Protein Unfolding and Aggregation near a Hydrophobic Interface



Abstract
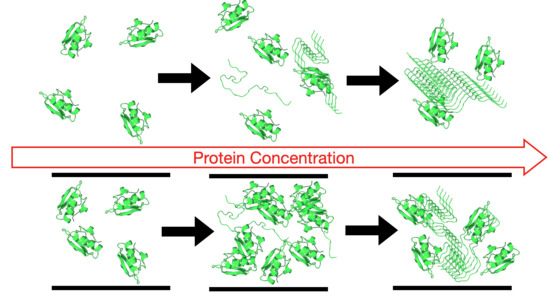
1. Introduction
2. Model
2.1. Franzese–Stanley Water Model
2.2. Protein and Interface Model
2.3. The Hydrophobic Surface
2.4. The Model’s Parameters
3. Method
3.1. The Protein
3.2. The Monte Carlo Simulation
- We choose randomly a global protein-move among shift, rotation, crankshaft, or pivot [51]. Then, we pick at random one of the proteins, and we attempt the selected global move. We repeat the random selection for times, updating on average all the proteins.
- We choose a random number m between 1 and . For m times, we select one of the cells. If it includes an amino acid, we attempt a corner flip, i.e., the local protein-move [51]. If it includes a water molecule, we select one of its four -variables and attempt to change its state, hence, breaking or forming a HB.
- We attempt a global change of the system volume.
3.3. The Observables
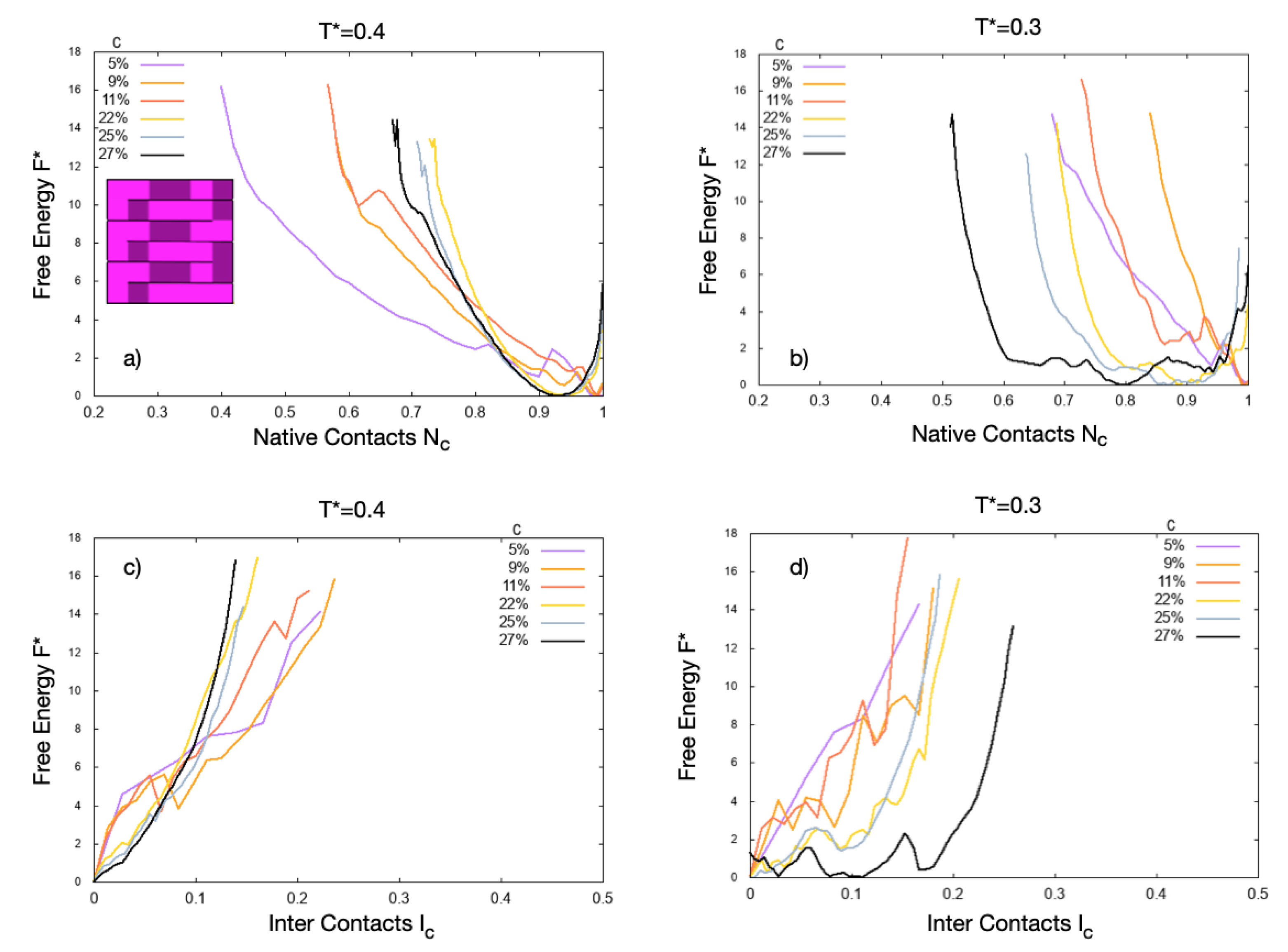
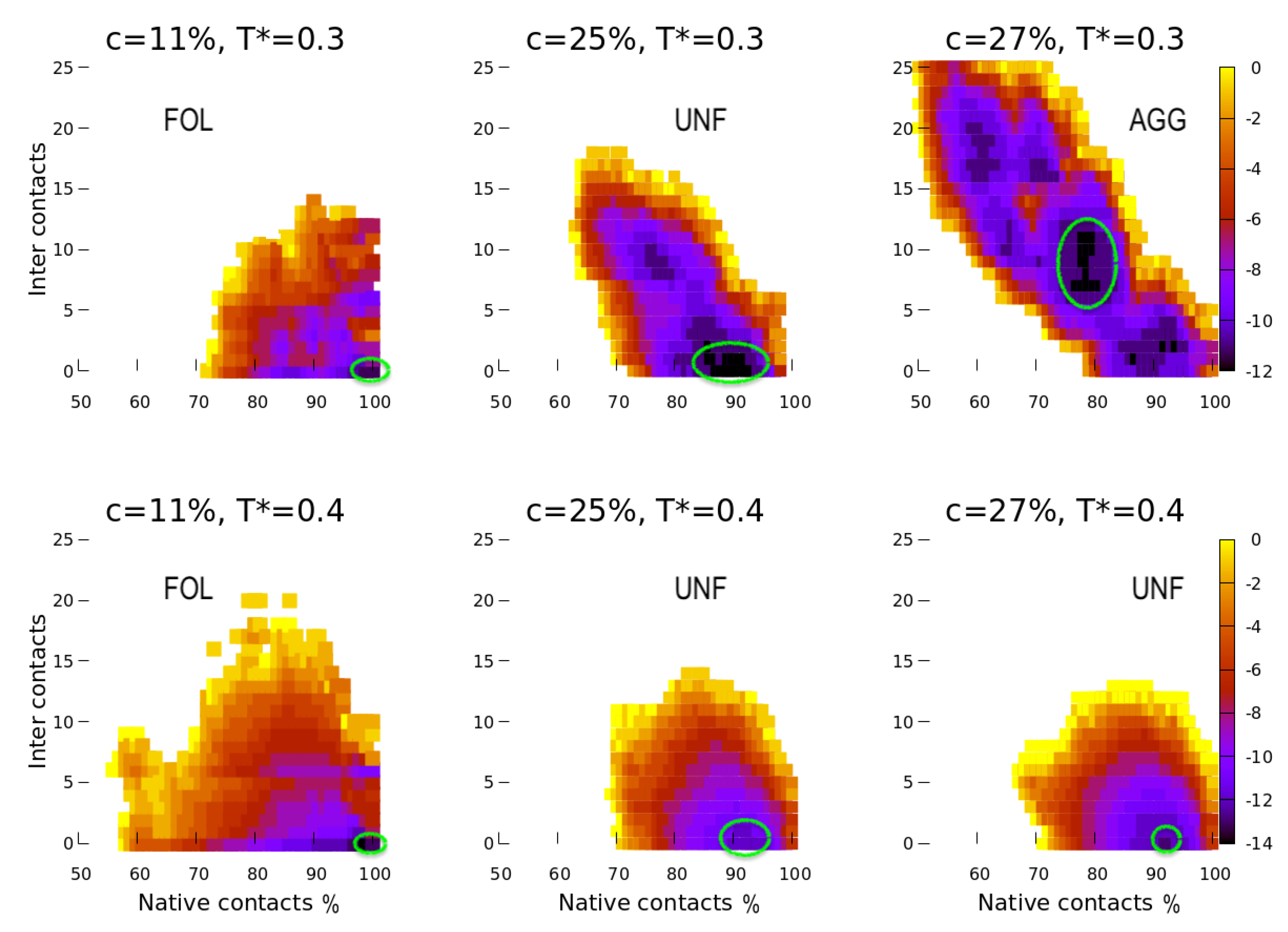
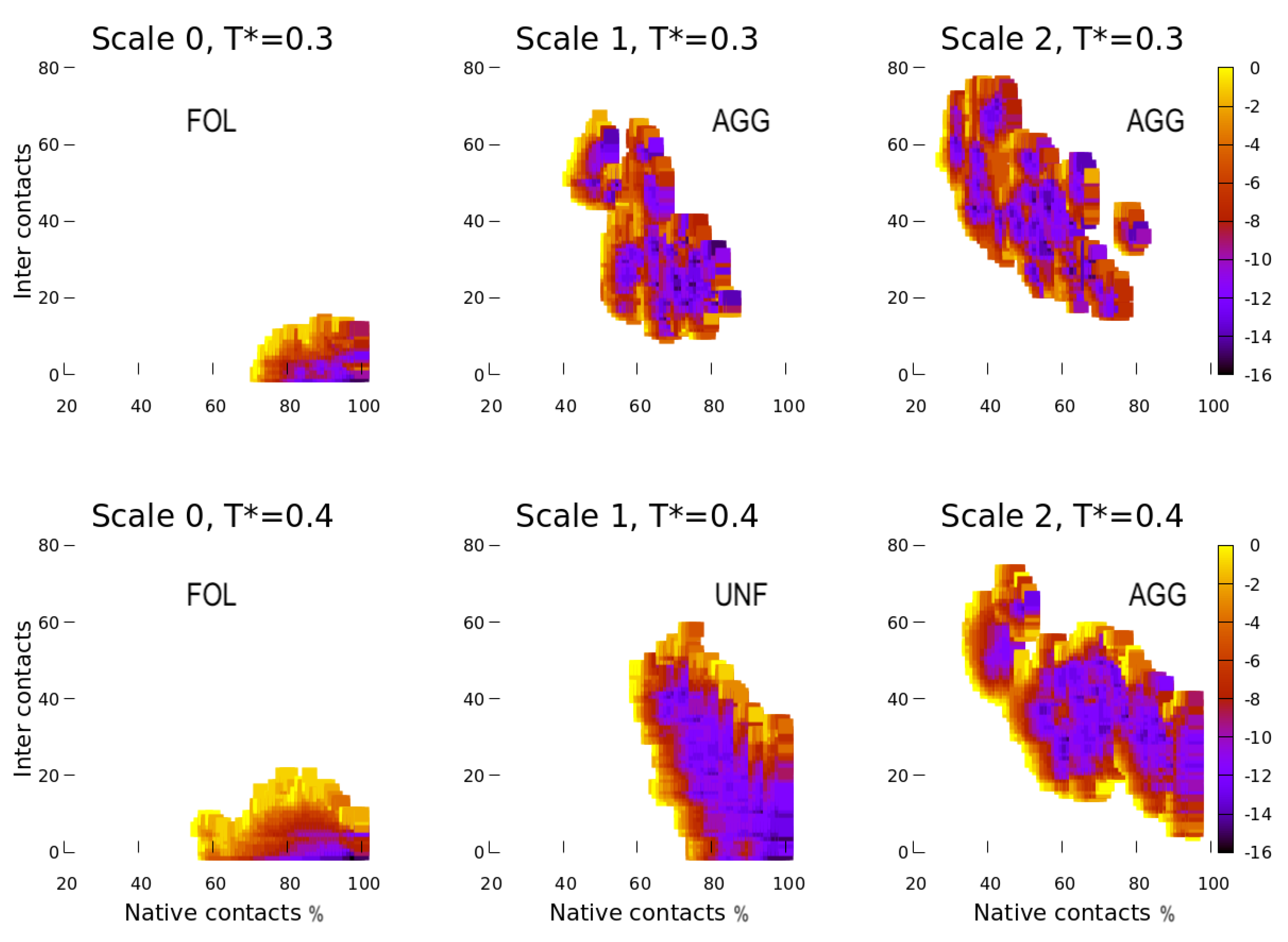
4. Results
4.1. Scale 0
4.2. Scale 1 and Scale 2
5. Discussion
5.1. Effect of the Hydrophobic Walls
5.2. Effect of the Temperature
5.3. Effect of the HB Strength near a Hydrophobic Interface
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGG | Aggregated |
| FOL | Folded |
| HB | Hydrogen Bond |
| MC | Monte Carlo |
| PBC | Periodic Boundary Conditions |
| UNF | Unfolded |
| Hydrophobic | |
| Hydrophilic | |
| Mixed |
References
- Finkelstein, A.V.; Ptitsyn, O.B. Protein Physics, 2nd ed.; Academic Press: Amsterdam, The Netherlands, 2016. [Google Scholar] [CrossRef]
- Kiefhaber, T.; Rudolph, R.; Kohler, H.H.; Buchner, J. Protein Aggregation in vitro and in vivo: A Quantitative Model of the Kinetic Competition between Folding and Aggregation. BioTechnology 1991, 9, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Chiba, K.; Tsuruta, H.; Doniach, S.; Hodgson, K.O.; Kihara, H. Evidence of an associative intermediate on the myoglobin refolding pathway. Biophys. J. 1993, 65, 912–917. [Google Scholar] [CrossRef][Green Version]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef]
- Roberts, C.J. Non-native protein aggregation kinetics. Biotechnol. Bioeng. 2007, 98, 927–938. [Google Scholar] [CrossRef]
- Neudecker, P.; Robustelli, P.; Cavalli, A.; Walsh, P.; Lundström, P.; Zarrine-Afsar, A.; Sharpe, S.; Vendruscolo, M.; Kay, L.E. Structure of an Intermediate State in Protein Folding and Aggregation. Science 2012, 336, 362–366. [Google Scholar] [CrossRef]
- De Simone, A.; Kitchen, C.; Kwan, A.H.; Sunde, M.; Dobson, C.M.; Frenkel, D. Intrinsic disorder modulates protein self-assembly and aggregation. Proc. Natl. Acad. Sci. USA 2012, 109, 6951–6956. [Google Scholar] [CrossRef]
- Tartaglia, G.G.; Vendruscolo, M. Correlation between mRNA expression levels and protein aggregation propensities in subcellular localisations. Mol. BioSyst. 2009, 5, 1873–1876. [Google Scholar] [CrossRef]
- Schröder, M.; Schäfer, R.; Friedl, P. Induction of protein aggregation in an early secretory compartment by elevation of expression level. Biotechnol. Bioeng. 2002, 78, 131–140. [Google Scholar] [CrossRef]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 2007, 32, 204–206. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Opinion: What is the role of protein aggregation in neurodegeneration? Nat. Reviews. Mol. Cell Biol. 2005, 6, 891–898. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Reviews. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Protein aggregation and its impact on product quality. Curr. Opin. Biotechnol. 2014, 30, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Ahmad, E.; Qadeer, A.; Rabbani, G.; Khan, R.H. Nanoparticles in relation to peptide and protein aggregation. Int. J. Nanomed. 2014, 9, 899–912. [Google Scholar] [CrossRef]
- Howes, P.D.; Chandrawati, R.; Stevens, M.M. Colloidal nanoparticles as advanced biological sensors. Science 2014, 346. [Google Scholar] [CrossRef]
- Vilanova, O.; Mittag, J.J.; Kelly, P.M.; Milani, S.; Dawson, K.A.; Rädler, J.O.; Franzese, G. Understanding the Kinetics of Protein–Nanoparticle Corona Formation. ACS Nano 2016, 10, 10842–10850. [Google Scholar] [CrossRef]
- Meesaragandla, B.; García, I.; Biedenweg, D.; Toro-Mendoza, J.; Coluzza, I.; Liz-Marzán, L.M.; Delcea, M. H-Bonding-mediated binding and charge reorganization of proteins on gold nanoparticles. Phys. Chem. Chem. Phys. 2020, 22, 4490–4500. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef]
- Jiao, Q.; Li, L.; Mu, Q.; Zhang, Q. Immunomodulation of Nanoparticles in Nanomedicine Applications. BioMed Res. Int. 2014, 2014, 426028. [Google Scholar] [CrossRef]
- Höök, F.; Rodahl, M.; Kasemo, B.; Brzezinski, P. Structural changes in hemoglobin during adsorption to solid surfaces: Effects of pH, ionic strength, and ligand binding. Proc. Natl. Acad. Sci. USA 1998, 95, 12271. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Dupont-Gillain, C.; Bertrand, P. Conformation Change of Albumin Adsorbed on Polycarbonate Membranes as Revealed by ToF-SIMS. Langmuir 2003, 19, 6271–6276. [Google Scholar] [CrossRef]
- Lacerda, S.H.D.P.; Park, J.J.; Meuse, C.; Pristinski, D.; Becker, M.L.; Karim, A.; Douglas, J.F. Interaction of Gold Nanoparticles with Common Human Blood Proteins. ACS Nano 2010, 4, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Cañaveras, F.; Madueño, R.; Sevilla, J.; Blázquez, M.; Pineda, T. Role of the Functionalization of the Gold Nanoparticle Surface on the Formation of Bioconjugates with Human Serum Albumin. J. Phys. Chem. C 2012, 116, 10430–10437. [Google Scholar] [CrossRef]
- Zhang, C.; Myers, J.N.; Chen, Z. Elucidation of molecular structures at buried polymer interfaces and biological interfaces using sum frequency generation vibrational spectroscopy. Soft Matter 2013, 9, 4738–4761. [Google Scholar] [CrossRef]
- Wei, S.; Ahlstrom, L.S.; Brooks III, C.L. Exploring Protein–Nanoparticle Interactions with Coarse-Grained Protein Folding Models. Small 2017, 13, 1603748. [Google Scholar] [CrossRef]
- Cellmer, T.; Bratko, D.; Prausnitz, J.M.; Blanch, H.W. Protein aggregation in silico. Trends Biotechnol. 2007, 25, 254–261. [Google Scholar] [CrossRef]
- Nasica-Labouze, J.; Nguyen, P.H.; Sterpone, F.; Berthoumieu, O.; Buchete, N.V.; Coté, S.; De Simone, A.; Doig, A.J.; Faller, P.; Garcia, A.; et al. Amyloid β Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev. 2015, 115, 3518–3563. [Google Scholar] [CrossRef]
- Bianco, V.; Franzese, G.; Coluzza, I. In Silico Evidence That Protein Unfolding is a Precursor of Protein Aggregation. ChemPhysChem 2020, 21, 377–384. [Google Scholar] [CrossRef]
- Bianco, V.; Alonso-Navarro, M.; Di Silvio, D.; Moya, S.; Cortajarena, A.L.; Coluzza, I. Proteins are Solitary! Pathways of Protein Folding and Aggregation in Protein Mixtures. J. Phys. Chem. Lett. 2019, 10, 4800–4804. [Google Scholar] [CrossRef]
- Franzese, G.; Bianco, V.; Iskrov, S. Water at Interface with Proteins. Food Biophys. 2011, 6, 186–198. [Google Scholar] [CrossRef]
- Bianco, V.; Iskrov, S.; Franzese, G. Understanding the role of hydrogen bonds in water dynamics and protein stability. J. Biol. Phys. 2012, 38, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Franzese, G.; Bianco, V. Water at Biological and Inorganic Interfaces. Food Biophys. 2013, 8, 153–169. [Google Scholar] [CrossRef]
- Bianco, V.; Franzese, G. Contribution of Water to Pressure and Cold Denaturation of Proteins. Phys. Rev. Lett. 2015, 115, 108101. [Google Scholar] [CrossRef]
- Bianco, V.; Pagès-Gelabert, N.; Coluzza, I.; Franzese, G. How the stability of a folded protein depends on interfacial water properties and residue-residue interactions. J. Mol. Liq. 2017, 245, 129–139. [Google Scholar] [CrossRef]
- Bianco, V.; Franzese, G.; Dellago, C.; Coluzza, I. Role of Water in the Selection of Stable Proteins at Ambient and Extreme Thermodynamic Conditions. Phys. Rev. X 2017, 7, 021047. [Google Scholar] [CrossRef]
- Franzese, G.; Stokely, K.; Chu, X.Q.; Kumar, P.; Mazza, M.G.; Chen, S.H.; Stanley, H.E. Pressure effects in supercooled water: Comparison between a 2D model of water and experiments for surface water on a protein. J. Phys. Condens. Matter 2008, 20, 494210. [Google Scholar] [CrossRef]
- Stokely, K.; Mazza, M.G.; Stanley, H.E.; Franzese, G. Effect of hydrogen bond cooperativity on the behavior of water. Proc. Natl. Acad. Sci. USA 2010, 107, 1301–1306. [Google Scholar] [CrossRef]
- Mazza, M.G.; Stokely, K.; Pagnotta, S.E.; Bruni, F.; Stanley, H.E.; Franzese, G. More than one dynamic crossover in protein hydration water. Proc. Natl. Acad. Sci. USA 2011, 108, 19873–19878. [Google Scholar] [CrossRef]
- De los Santos, F.; Franzese, G. Understanding Diffusion and Density Anomaly in a Coarse-Grained Model for Water Confined between Hydrophobic Walls. J. Phys. Chem. B 2011, 115, 14311–14320. [Google Scholar] [CrossRef]
- Bianco, V.; Franzese, G. Critical behavior of a water monolayer under hydrophobic confinement. Sci. Rep. 2014, 4, 4440. [Google Scholar] [CrossRef] [PubMed]
- Bianco, V.; Franzese, G. Hydrogen bond correlated percolation in a supercooled water monolayer as a hallmark of the critical region. J. Mol. Liq. 2019, 285, 727–739. [Google Scholar] [CrossRef]
- Gopinadhan, K.; Hu, S.; Esfandiar, A.; Lozada-Hidalgo, M.; Wang, F.C.; Yang, Q.; Tyurnina, A.V.; Keerthi, A.; Radha, B.; Geim, A.K. Complete steric exclusion of ions and proton transport through confined monolayer water. Science 2019, 363, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Calero, C.; Franzese, G. Water under extreme confinement in graphene: Oscillatory dynamics, structure, and hydration pressure explained as a function of the confinement width. J. Mol. Liq. 2020, 317, 114027. [Google Scholar] [CrossRef]
- Coronas, L.E.; Bianco, V.; Zantop, A.; Franzese, G. Liquid-Liquid Critical Point in 3D Many-Body Water Model. arXiv 2016, arXiv:1610.00419. [Google Scholar]
- Luzar, A.; Chandler, D. Effect of Environment on Hydrogen Bond Dynamics in Liquid Water. Phys. Rev. Lett. 1996, 76, 928–931. [Google Scholar] [CrossRef]
- Coronas, L.E.; Vilanova, O.; Bianco, V.; de los Santos, F.; Franzese, G. Properties of Water from Numerical and Experimental Perspectives; Chapter The Franzese-Stanley Coarse Grained Model for HydrationWater; Martelli, F., Ed.; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar]
- Miyazawa, S.; Jernigan, R.L. Estimation of effective interresidue contact energies from protein crystal structures: Quasi-chemical approximation. Macromolecules 1985, 18, 534–552. [Google Scholar] [CrossRef]
- Sarupria, S.; Garde, S. Quantifying Water Density Fluctuations and Compressibility of Hydration Shells of Hydrophobic Solutes and Proteins. Phys. Rev. Lett. 2009, 103, 037803. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understand Molecular Simulations; Academic Press: San Diego, CA, USA; London, UK, 2002. [Google Scholar]
- Brini, E.; Simmerling, C.; Dill, K. Protein storytelling through physics. Science 2020, 370. [Google Scholar] [CrossRef]
- Mathieu, C.; Pappu, R.V.; Taylor, J.P. Beyond aggregation: Pathological phase transitions in neurodegenerative disease. Science 2020, 370, 56. [Google Scholar] [CrossRef]
- Vilanova, O.; Bianco, V.; Franzese, G. Multi-Scale Approach for Self-Assembly and Protein Folding. In Design of Self-Assembling Materials; Coluzza, I., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 107–128. [Google Scholar] [CrossRef][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scale | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 0.48 | ||||||
| 1 | 2 | 4 | 0.24 | ||||||
| 2 | 2 | 4 | 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
March, D.; Bianco, V.; Franzese, G. Protein Unfolding and Aggregation near a Hydrophobic Interface. Polymers 2021, 13, 156. https://doi.org/10.3390/polym13010156
March D, Bianco V, Franzese G. Protein Unfolding and Aggregation near a Hydrophobic Interface. Polymers. 2021; 13(1):156. https://doi.org/10.3390/polym13010156
Chicago/Turabian StyleMarch, David, Valentino Bianco, and Giancarlo Franzese. 2021. "Protein Unfolding and Aggregation near a Hydrophobic Interface" Polymers 13, no. 1: 156. https://doi.org/10.3390/polym13010156
APA StyleMarch, D., Bianco, V., & Franzese, G. (2021). Protein Unfolding and Aggregation near a Hydrophobic Interface. Polymers, 13(1), 156. https://doi.org/10.3390/polym13010156