexible Force Constants of the Two Weakest Bonds in the Carbon Chain (mdyn/Å) | Fuzzy Bond Order that Corresponds to the Weakest Two Bonds | Single Point Energy (kcal/mol) | ||
|---|---|---|---|---|---|---|
| 1 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 3.94 | 4.02 | 1.12 | 1.17 | - |
| 2 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 2.81 | 1.77 | 1.11 | 0.92 | - |
| 3 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 1.54 | 1.66 | 0.94 | 0.93 | 0 |
| 4 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 1.08 | 1.13 | 0.86 | 0.88 | 7.61 |
| 5 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 1.25 | 1.24 | 0.87 | 0.87 | −5.32 |
| 6 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 1.22 | 1.33 | 0.85 | 0.87 | −17.73 |
| 7 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 1.48 | 1.43 | 0.89 | 0.88 | −26.54 |
| 8 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 1.60 | 1.52 | 0.90 | 0.89 | −33.81 |
| 9 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 1.63 | 1.59 | 0.91 | 0.90 | −39.42 |
| 10 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 1.70 | 1.66 | 0.91 | 0.90 | −43.66 |
| 11 | C1–C2–C3–C4–C5–C6–C7–C8–C9–C10–C11–C12–R | 0.79 | 1.00 | 0.79 | 0.85 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Q.; Li, Z.; Cui, Z.; Wei, R.; Nie, K.; Xu, H.; Liu, L. Quantum Mechanical Investigation of the Oxidative Cleavage of the C–C Backbone Bonds in Polyethylene Model Molecules. Polymers 2021, 13, 2730. https://doi.org/10.3390/polym13162730
Jiang Q, Li Z, Cui Z, Wei R, Nie K, Xu H, Liu L. Quantum Mechanical Investigation of the Oxidative Cleavage of the C–C Backbone Bonds in Polyethylene Model Molecules. Polymers. 2021; 13(16):2730. https://doi.org/10.3390/polym13162730
Chicago/Turabian StyleJiang, Qixuan, Zhongyu Li, Ziheng Cui, Ren Wei, Kaili Nie, Haijun Xu, and Luo Liu. 2021. "Quantum Mechanical Investigation of the Oxidative Cleavage of the C–C Backbone Bonds in Polyethylene Model Molecules" Polymers 13, no. 16: 2730. https://doi.org/10.3390/polym13162730
APA StyleJiang, Q., Li, Z., Cui, Z., Wei, R., Nie, K., Xu, H., & Liu, L. (2021). Quantum Mechanical Investigation of the Oxidative Cleavage of the C–C Backbone Bonds in Polyethylene Model Molecules. Polymers, 13(16), 2730. https://doi.org/10.3390/polym13162730