Polyaniline Based Pt-Electrocatalyst for a Proton Exchanged Membrane Fuel Cell



 ,
,
Abstract
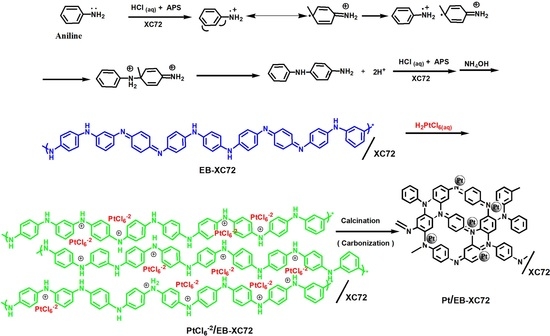
1. Introduction
2. Material and Methods
2.1. Materials
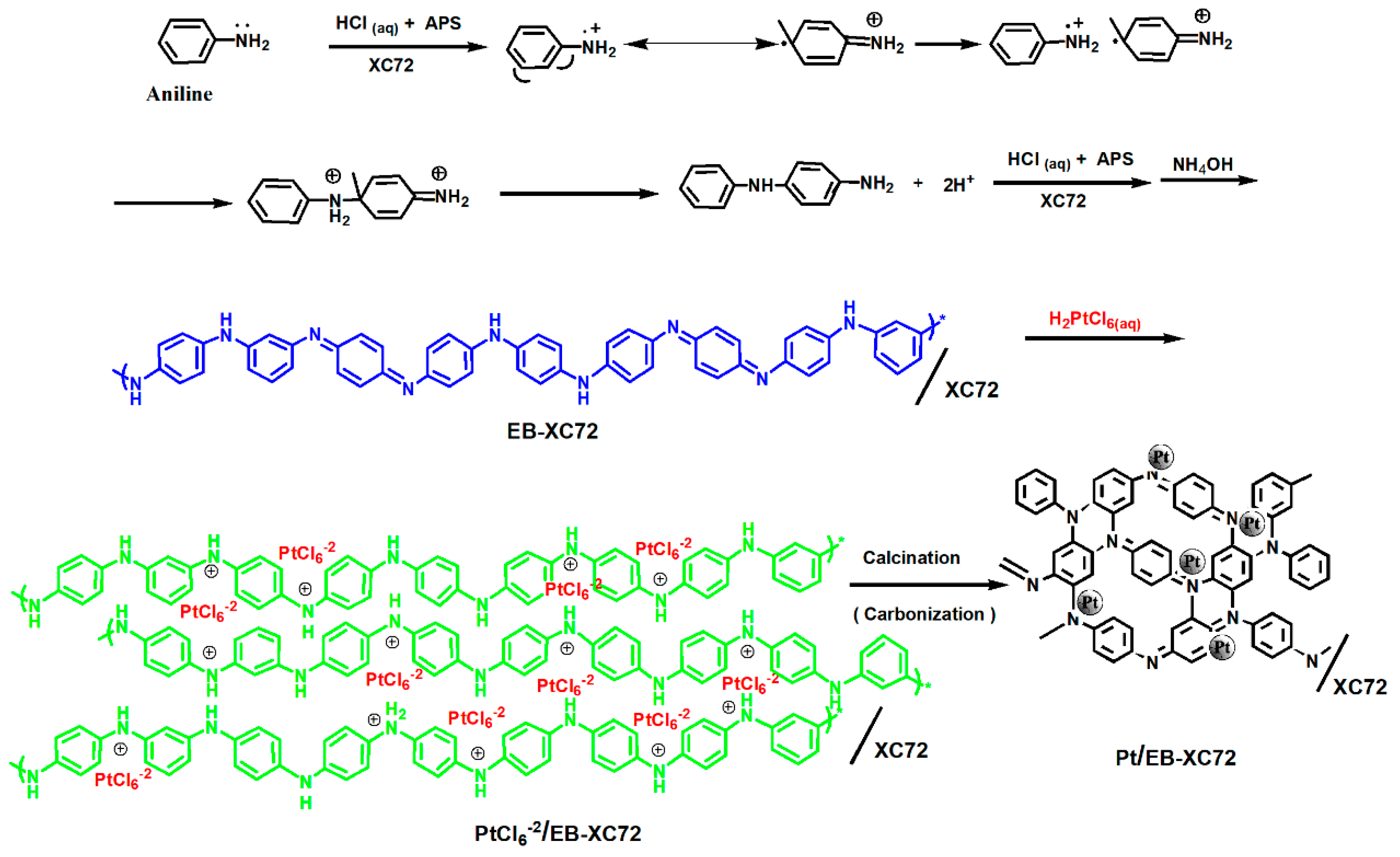
2.2. Preparation of EB-XC72
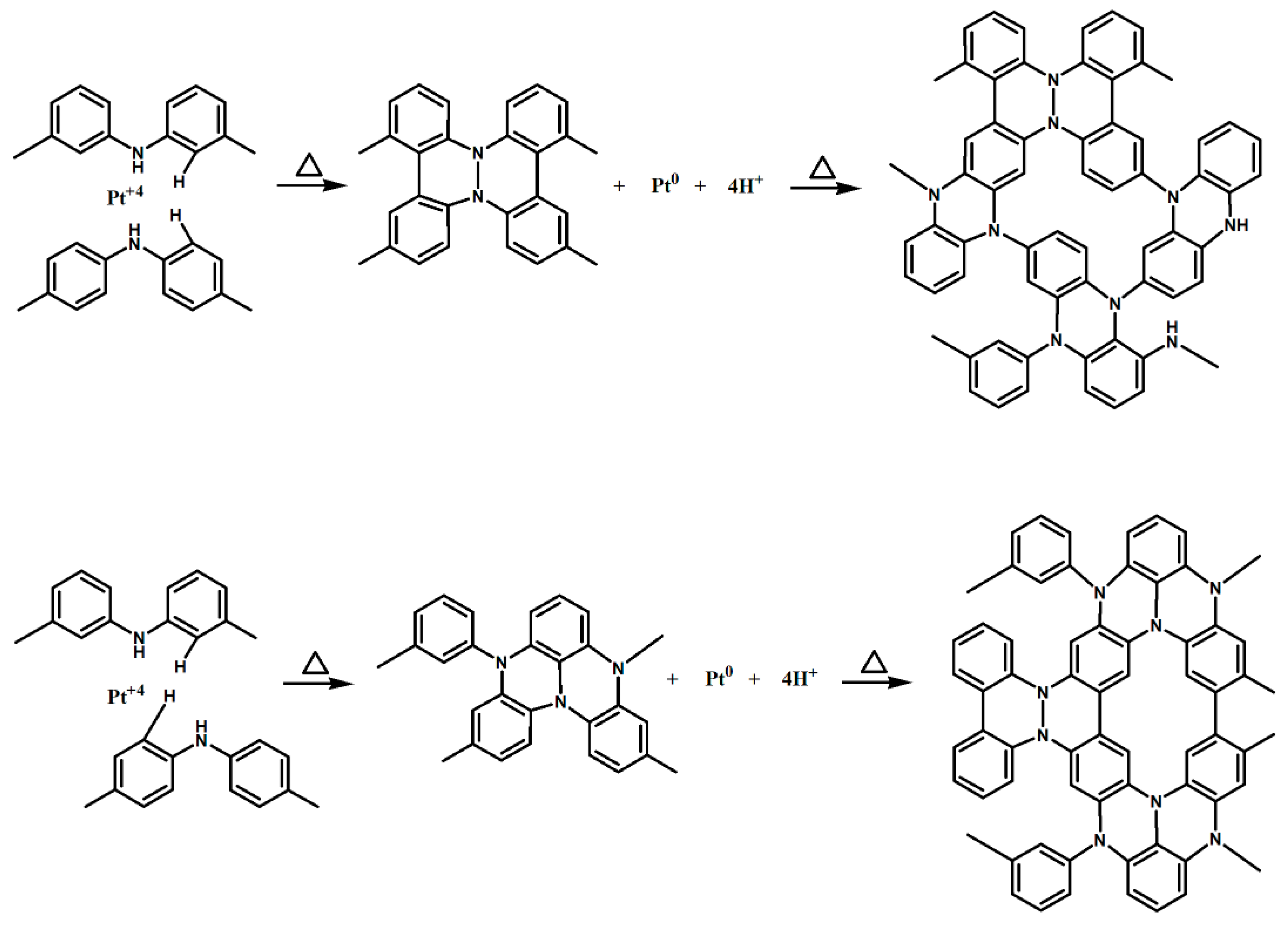
2.3. Preparation of Pt/XC72 and Pt/EB-XC72
2.4. Characterization
2.4.1. WXRD (Wide Angle X-Ray Diffraction)
2.4.2. TEM (Transmission Electron Microscopy)
2.4.3. SEM (Scanning Electron Microscopy)
2.4.4. TGA (Thermogravimetric Analysis)
2.4.5. Raman Spectroscopy
2.4.6. EDS (Energy Dispersive X-Ray Spectra)
2.5. Electrochemical Characterization
2.6. MEA Preparation
2.7. Single-Cell Performance Testing
3. Results and Discussion
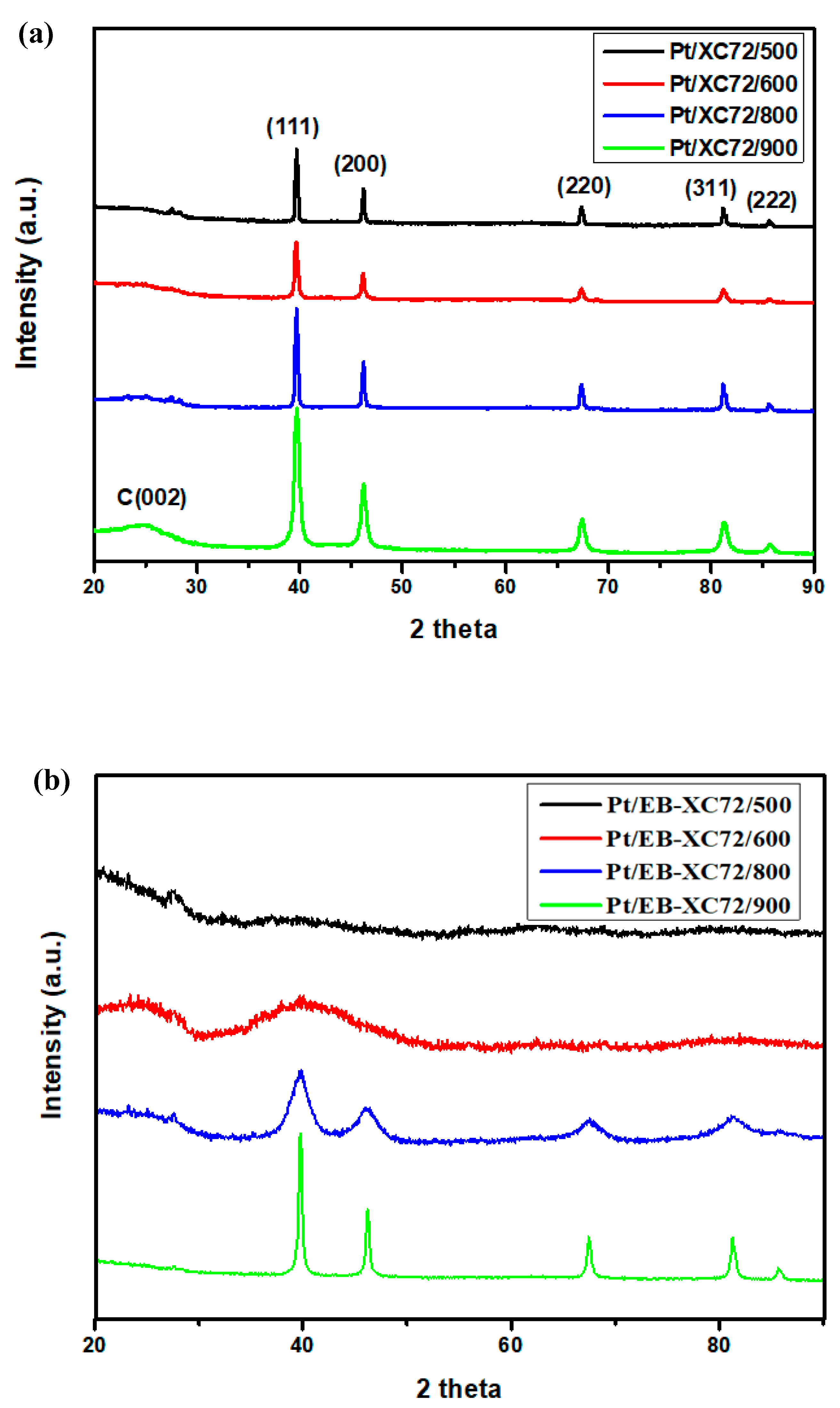
3.1. XRD Pattern of the Electrocatalyst Electrode Materials
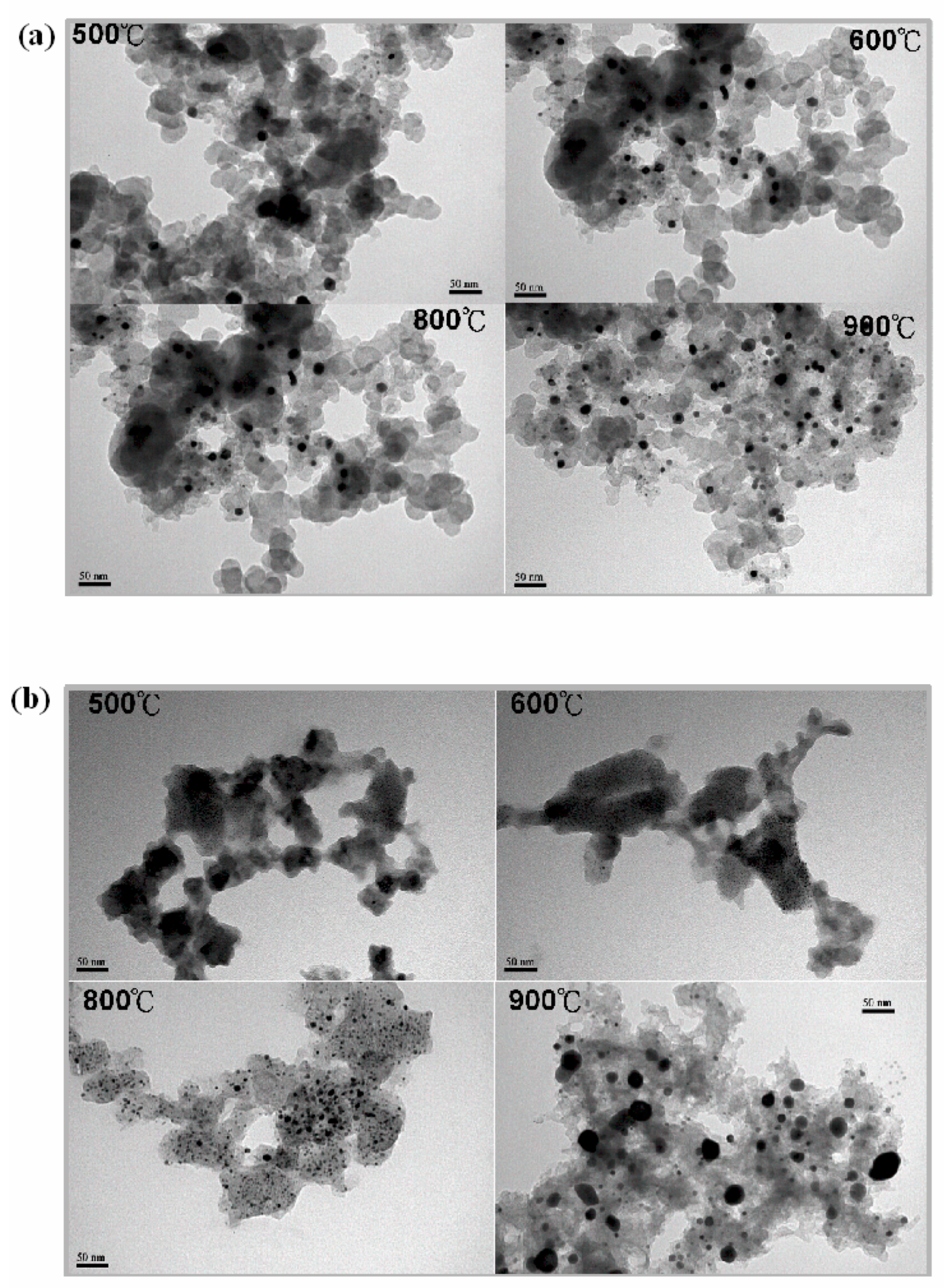
3.2. Transmission and Scanning Electron Microscopy (TEM and SEM)
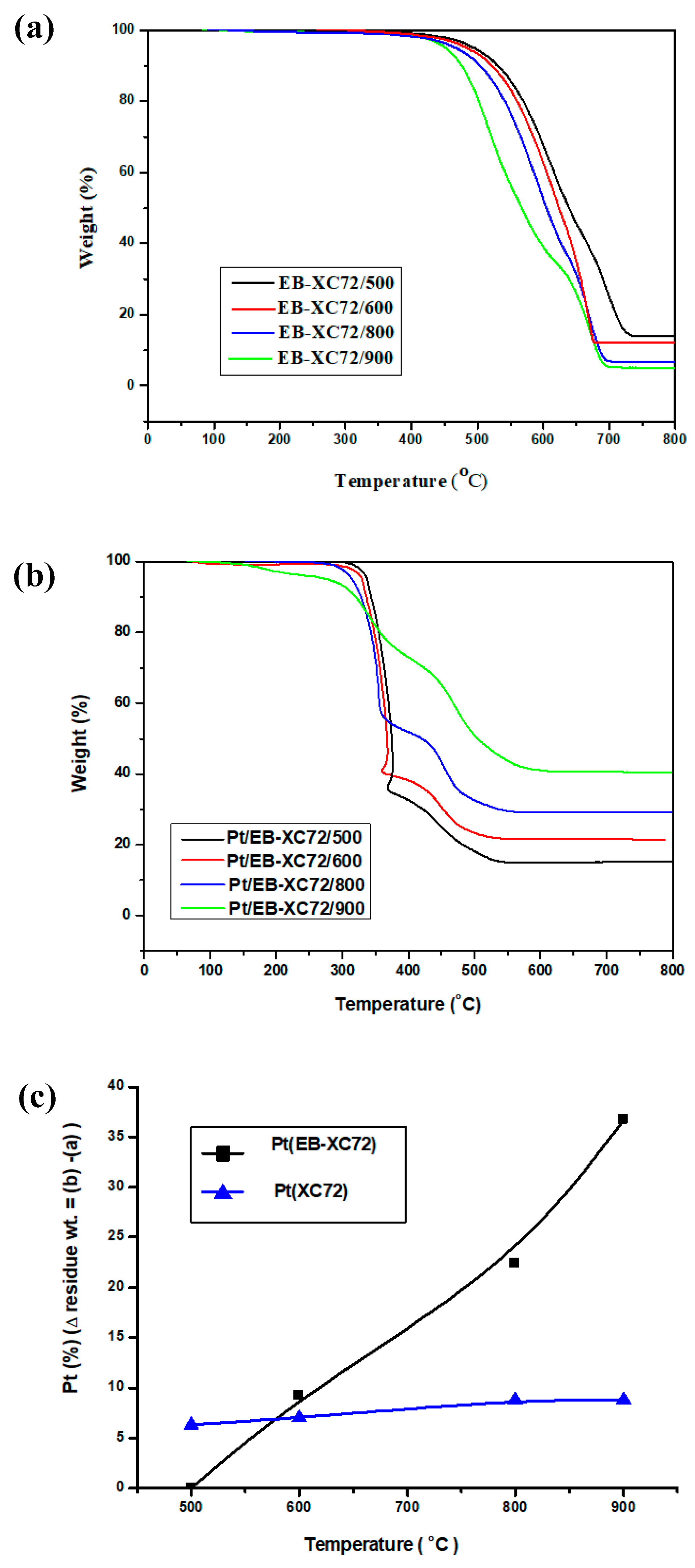
3.3. Thermo Gravimetric Analysis (TGA)
3.4. Raman Spectroscopy
3.5. Energy Dispersive X-Ray Spectroscopy (EDS)
3.6. Electrochemical Analysis
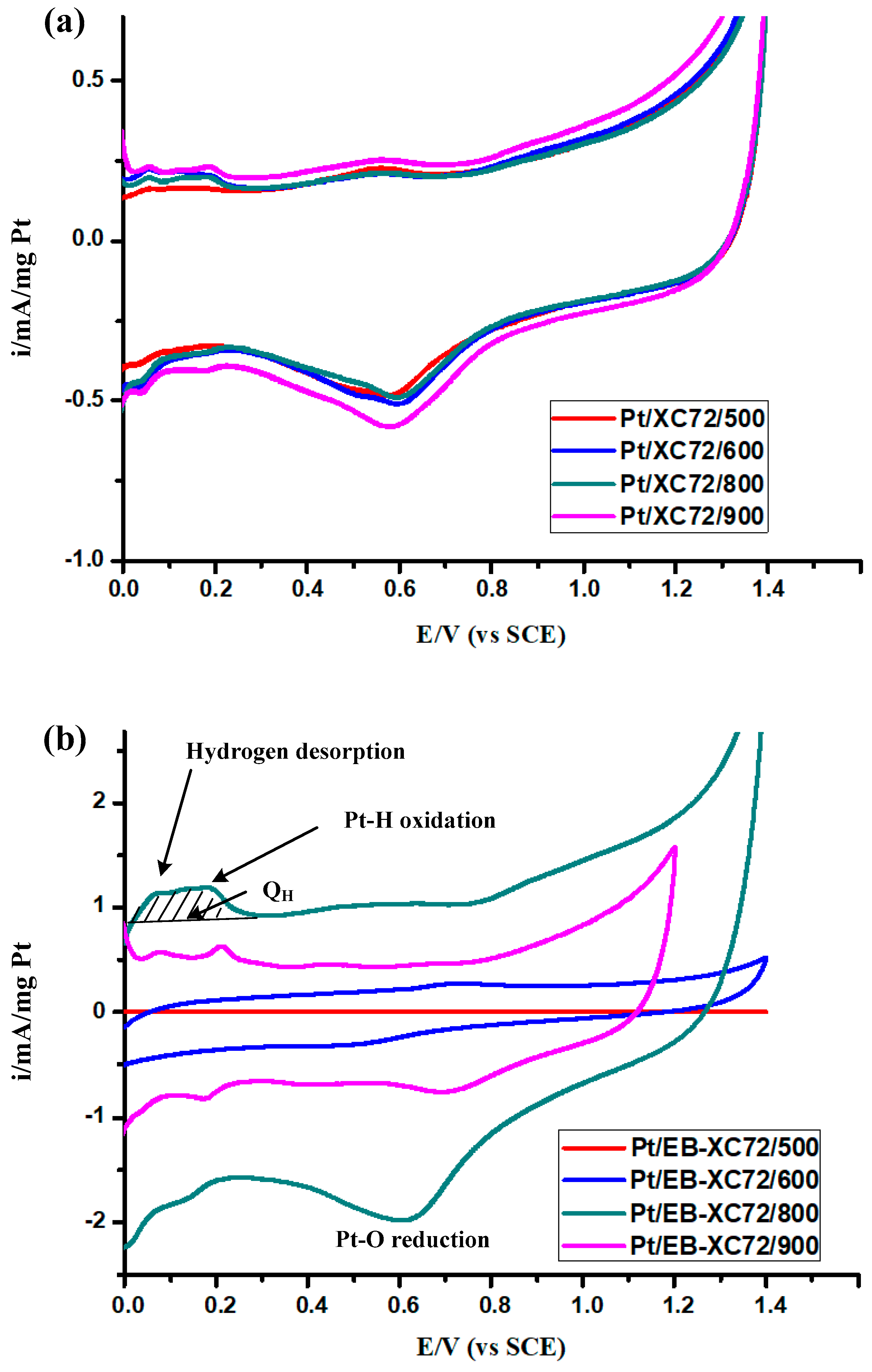
3.6.1. Cyclic Voltammetry (C-V)
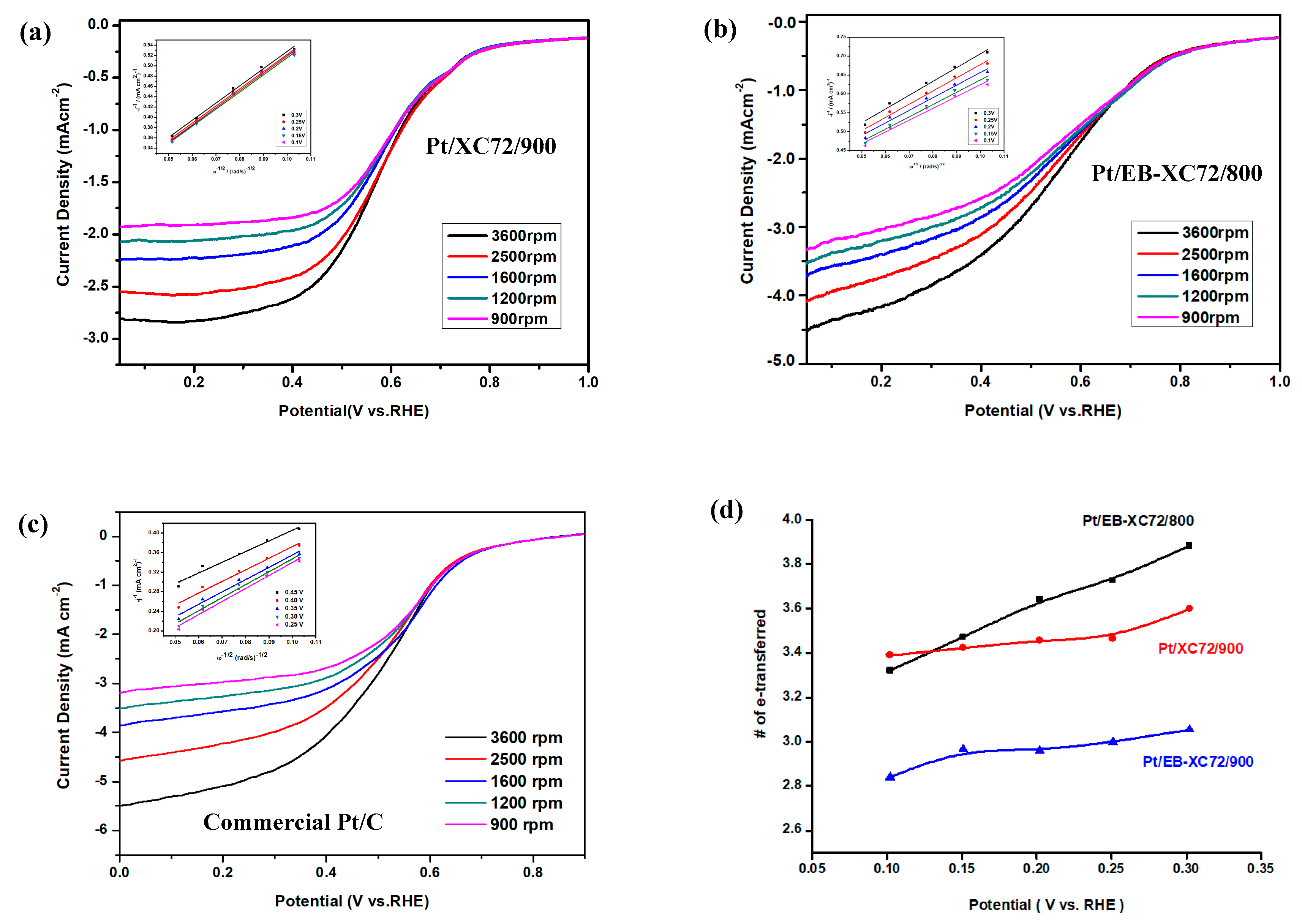
3.6.2. Linear Sweep Voltammetry (LSV) and Koutecky-Levich (K-L Plot)
3.6.3. Single Cell Performance Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brentner, L.B.; Peccia, J.; Zimmerman, J.B. Challenges in Developing Biohydrogen as a Sustainable Energy Source. Env. Sci. Tech. 2010, 44, 2243–2254. [Google Scholar] [CrossRef]
- Das, D. Advances in bio-hydrogen production processes: An approach towards commercialization. Inter. J. Hydrog. Energy 2009, 34, 7349–7357. [Google Scholar] [CrossRef]
- Logan, B.E. Biologically extracting energy from wastewater: Biohydrogen production and microbial fuel cells. Env. Sci. Tech. 2004, 38, 160–167. [Google Scholar] [CrossRef]
- Ho, L.Q.; Sugano, Y.; Yoshikawa, H.; Saito, M.; Tamiya, E. Structural assembly effects of Pt nanoparticle-carbon nanotube-polyaniline nanocomposites on the enhancement of biohydrogen fuel cell performance. Electrochim. Acta. 2011, 56, 9875–9882. [Google Scholar] [CrossRef]
- He, D.; Zeng, C.; Xu, C.; Cheng, N.; Li, H.; Mu, S.; Pan, M. Polyaniline-functionalized carbon nanotube supported platinum catalysts. Langmuir 2011, 27, 5582–5588. [Google Scholar] [CrossRef] [PubMed]
- Cindrella, L.; Kannan, A.M. Membrane electrode assembly with doped polyaniline interlayer for proton exchange membrane fuel cells under low relative humidity conditions. J. Power Sources 2009, 193, 447–453. [Google Scholar] [CrossRef]
- Michel, M.; Ettingshausen, F.; Scheiba, F.; Wolz, A.; Roth, C. Using layer-by-layer assembly of polyaniline fibers in the fast preparation of high performance fuel cell nanostructured membrane electrodes. Phys. Chem. Chem. Phys. 2008, 10, 3796–3801. [Google Scholar] [CrossRef]
- Kinoshita, K. Carbon: Electrochemical and Physicochemical Properties; Wiley: New York, NY, USA, 1988; pp. 5–10. [Google Scholar]
- Kinoshita, K.; Bett, J.A.S. Potentiodynamic analysis of surface oxides on carbon blacks. Carbon 1973, 11, 403–411. [Google Scholar] [CrossRef]
- Pyun, S.I.; Lee, E.J.; Kim, T.Y.; Lee, S.J.; Ryu, Y.G.; Kim, C.S. Role of surface oxides in corrosion of carbon black in phosphoric acid solution at elevated temperature. Carbon 1994, 32, 155–159. [Google Scholar]
- Liu, G.; Yang, Z.; Halim, M.; Li, X.; Wang, M.; Mei, Q.; Wang, X.; Lee, J.K. A gradient activation method for direct methanol fuel cells. Energy Convers. Manag. 2017, 138, 54–60. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, K.Y.; Kim, H.J.; Cho, E.A.; Lee, S.Y.; Lim, T.H.; Yoon, S.P.; Hwang, I.C.; Jang, J.H. The effects of Nafion® ionomer content in PEMFC MEAs prepared by a catalyst-coated membrane (CCM) spraying method. Int. J. Hydrog. Energy 2010, 35, 2119–2126. [Google Scholar] [CrossRef]
- Gavrilov, N.; Dašić-Tomić, M.; Pašti, I.; Ćirić-Marjanović, G.S. Carbonized polyaniline nanotubes/nanosheets-supported Pt nanoparticles: Synthesis, characterization and electrocatalysis. Mater. Lett. 2011, 65, 962–965. [Google Scholar] [CrossRef]
- Higgins, D.C.; Meza, D.; Chen, Z. Nitrogen-doped carbon nanotubes as platinum catalyst supports for oxygen reduction reaction in proton exchange membrane fuel cells. J. Phys. Chem. C. 2010, 114, 21982–21988. [Google Scholar] [CrossRef]
- Gong, K.; Du, F.; Xia, Z.; Durstock, M.; Dai, L. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 2009, 323, 760–764. [Google Scholar] [CrossRef]
- Khomenko, V.G.; Barsukov, V.Z.; Katashinskii, A.S. The catalytic activity of conducting polymers toward oxygen reduction. Electrochim. Acta 2005, 50, 1675–1683. [Google Scholar] [CrossRef]
- Chen, Z.; Higgins, D.; Chen, Z. Electrocatalystic activity of nitrogen doped carbon nanotubes with different morphologies for oxygen reduction reaction. Electro. Acta 2010, 55, 4799–4804. [Google Scholar] [CrossRef]
- Chen, Z.; Higgins, D.; Tao, H.; Hsu, R.S.; Chen, Z. Highly active nitrogen-doped carbon nanotubes for oxygen reduction reaction reaction in fuel cell applications. J. Phys. Chem. C 2009, 113, 21008–21013. [Google Scholar] [CrossRef]
- Qu, B.; Xu, Y.; Deng, Y.; Peng, X.; Chen, J.; Dai, L. Polyaniline/carbon black composite as Pt electrocatalyst supports for methanol oxidation: Synthesis and characterization. J. Appl. Polym. Sci. 2010, 118, 2034–2042. [Google Scholar] [CrossRef]
- Gharibi, H.; Kakaei, K.; Zhiani, M. Platinum Nanoparticles Supported by a Vulcan XC-72 and PANI Doped with Trifluoromethane Sulfonic Acid Substrate as a New Electrocatalyst for Direct Methanol Fuel Cells. J. Phys. Chem. C 2010, 114, 5233–5240. [Google Scholar] [CrossRef]
- Kakaei, K. Electrochemical Characteristics and Performance of Platinum Nanoparticles Supported by Vulcan/Polyaniline for Oxygen Reduction in PEMFC. Fuel Cell 2012, 12, 939–945. [Google Scholar] [CrossRef]
- Li, P.; Ding, X.; Yang, Z.; Chen, M.; Wang, M.; Wang, X. Electrochemical synthesis and characterization of polyaniline-coated PEMFC metal bipolar plates with improved corrosion resistance. Ionics 2018, 24, 1129–1137. [Google Scholar] [CrossRef]
- Tsai, M.J.; Hsieh, T.H.; Wang, Y.; Ho, K.S.; Chang, C. Microwave Assisted Reduction of Pt-Catalyst by N-Phenyl-p-Phenylenediamine for Proton Exchange Membrane Fuel Cells. Polymers 2017, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ko, T.H.; Huang, W.; Hsieh, T.H.; Ho, K.S.; Chen, Y.; Hsieh, S.J. Preparation of Pt-catalyst by poly (p-phenylenediamine) nanocomposites assisted by microwave radiation for proton exchange membrane fuel cell. Polymers 2018, 10, 1388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tsai, M.J.; Hsieh, T.H.; Tseng, P.H.; Ho, K.S. Studies on the one-dimensional polyanilines prepared with n-dodecylbenzenesulfonic and camphorsulfonic acids. Polym. Inter. 2015, 64, 1568–1577. [Google Scholar] [CrossRef]
- Wu, R.; Tsai, M.J.; Ho, K.S.; Wei, T.; Hsieh, T.H.; Han, Y.; Kuo, C.; Tseng, P.H.; Wang, Y. Sulfonated Polyaniline Nanofiber as Pt-Catalyst Conducting Support for Proton Exchange Membrane Fuel Cell. Polymer 2014, 55, 2035–2043. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, K.; Hung, L.F.; Ho, K.S.; Chen, J.; Hsieh, T.H.; Chao, L. Carboxylated Carbonized Polyaniline nanofibers as Pt-Catalyst Conducting Support for Proton Exchange Membrane Fuel Cell. Synth. Met. 2014, 188, 21–29. [Google Scholar] [CrossRef]
- Ho, K.S.; Han, Y.; Tuan, Y.; Huang, Y.; Wang, Y.; Ho, H.H.; Hsieh, T.H.; Lin, J.; Lin, S. Formation and Degradation Mechanism of a Novel Nanofibrous polyaniline. Synth. Met. 2009, 159, 1202–1209. [Google Scholar] [CrossRef]
- Hubert, A.; Gasteiger, S.S.; Kocha, B.S.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar]
- Qiu, L.; Liu, B.; Peng, Y.; Yan, F. Fabrication of ionic liquid-functionalized polypyrrole nanotubes decorated with platinum nanoparticles and their electrocatalytic oxidation of methanol. Chem. Commun. 2011, 47, 2934–2936. [Google Scholar] [CrossRef]
- Kong, L.B.; Li, H.; Zhang, J.; Luo, Y.C.; Kang, L. Platinum catalyst on ordered mesoporous carbon with controlled morphology for methanol electrochemical oxidation. Appl. Surf. Sci. 2010, 256, 6688–6693. [Google Scholar] [CrossRef]
- Shinozaki, K.; Zack, J.W.; Richards, R.M.; Pivovar, R.S.; Kocha, S.S. Oxygen Reduction Reaction Measurements on Platinum Electrocatalysts Utilizing Rotating Disk Electrode Technique I Impact of Impurities, measurement Protocols and Applied Corrections. J. Electrochem. Soc. 2015, 162, 1144–1158. [Google Scholar] [CrossRef]
- Do, C.L.; Pham, T.S.; Nguyen, N.P.; Tran, V.Q. Properties of Pt/C nanoparticle catalysts synthesized by electroless deposition for proton exchange membrane fuel cell. Adv. Nat. Sci. Nanosci. Nanotechnol. 2013, 4, 035011. [Google Scholar] [CrossRef]
- Yeager, E. Dioxygen electrocatalysis: Mechanisms in relation to catalyst structure. J. Mol. Catal. 1986, 38, 5–25. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
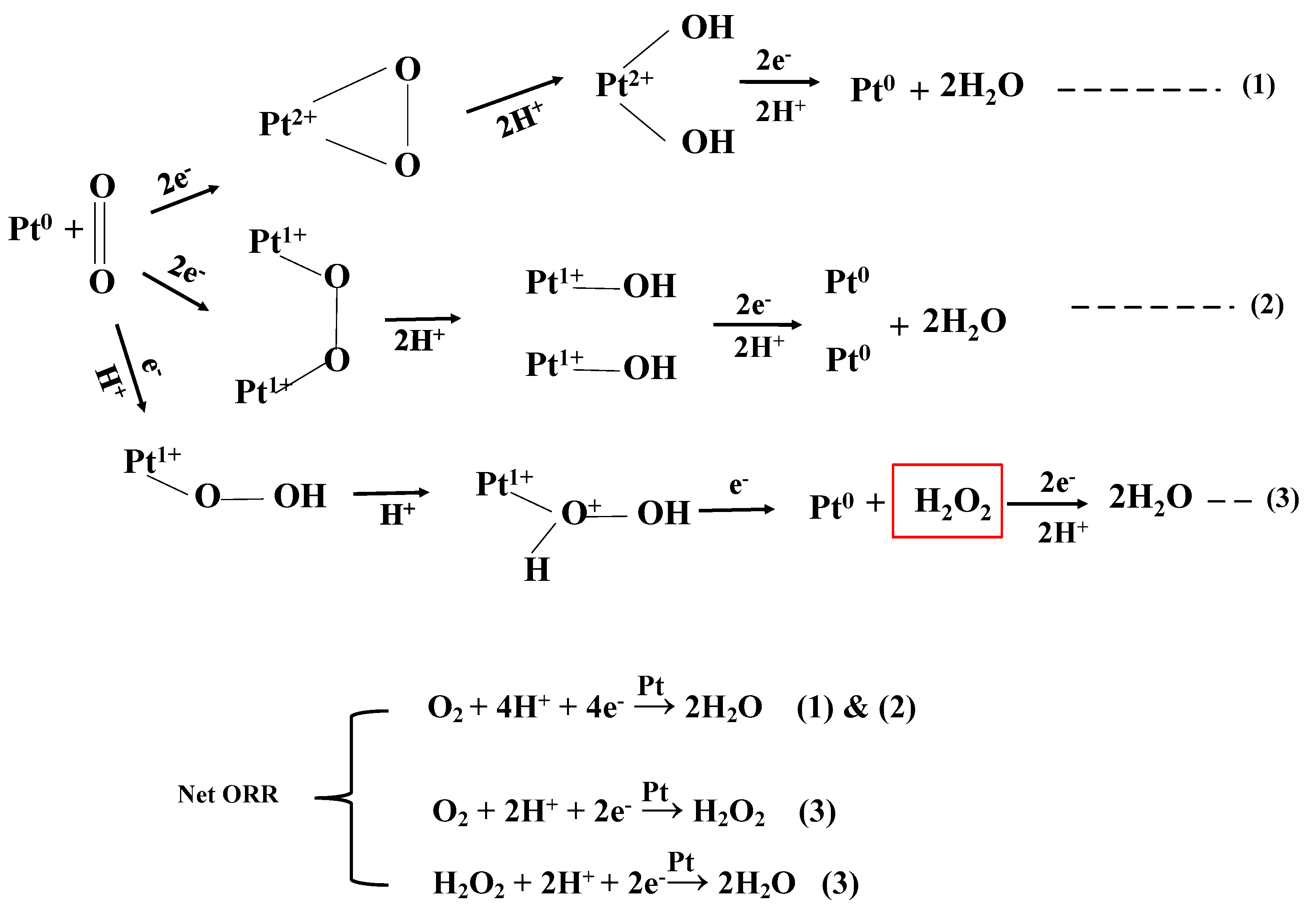{kind=link}
| Substrate | XC72 | EB-XC72 | |
|---|---|---|---|
| Temperature (°C) | |||
| 500 | 3.01 | - | |
| 600 | 4.42 | - | |
| 800 | 4.86 | 1.81 | |
| 900 | 5.20 | 3.86 | |
| 500 °C | 600 °C | 800 °C | 900 °C | |
|---|---|---|---|---|
| Pt/EB-XC72 | 0.93 | 0.95 | 1.01 | 1.06 |
| Pt/XC72 | 0.90 | 0.90 | 0.91 | 0.95 |
| T (°C) | 500 | 800 | 900 | |||
|---|---|---|---|---|---|---|
| Element | Pt/XC72 | Pt/EB-XC72 | Pt/XC72 | Pt/EB-XC72 | Pt/EB-XC72 | |
| C | 99.23 | 82.75 | 97.04 | 86.32 | 86.25 | |
| N | 0.00 | 15.31 | 0.00 | 10.72 | 5.11 | |
| O | 0.00 | 0.00 | 1.93 | 0.00 | 4.96 | |
| S | 0.28 | 0.54 | 0.40 | 0.51 | 0.34 | |
| Pt | 0.49 | 1.40 | 0.63 | 2.45 | 3.34 | |
| ESA a (cm2) | Pt-Weight b (%) | ECS c (m2 g−1Pt) | Pt Surface Area Per Unit Support d (cm2 mg−1) | No of E-transferred e | |
|---|---|---|---|---|---|
| t/EB-XC72/800 | 647 | 22.2 | 29.14 | 798.17 | 3.88 |
| Pt/EB-XC72/900 | 74 | 36.6 | 2.02 | 41.83 | 3.05 |
| Pt/XC72/900 | 2 | 8.7 | 2.29 | 88.55 | 3.58 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.-Y.; Chang, M.-Y.; Wang, Y.-Z.; Huang, Y.-C.; Ho, K.-S.; Hsieh, T.-H.; Kuo, Y.-C. Polyaniline Based Pt-Electrocatalyst for a Proton Exchanged Membrane Fuel Cell. Polymers 2020, 12, 617. https://doi.org/10.3390/polym12030617
Huang W-Y, Chang M-Y, Wang Y-Z, Huang Y-C, Ho K-S, Hsieh T-H, Kuo Y-C. Polyaniline Based Pt-Electrocatalyst for a Proton Exchanged Membrane Fuel Cell. Polymers. 2020; 12(3):617. https://doi.org/10.3390/polym12030617
Chicago/Turabian StyleHuang, Wen-Yao, Mei-Ying Chang, Yen-Zen Wang, Yu-Chang Huang, Ko-Shan Ho, Tar-Hwa Hsieh, and Yu-Chun Kuo. 2020. "Polyaniline Based Pt-Electrocatalyst for a Proton Exchanged Membrane Fuel Cell" Polymers 12, no. 3: 617. https://doi.org/10.3390/polym12030617
APA StyleHuang, W.-Y., Chang, M.-Y., Wang, Y.-Z., Huang, Y.-C., Ho, K.-S., Hsieh, T.-H., & Kuo, Y.-C. (2020). Polyaniline Based Pt-Electrocatalyst for a Proton Exchanged Membrane Fuel Cell. Polymers, 12(3), 617. https://doi.org/10.3390/polym12030617