
Thermally Remendable Polyurethane Network Cross-Linked via Reversible Diels–Alder Reaction


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Difurfurylamine
2.3. Synthesis of PU-V0
2.4. Synthesis of Crosslinked PUs
2.5. Film Preparation
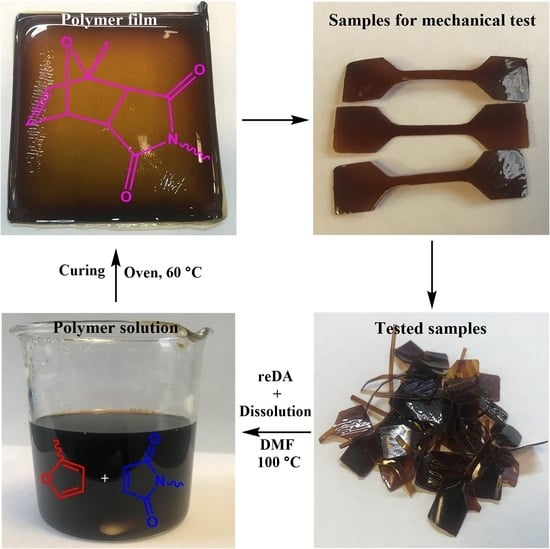
2.6. Recycling of PU-V1.0-3.0
2.7. Characterization
3. Results and Discussion
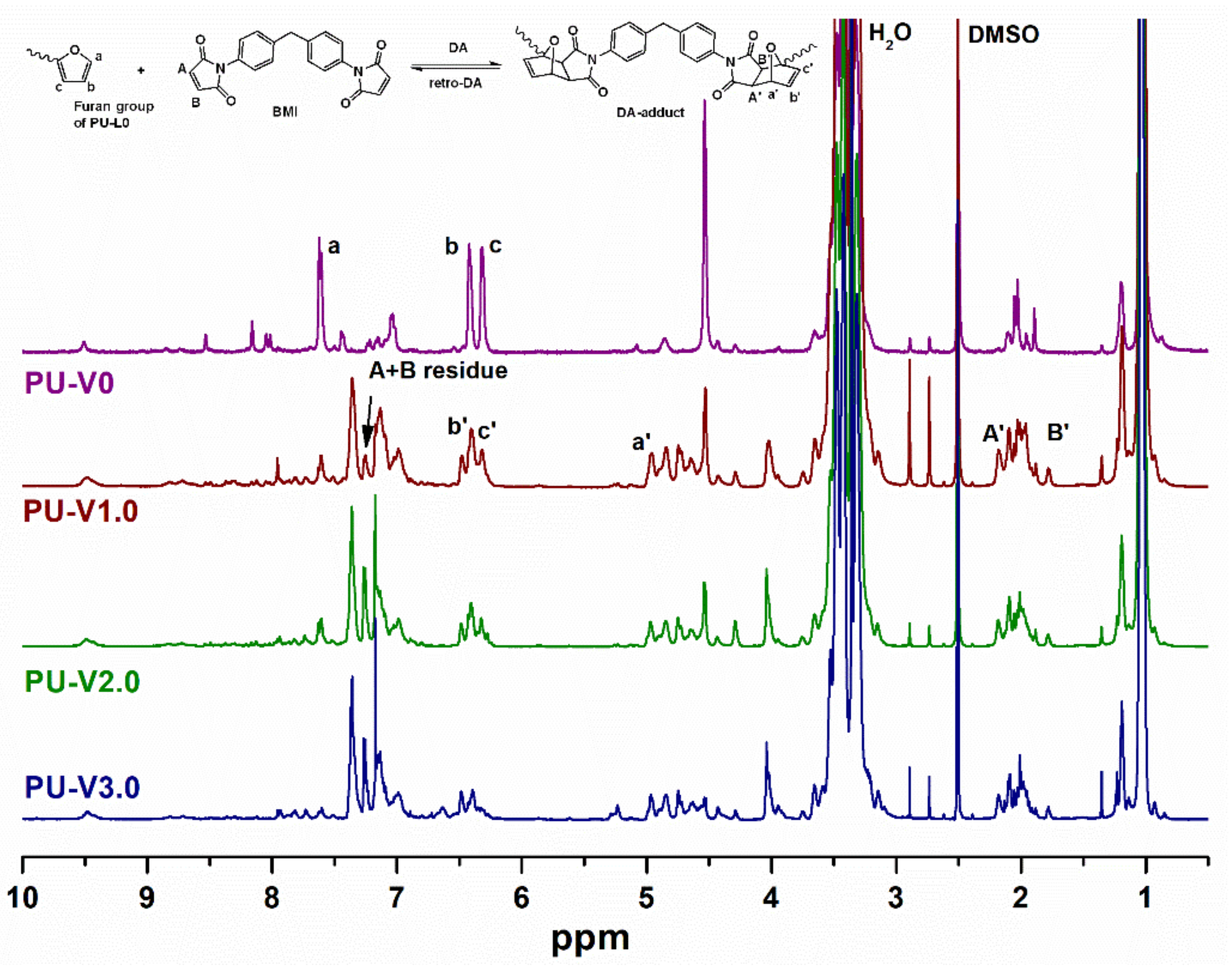
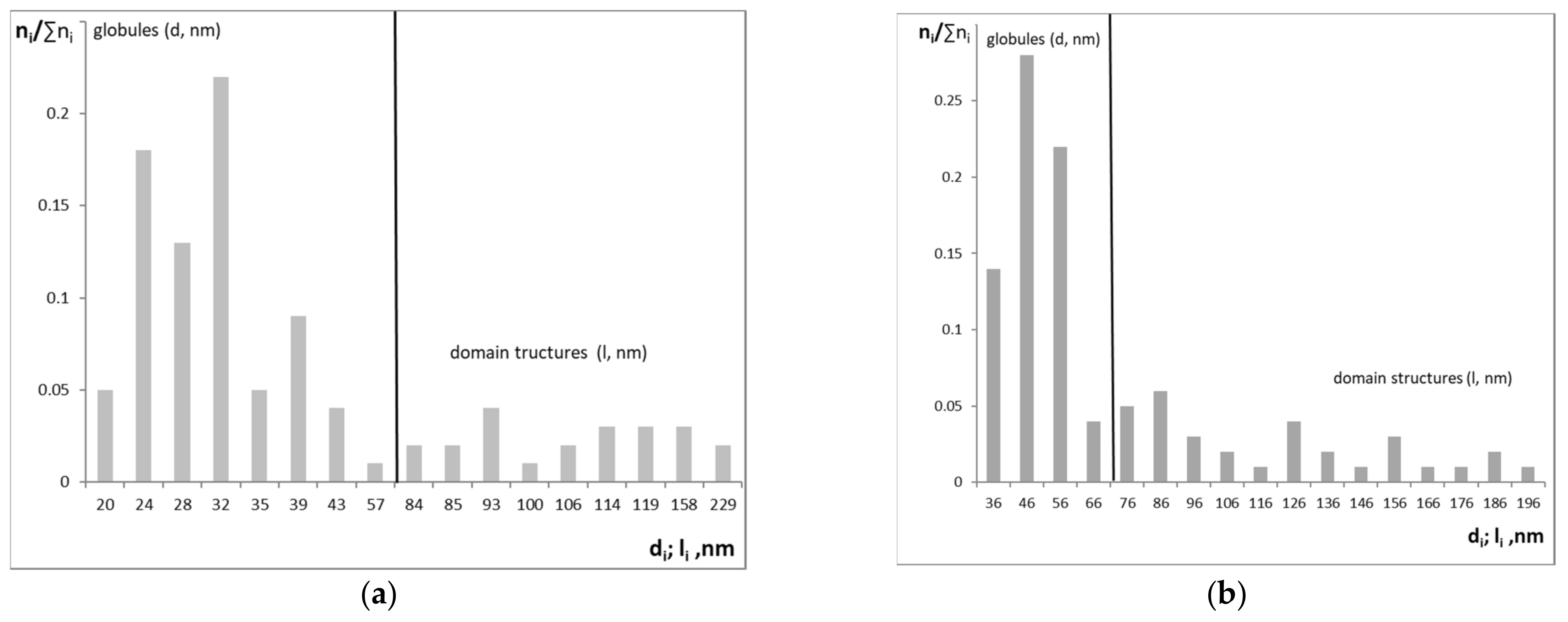
3.1. Synthesis and Characterization of PU-V0 and PU-V1.0-3.0
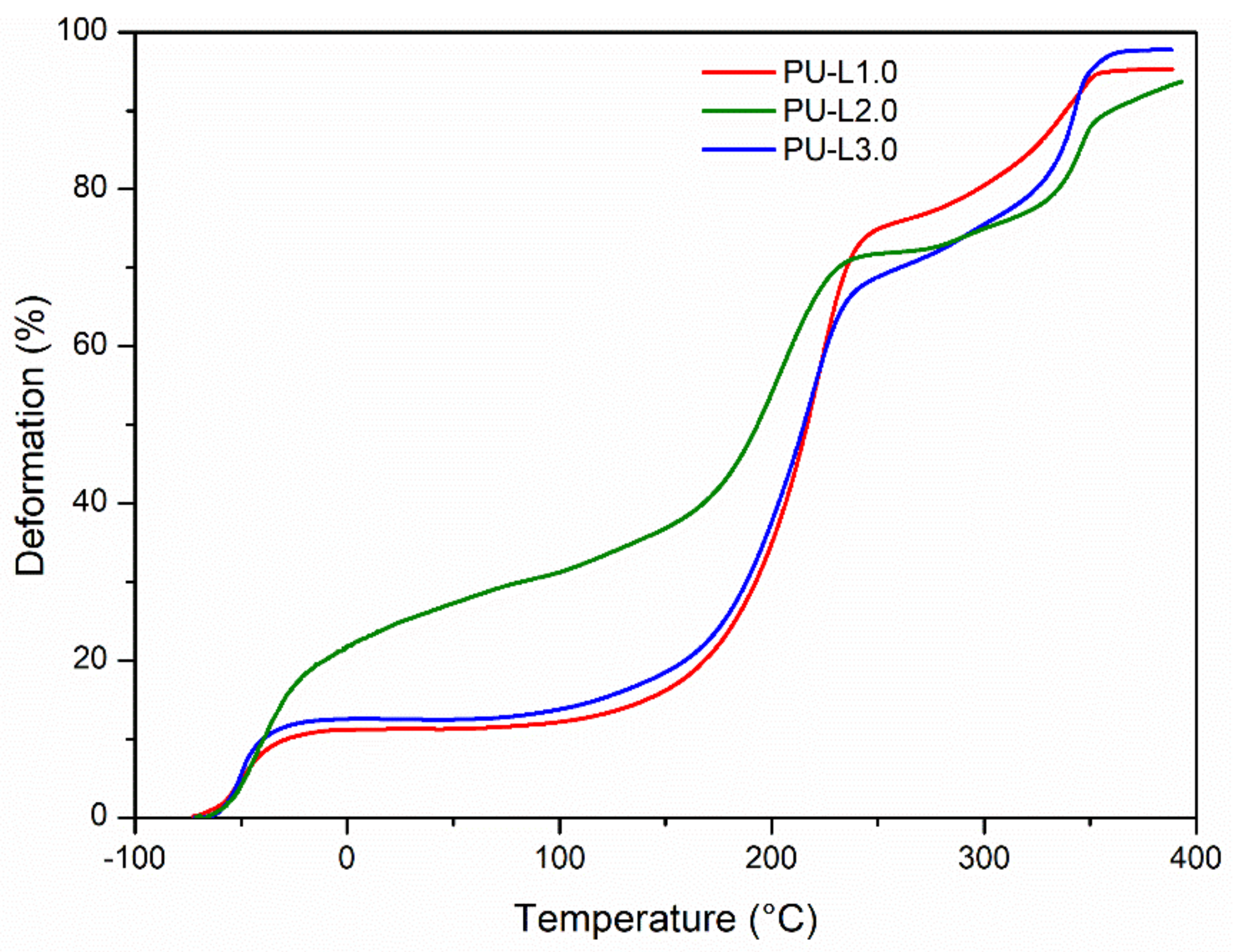
3.2. Thermal Properties and Reversibility of the DA Bonds
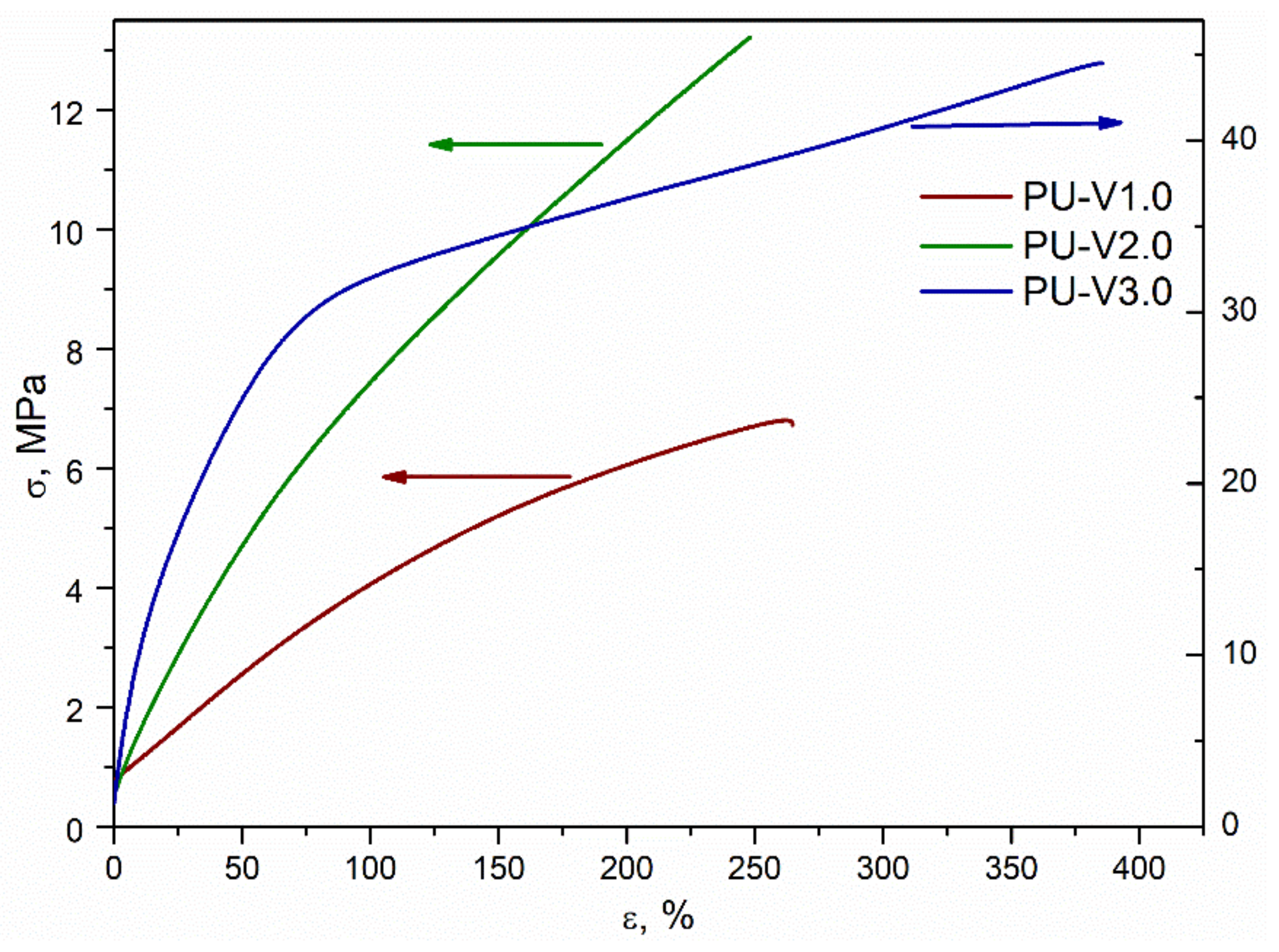
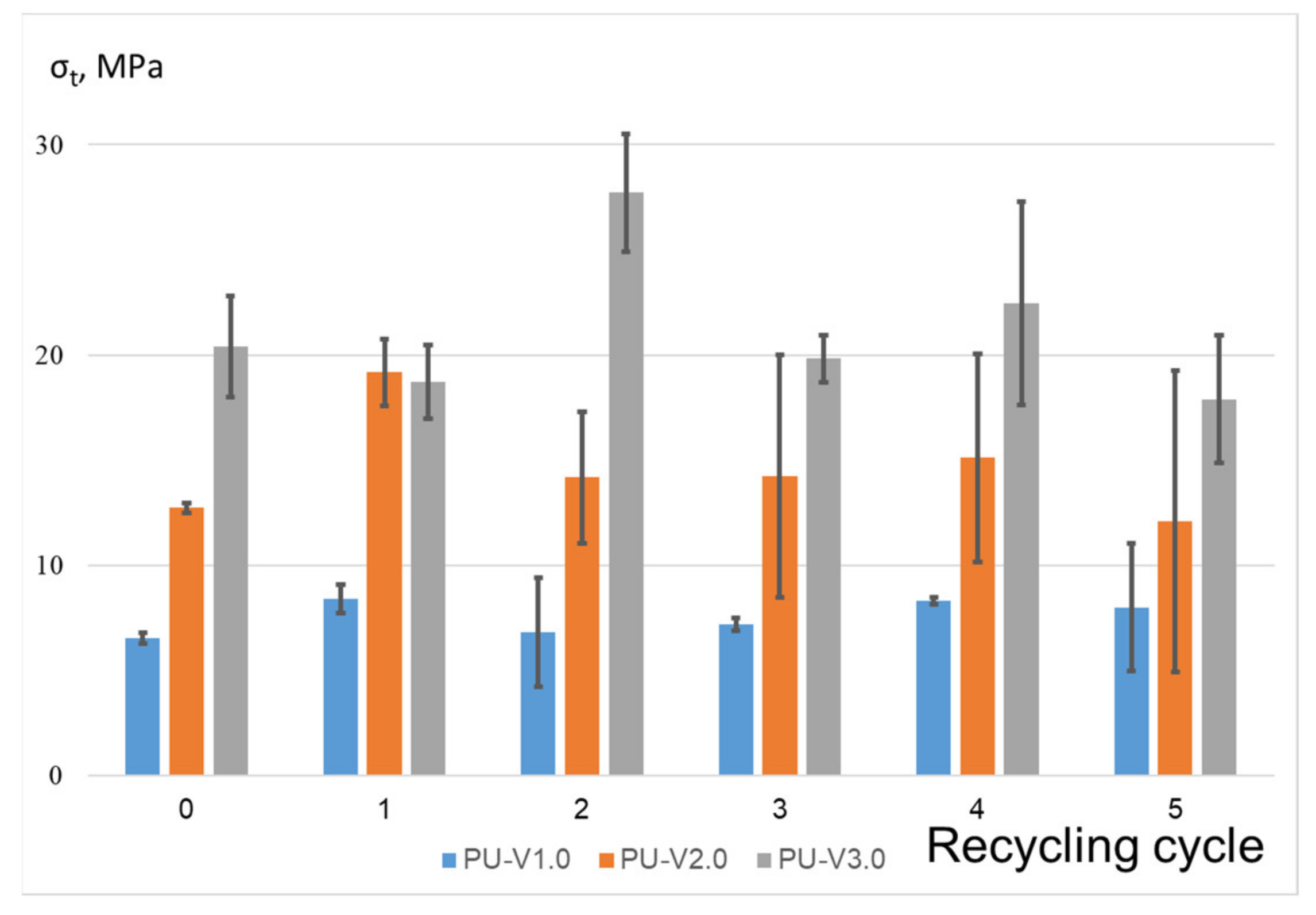
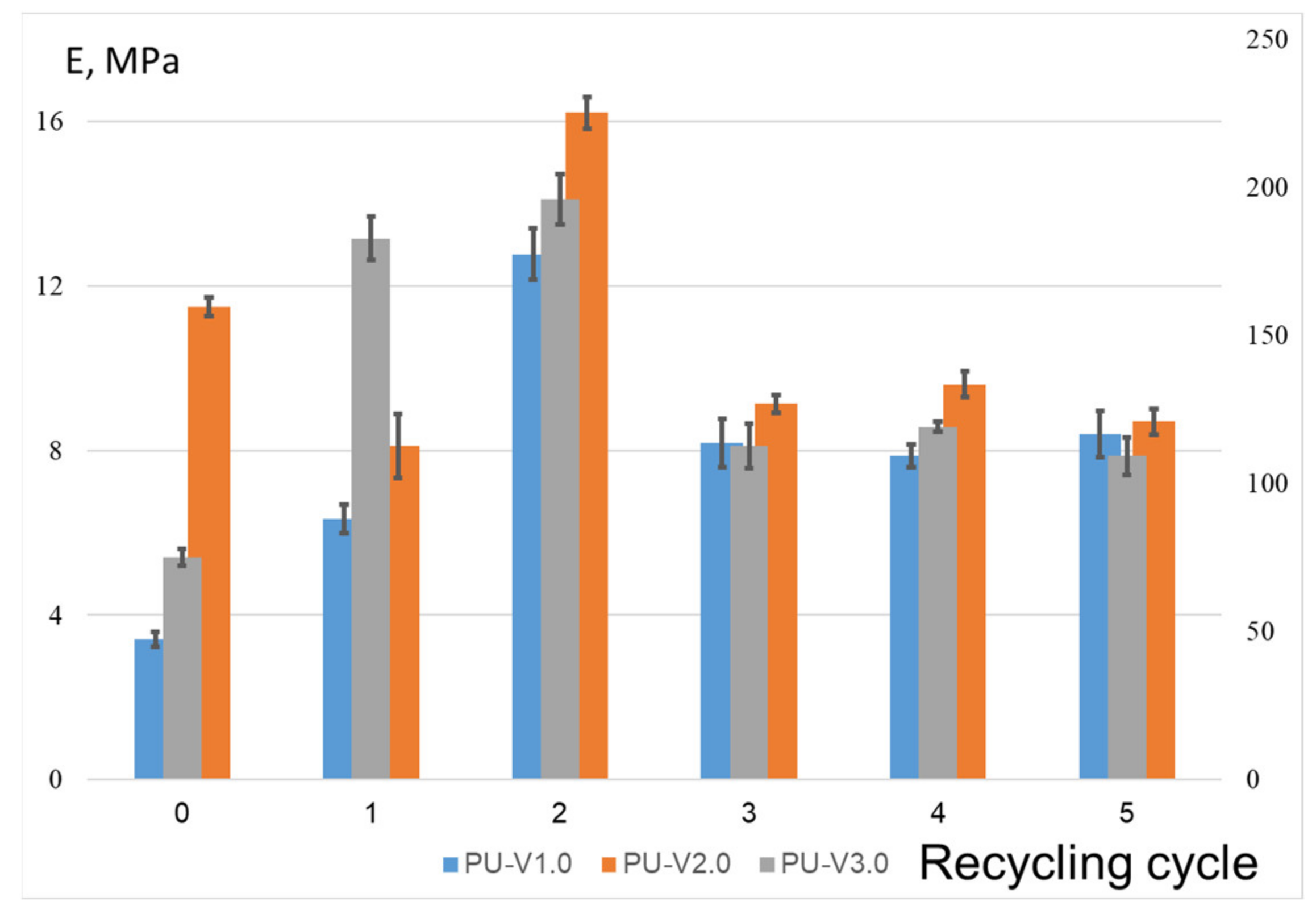
3.3. Mechanical Properties and Recycling Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane types, synthesis and applications—A review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef]
- Gama, N.V.; Ferreira, A.; Barros-Timmons, A. Polyurethane Foams: Past, Present, and Future. Materials 2018, 11, 1841. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Perkins, J.H.; Jackson, D.C.; Trammel, N.E.; Hunt, M.A.; Naskar, A.K. Development of lignin-based polyurethane thermoplastics. RSC Adv. 2013, 3, 21832–21840. [Google Scholar] [CrossRef]
- Kumari, S.; Chauhan, G.S.; Monga, S.; Kaushik, A.; Ahn, J.-H. New lignin-based polyurethane foam for wastewater treatment. RSC Adv. 2016, 6, 77768–77776. [Google Scholar] [CrossRef]
- Galbis, J.A.; García-Martín, M.G.; De Paz, M.V.; Galbis, E. Synthetic Polymers from Sugar-Based Monomers. Chem. Rev. 2016, 116, 1600–1636. [Google Scholar] [CrossRef]
- Begines, B.F.; Zamora, I.; Roffé, M.; Mancera, J.A. Sugar-based hydrophilic polyurethanes and polyureas. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 1953–1961. [Google Scholar] [CrossRef]
- Schneiderman, D.K.; Vanderlaan, M.E.; Mannion, A.M.; Panthani, T.R.; Batiste, D.C.; Wang, J.Z.; Bates, F.S.; Macosko, C.W.; Hillmyer, M.A. Chemically Recyclable Biobased Polyurethanes. ACS Macro Lett. 2016, 5, 515–518. [Google Scholar] [CrossRef]
- Shin, S.; Kim, H.; Liang, J.; Lee, S.; Lee, D. Sustainable rigid polyurethane foams based on recycled polyols from chemical recycling of waste polyurethane foams. J. Appl. Polym. Sci. 2019, 136, 47916. [Google Scholar] [CrossRef]
- Howard, G.T. Biodegradation of polyurethane: A review. Int. Biodeterior. Biodegrad. 2002, 49, 245–252. [Google Scholar] [CrossRef]
- Simón, D.; Borreguero, A.; de Lucas, A.; Rodríguez, J. Recycling of polyurethanes from laboratory to industry, a journey towards the sustainability. Waste Manag. 2018, 76, 147–171. [Google Scholar] [CrossRef]
- Gu, L.; Wu, Q.-Y. Recyclable bio-based crosslinked polyurethanes with self-healing ability. J. Appl. Polym. Sci. 2018, 135, 46272. [Google Scholar] [CrossRef]
- Zheng, K.; Tian, Y.; Fan, M.; Zhang, J.; Cheng, J. Recyclable, shape-memory, and self-healing soy oil-based polyurethane crosslinked by a thermoreversible Diels-Alder reaction. J. Appl. Polym. Sci. 2018, 135, 46049. [Google Scholar] [CrossRef]
- Vlad, S.C.; Ciobanu, D.; Macocinschi, D.; Filip, D.; Spiridon, I. Evaluation of some polyetherurethane elastomers for chemicals, oils and solvents resistance. J. Optoelectron. Adv. Mater. 2009, 11, 1160–1168. [Google Scholar]
- Li, K.; Wei, P.; Huang, J.; Xu, D.; Zhong, Y.; Hu, L.; Zhang, L.; Cai, J. Mechanically Strong Shape-Memory and Solvent-Resistant Double-Network Polyurethane/Nanoporous Cellulose Gel Nanocomposites. ACS Sustain. Chem. Eng. 2019, 7, 15974–15982. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Liao, X.; Fang, F.; Bai, K.; Qiao, L.; Wang, L. Renewable High-Performance Polyurethane Bioplastics Derived from Lignin–Poly(ε-caprolactone). ACS Sustain. Chem. Eng. 2017, 5, 4276–4284. [Google Scholar] [CrossRef]
- Chattopadhyay, D.; Raju, K. Structural engineering of polyurethane coatings for high performance applications. Prog. Polym. Sci. 2007, 32, 352–418. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, G.; Huang, T.; Yu, W.; Gao, Y. Synthesis and performance of polyurethane/silicon oxide nano-composite coatings. Sci. Eng. Compos. Mater. 2019, 26, 301–307. [Google Scholar] [CrossRef]
- Fortman, D.J.; Sheppard, D.T.; Dichtel, W.R. Reprocessing Cross-Linked Polyurethanes by Catalyzing Carbamate Exchange. Macromolecules 2019, 52, 6330–6335. [Google Scholar] [CrossRef]
- Bergman, S.D.; Wudl, F. Mendable polymers. J. Mater. Chem. 2007, 18, 41–62. [Google Scholar] [CrossRef]
- Datta, J.M.; Włoch, M. Chapter 14—Recycling of Polyurethanes. In Polyurethane Polymers; Thomas, S.J., Datta, J., Haponiuk, J.T., Reghunadhan, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 323–358. [Google Scholar]
- Zia, K.M.; Bhatti, H.N.; Bhatti, I.A. Methods for polyurethane and polyurethane composites, recycling and recovery: A review. React. Funct. Polym. 2007, 67, 675–692. [Google Scholar] [CrossRef]
- Fang, Y.; Du, X.; Jiang, Y.; Du, Z.; Pan, P.; Cheng, X.; Wang, H. Thermal-Driven Self-Healing and Recyclable Waterborne Polyurethane Films Based on Reversible Covalent Interaction. ACS Sustain. Chem. Eng. 2018, 6, 14490–14500. [Google Scholar] [CrossRef]
- Chen, S.; Wang, F.; Peng, Y.; Chen, T.; Wu, Q.; Sun, P. A Single Molecular Diels-Alder Crosslinker for Achieving Recyclable Cross-Linked Polymers. Macromol. Rapid Commun. 2015, 36, 1687–1692. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Polymer engineering based on reversible covalent chemistry: A promising innovative pathway towards new materials and new functionalities. Prog. Polym. Sci. 2018, 80, 39–93. [Google Scholar] [CrossRef]
- Bessonov, I.V.; Polezhaev, A.V.; Kuznetsova, M.N.; Nelub, V.A.; Buyanov, I.A.; Chudnov, I.V.; Borodulin, A.S. Rheological and thermal analysis of low-viscosity epoxy-furan composites. Polym. Sci. Ser. D 2013, 6, 308–311. [Google Scholar] [CrossRef]
- Bessonov, I.V.; Kopitsyna, M.N.; Polezhaev, A.V.; Nelyub, V.A. A mechanistic study of the reaction between furfural-acetone resins and polyamines. Polym. Sci. Ser. D 2016, 9, 17–21. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M.; Carvalho, A.; Trovatti, E. Progress of Polymers from Renewable Resources: Furans, Vegetable Oils, and Polysaccharides. Chem. Rev. 2016, 116, 1637–1669. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-L.; Chuo, T.-W. Self-healing polymers based on thermally reversible Diels–Alder chemistry. Polym. Chem. 2013, 4, 2194–2205. [Google Scholar] [CrossRef]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nutt, S.R.; Sheran, K.; Wudl, F. A Thermally Remendable Cross-Linked Polymeric Material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, X.; Zheng, Z.; Du, P. Polyether-maleimide-based crosslinked self-healing polyurethane with Diels-Alder bonds. J. Appl. Polym. Sci. 2015, 132, 19. [Google Scholar] [CrossRef]
- Platonova, E.O.; Vlasov, E.; Pavlov, A.A.; Kireynov, A.; Nelyub, V.A.; Polezhaev, A.V. Self-healing polyurethane based on a difuranic monomer from biorenewable source. J. Appl. Polym. Sci. 2019, 136, 47869. [Google Scholar] [CrossRef]
- Zhang, C.; Madbouly, S.A.; Kessler, M. Biobased Polyurethanes Prepared from Different Vegetable Oils. ACS Appl. Mater. Interfaces 2015, 7, 1226–1233. [Google Scholar] [CrossRef]
- Furtwengler, P.; Avérous, L. Renewable polyols for advanced polyurethane foams from diverse biomass resources. Polym. Chem. 2018, 9, 4258–4287. [Google Scholar] [CrossRef]
- Khanderay, J.C.; Gite, V.V. Fully biobased polyester polyols derived from renewable resources toward preparation of polyurethane and their application for coatings. J. Appl. Polym. Sci. 2019, 136, 47558. [Google Scholar] [CrossRef]
- Du, P.; Wu, M.; Liu, X.; Zheng, Z.; Wang, X.; Sun, P.; Joncheray, T.; Zhang, Y. Synthesis of linear polyurethane bearing pendant furan and cross-linked healable polyurethane containing Diels–Alder bonds. N. J. Chem. 2014, 38, 770–776. [Google Scholar] [CrossRef]
- Du, P.; Jia, H.; Chen, Q.; Zheng, Z.; Wang, X.; Chen, D. Slightly crosslinked polyurethane with Diels-Alder adducts from trimethylolpropane. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Montano, V.; Wempe, M.M.B.; Does, S.M.H.; Bijleveld, J.C.; Van Der Zwaag, S.; Garcia, S.J. Controlling Healing and Toughness in Polyurethanes by Branch-Mediated Tube Dilation. Macromolecules 2019, 52, 8067–8078. [Google Scholar] [CrossRef]
- Feng, L.; Yu, Z.; Bian, Y.; Lu, J.; Shi, X.; Chai, C. Self-healing behavior of polyurethanes based on dual actions of thermo-reversible Diels-Alder reaction and thermal movement of molecular chains. Polymer 2017, 124, 48–59. [Google Scholar] [CrossRef]
- Pikhurov, D.V.; Zuev, V.V. Kinetics of formation of microstructure in polyurethane foams infused with micro and nanosized carbonaceous fillers. Polym. Eng. Sci. 2019, 59, 941–948. [Google Scholar] [CrossRef]
- Höhne, C.-C.; Hanich, R.; Kroke, E. Intrinsic flame resistance of polyurethane flexible foams: Unexpectedly low flammability without any flame retardant. Fire Mater. 2018, 42, 394–402. [Google Scholar] [CrossRef]
- Arshad, M.; Saied, S.; Ullah, A. PEG–lipid telechelics incorporating fatty acids from canola oil: Synthesis, characterization and solution self-assembly. RSC Adv. 2014, 4, 26439–26446. [Google Scholar] [CrossRef]
- Szycher, M. Structure–Property Relations in Polyurethanes. In Handbook of Polyurethanes; Szycher, M., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 41–85. [Google Scholar]
- Hunter, D.H.; Sim, S.K. 2,4-Diazapentadienes. I. Prototropy, Cyclization, and Addition–Elimination. Can. J. Chem. 1972, 50, 669–677. [Google Scholar] [CrossRef]
- O’Platonova, E.; Vlasov, E.; Kireynov, A.; Polezhaev, A.V. Synthesis of cross-linked polyurethane with self-healing properties. Mater. Sci. Eng. 2019, 683, 012001. [Google Scholar] [CrossRef]
- Tan, C.; Tirri, T.; Wilen, C.E. Investigation on the Influence of Chain Extenders on the Performance of One-Component Moisture-Curable Polyurethane Adhesives. Polymers 2017, 9, 184. [Google Scholar] [CrossRef]
- Goussé, C.; Gandini, A.; Hodge, P. Application of the Diels–Alder Reaction to Polymers Bearing Furan Moieties. 2. Diels–Alder and Retro-Diels–Alder Reactions Involving Furan Rings in Some Styrene Copolymers. Macromolecules 1998, 31, 314–321. [Google Scholar] [CrossRef]
- Petrova, T.V.; Solodilov, V.I.; Kabantseva, E.V.; Karelina, N.V.; Polezhaev, A.V. Furfurylglycidyl ether: A new effective active diluent for epoxy resins from bio-renewable raw materials. Mater. Sci. Eng. 2019, 683, 012070. [Google Scholar] [CrossRef]
- Du, P.; Liu, X.; Zheng, Z.; Wang, X.; Joncheray, T.; Zhang, Y. Synthesis and characterization of linear self-healing polyurethane based on thermally reversible Diels–Alder reaction. RSC Adv. 2013, 3, 15475–15482. [Google Scholar] [CrossRef]
- Froidevaux, V.; Borne, M.; Laborbe, E.; Auvergne, R.; Gandini, A.; Boutevin, B. Study of the Diels-Alder and retro-Diels-Alder reaction between furan derivatives and maleimide for the creation of new materials. RSC Adv. 2015, 5, 37742–37754. [Google Scholar] [CrossRef]
- Cuvellier, A.; Verhelle, R.; Brancart, J.; Vanderborght, B.; Van Assche, G.; Rahier, H. The influence of stereochemistry on the reactivity of the Diels–Alder cycloaddition and the implications for reversible network polymerization. Polym. Chem. 2018, 10, 473–485. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, R.; Wu, Q.; Chen, T.; Sun, P. Bio-Inspired High-Performance and Recyclable Cross-Linked Polymers. Adv. Mater. 2013, 25, 4912–4917. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Td5% | Td10% | Tdmax | Residue (wt%) |
|---|---|---|---|---|
| PU-V1.0 | 267 | 300 | 380 | 14.1 |
| PU-V2.0 | 279 | 307 | 382 | 16.8 |
| PU-V3.0 | 285 | 330 | 383 | 25.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Platonova, E.; Chechenov, I.; Pavlov, A.; Solodilov, V.; Afanasyev, E.; Shapagin, A.; Polezhaev, A. Thermally Remendable Polyurethane Network Cross-Linked via Reversible Diels–Alder Reaction. Polymers 2021, 13, 1935. https://doi.org/10.3390/polym13121935
Platonova E, Chechenov I, Pavlov A, Solodilov V, Afanasyev E, Shapagin A, Polezhaev A. Thermally Remendable Polyurethane Network Cross-Linked via Reversible Diels–Alder Reaction. Polymers. 2021; 13(12):1935. https://doi.org/10.3390/polym13121935
Chicago/Turabian StylePlatonova, Elena, Islam Chechenov, Alexander Pavlov, Vitaliy Solodilov, Egor Afanasyev, Alexey Shapagin, and Alexander Polezhaev. 2021. "Thermally Remendable Polyurethane Network Cross-Linked via Reversible Diels–Alder Reaction" Polymers 13, no. 12: 1935. https://doi.org/10.3390/polym13121935
APA StylePlatonova, E., Chechenov, I., Pavlov, A., Solodilov, V., Afanasyev, E., Shapagin, A., & Polezhaev, A. (2021). Thermally Remendable Polyurethane Network Cross-Linked via Reversible Diels–Alder Reaction. Polymers, 13(12), 1935. https://doi.org/10.3390/polym13121935